

1. Introduction
β-Lactam or azetidin-2-one is an important structural motif of the penicillin, cephalosporin, carbapenem and carbecephem classes of antibiotics.1 Naturally occurring as well as synthetic monobactams, such as nocardicins and tabtoxin, are also known for their unique antibacterial activities.2–4 Besides their importance as the key structural component of β-lactam antibiotics, β-lactams have been attracting considerable interest in organic synthesis as versatile synthetic intermediates and chiral synthons.5–13 In addition, the β-lactam scaffold has found new pharmaceutical applications other than its use as antibiotics, such as LHRH antagonists,14 cholesterol-absorption inhibitors,15 and anticancer agents.16–19 The ring strain of the β-lactam skeleton facilitates ring-opening reactions,8,20,21,22 and this unique property has been exploited for the synthesis of a variety of medicinally active compounds.
For the last couple of decades, a large number of β-lactam-based synthetic methods, collectively termed as “β-lactam-synthon method”, has been developed. This method has provided highly efficient routes to a variety of non-protein amino acids, oligopeptides, peptidomimetics and nitrogen-heterocycles, as well as biologically active natural and unnatural products of medicinal interest, such as indolizidine alkaloids, paclitaxel, docetaxel, taxoids, cyptophycins, lankacidins, etc.5,7–13
In this report, we present an overview of the evolution of the methods for the synthesis of enantiopure β-lactams, and the applications of the “β-lactam synthon method” for the synthesis of biologically active compounds of medicinal interest. Examples of the use of the rigid β-lactam scaffold for drug design and discovery are also described.
2. Asymmetric synthesis of β-lactams
The Staudinger ketene-imine [2+2] cycloaddition and the chiral ester enolate-imine cyclocondensation are the two methods which are most commonly used for the synthesis of β-lactams with excellent enantiopurity. Thus, these two synthetic methods are discussed in this section.
2.1 Staudinger ketene-imine [2+2] cycloaddition
In 1907, long before the antibacterial activity of penicillin was discovered, Staudinger reported the first synthesis of a β-lactam, 1,3,3,4-tetraphenylazetidin-2-one, through the [2+2] cycloaddition of diphenylketene with a Schiff base derived from aniline and benzaldehyde.23 Subsequently, the scope of this reaction has been extended to alkyl-, amino-, halo-, alkoxy- and siloxy-ketenes, as well as imino esters. Because of its broad scope in substrate structures and simplicity in experimental procedure, this reaction is regarded as one of the most reliable routes to β-lactams.1 However, in spite of common practice in laboratory synthesis for decades, the mechanistic details of this reaction have been disputed and subjected to theoretical investigations since its inception.24,25 The most widely accepted mechanism is a two-step reaction process, which involves the nucleophilic attack of the imine nitrogen on the electrophilic central carbon of a ketene, generated in situ from an acid chloride and a base, to form a zwitterionic intermediate, followed by conrotatory ring closure to give the 4-membered ring system (Figure 1).24,25
Figure 1.

Mechanism of Staudinger [2+2] ketene-imine cycloaddition
In the reaction of a monosubstituted ketene with an aldimine, two chiral centers are introduced to the cycloadduct. Thus, the reaction would give either a single stereoisomer (i.e. cis- or trans-) of β-lactam or a mixture of cis- and trans-β-lactams, depending on the reactants and reaction conditions.25,24 As Figure 1 shows, cis-β-lactam should be formed from the zwitterionic intermediate bearing the (E)-imine moiety, but if isomerization to the zwitterionic intermediate bearing the (Z)-imine moiety takes place, the reaction should give trans-β-lactam.26–29 This mechanism also indicates that the reaction with a (Z)-imine would give the corresponding trans-β-lactam selectively. Theoretical studies on the origin of the stereoselectivity suggest that the relative transition-state energy in the rate-determining step is dictated by electronic torque selectivity.30,31
The stereoselectivity of the reaction is influenced by reaction variables such as temperature, solvent, base, additives, chiral pendant groups, order of addition of reagents, microwave irradiation, etc.32–34 For example, it has been shown that non-polar solvents favor the formation of cis-β-lactam whereas polar solvents facilitate trans-β-lactam formation. The result was explained by the stabilization of the zwitterionic intermediate in a polar solvent, promoting isomerization to the energetically more stable intermediate bearing the (Z)-imine moiety before conrotatory ring closure.32 Asymmetric ketene-imine [2+2] cycloaddition can be performed using conbinations of (i) a chiral imine with an achiral ketene, (ii) a chiral ketene with an achiral amine or (iii) a chiral ketene with a chiral imine.
2.1.1 Asymmetric [2 + 2] cycloaddition of achiral ketenes and chiral imines
Chiral imines can be prepared either from chiral amines or from aldehydes. However, the diastereoselectivity of the reaction is lower, in most cases, using a chiral imine from a chiral amine, as compared to that from a chiral aldehyde.35 Nevertheless, chiral amine sources are widely used for the asymmetric synthesis of β-lactams.9,36 For example, the reaction of phthalimidoacetyl chloride with chiral imine 1, derived from (R)-1-phenylethylamine, in the presence of triethylamine gave β-lactams 2 and 3 in 74% combined yield with high stereoselectivity (2:3 = 9:1) (Scheme 1).36
Scheme 1.

Commercially available enantiopure esters of α-amino acids are common sources of chiral amines. For example, 3-azido-β-lactam 5a (R = triphenylsilyl = TPS) was obtained in 64% yield and 19:1 dr (5a:6a) using imine 4a derived from (R)-O-TPS-threonine benzyl ester and cinnamaldehyde (Scheme 2).37 The stereoselectivity of this reaction was found to depend critically on the bulkiness of the hydroxyl protecting group. Thus, the reaction of 4b (R = t-butyldimethylsilyl = TBS) gave 5b with 9:1 dr (5b:6b) and that of 4c (R = H) afforded a 1:1 mixture of 5c and 6c.37
Scheme 2.

Alternatively, one of the most effective routes for a large-scale synthesis of β-lactams with high enantiopurity is to use chiral imines derived from chiral aldehydes such as α-oxyaldehydes and sugar-derived aldehydes (Scheme 3).35,38–40 For example, the [2+2] cycloaddition of various ketenes with chiral imines 9 of D-glyceraldehyde acetonide (8), derived from D-mannitol via 7, afforded cis-β-lactams 10 exclusively in 45–70% yield.41,42 The acetonide moiety of β-lactams 10 was deprotected to the corresponding β-lactam diols, which were oxidized by ruthenium tetroxide, followed by diazomethane esterification, to afford the 4-carbomethoxy-β-lactams.41 The β-lactam diols was also converted into the corresponding 4-formyl-β-lactam 11, which are versatile synthetic intermediates for further manipulations. 13,42–44
Scheme 3.

When a chiral aldimine derived from a chiral aldehyde as well as a chiral amine was used, clear double asymmetric induction was observed, i.e., a matching pair gave the corresponding β-lactam as a single stereoisomer, while a mismatching pair afforded the β-lactam with only modest stereoselectivity. As Scheme 4 illustrates, the reaction of chiral aldimine 13, derived from (R)-threonine and chiral aldehyde 8 via 12, with a ketene, generated in situ from diphenyloxazolylacetyl chloride and triethylamine, gave a 1:2.9 mixture of β-lactams 14 and 15 (i.e. mismatching pair reaction), while the reaction using chiral aldimine 16, derived from (S)-threonine and 8, afforded β-lactam 17 exclusively in 69% yield for two steps (i.e. matching pair reaction).45
Scheme 4.

2.1.2 [2 + 2] Cycloaddition of chiral ketenes and achiral imines
The ketene–imine [2 + 2] cycloaddition using chiral ketenes has been very successful for the asymmetric synthesis of 3-amino-β-lactams. Enantiopure oxazolidinones derived from (S)- and (R)-phenylglycine are excellent chiral auxiliaries for this reaction and have been extensively utilized.8,9,46 For example, (S)-4-phenyloxazolidinon-3-ylacetyl chloride (18) with N-benzylaldimines 20 gave β-lactams 21 with 95:5~97:3 dr in 80~90% yield (Scheme 5).46 This methodology has been successfully applied to the solid-phase synthesis of a library of β-lactams.47,48 Even a fused nitrogen aromatic compound, phenanthridine (22) was found to react with ketene 19 to afford fused tetracyclic trans-β-lactam 23 exclusively in 53% yield, wherein 22 apparently reacted as a (Z)-imine (Scheme 5).49
Scheme 5.

It has been shown that the [2+2] cycloaddition of polyaromatic imines and ketenes, generated from acetoxy, phenoxy, and phthalimidoacetyl chloride and an amine base, exclusively gives trans-β-lactams, which makes a sharp contract to the normal reaction pattern, yielding cis-β-lactams from the same ketene sources and diarylaldimines.16,17 In a similar manner, the reaction of a chiral ketene, generated from alkoxyacetic acid 24 bearing an α-glycoside group, with chresenylaldimine 25 gave a 45:55 mixture of 26 and 27 in 70% combined yield (Scheme 6). These two β-lactams were readily separated by column chromatography and the hydrolysis of the sugar chiral auxiliary afforded the corresponding enantiopure trans-3-hydroxy-4-phenyl-β-lactams.18
Scheme 6.

2.1.3 Synthesis of enantiopure β-lactams via enzymatic kinetic resolution
Enzymatic kinetic optical resolution of racemic β-lactams provides a practical route to enantiopure β-lactams. This method has been successfully applied to the synthesis of 3-hydroxy-β-lactams required for the semisynthesis of paclitaxel, docetaxel and taxoids.43,50–55 Hydrolytic enzymes such as PS-Amano lipase and the esterase in pig liver acetone powder (PLAP) have been shown to give excellent results for the optical resolution of racemic cis-3-acetoxy-β-lactams. For example, the kinetic resolution of racemic cis-3-acetoxy-4-phenyl-β-lactam 29, obtained from 28, with PS Amano lipase afforded (3R,4S)-3-acetoxy-β-lactam 30 with >99.9% ee and (3S,4R)-3-hydroxy-β-lactam 31 with >99.0% ee in good yields (Scheme 7).54
Scheme 7.

This protocol has also been successfully employed for the kinetic optical resolution of cis-3-acetoxy-4-isobutenyl-β-lactam.43,50,55 Although kinetic optical resolution does not have good “atom economy”, it should be noted that the method is still quite economical and easily scalable. In addition, both enantiomers serve as useful chiral building blocks in organic synthesis, especially for the synthesis of non-protein amino acids, peptides and peptidomimetics.
2.1.4 Catalytic asymmetric reactions for β-lactam synthesis
A catalytic process applicable to the asymmetric [2+2] ketene-imine cycloaddition has been developed56. Cobalticenium tetracarbonyl cobaltate (32) contains both a nucleophilic Co(CO)4 anion and a Cp2Co cation that could act in a concerted manner. Thus, in the [2+2] cycloaddition of diphenylketene (34), generated from 33 and NaH, with N-tosylimino ester 36 via dicobalt species 35, dicobalt complex 32 showed a good catalyst activity, giving β-lactam 37 in up to 85% yield in 5 min at room temperature (Scheme 8) 57. It should be noted that, in this process, the electronic demand of the two reactants is reversed from that in the normal ketene-imine [2+2] cycloaddition process. Thus, in this process, the imine component is made highly electrophilic by incorporating an electron-withdrawing group into the imine nitrogen and a carbalkoxy substituent to the imine carbon. The electrophilic ketene component is transformed into a nucleophilic zwitterionic enolate through the addition of a nucleophilic catalyst to the central carbon of the ketene component. Then, the nucleophilic attack of the zwitterion on the α-carbon of the imine leads to the C-C bond formation, followed by cyclization to give β-lactam 37 and regenerates the catalyst 32. This reaction mechanism logically led to the realization of the fact that a catalyst for this reaction does not have to be a metal ion or charged and thus nucleophilic organocatalysts should be effective as well.58 It was also hypothesized and proven that an organocatalyst, containing a nucleophilic center (e.g. tertiary amine) as well as an electrophilic center (e.g. hydrogen-bonding acceptor), would rigidify the key intermediate and the transition state to yield β-lactams with higher diastereoselectivity than that without the electrophilic center.58
Scheme 8.

In order to accomplish the catalytic asymmetric synthesis of β-lactams based on this process, a number of optically active cinchona alkaloids were screened as potential chiral organocatalysts, and benzoylquinine (38) was found to give best results. Thus, the reaction of acid chlorides 39 with imine 36 in the presence of 38 as catalyst (10 mol%) and a proton sponge gave β-lactams 40 with 95–99% ee and 96:4–99:1 dr in 36–65% yield (Scheme 9).58
Scheme 9.

In order to improve the chemical yield, metal salts were explored as effective co-catalysts to further activate imines.59 Then, it was found that the use of triflates of Sc(III), Al(III), Zn(II) and In(III) (10 mol%) with 38 (10 mol%) gave β-lactams 40 in excellent yield and enantioselectivity (96–98% ee) as well as high diastereoselectivity (9:1–60:1 dr) (Scheme 10). Among the metal salts examined, In(OTf)3 gave the best results followed by Zn(OTf)2, while Al(OTf)3 and Sc(OTf)3 were less effective.59.
Scheme 10.

It has been shown that fused DMAP/ferrocene-based ”planar-chiral” nucleophilic catalysts, such as 41, are effective for the asymmetric synthesis of β-lactams through ketene-imine [2+2] cycloaddition. For example, the reaction of symmetrical ketenes 42 with electron-deficient N-tosylaldimines 43 catalyzed by 41 (0.1 mol%) gave β-lactams 44 in 76–93% yield and 81–94% ee, while the reaction of unsymmetrical ketenes 42 (R = Ph, R1= i-Bu or Et) with 43 under the same conditions afforded β-lactams 44 in 88–98% yield with 8:1–15:1 dr and 89–98% ee (Scheme 11).60
Scheme 11.

Recently, N-heterocyclic carbenes (NHCs) have been shown to serve as excellent catalysts for the asymmetric synthesis of β-lactams through ketene-imine [2+2] cycloaddition.61,62 For example, NHC 45a derived from (S)-pyroglutamic acid catalyzed the reactions of various arylketenes 46 with N-Boc-arylaldimines 47 to give the corresponding β-lactams 48 in moderate-to-good yields and excellent enantioselectivity61 (Scheme 12). Another NHC 45b was used as a catalyst for the reaction of arylketenes 49 with azodicarboxylate 50, which afforded aza-β-lactams 51 in fairly good-to-excellent yields and 33~91% ee (Scheme 12).62
Scheme 12.

C-2 symmetric imidazolinium catalyst 52 and chiral triazolium catalyst 53 have been used in the reaction of diphenylketene (34) with N-tosylaldimines 54 to give the corresponding β-lactams 55 in high yields and 55–75% ee (Scheme 13).63 It has also been shown that chiral imidazoliniumdithiocarboxylates, e.g. 56, act as efficient organocatalysts for Staudinger [2+2] cycloaddition reactions of arylketenes 57 with N-tosylaldimine 58, giving the corresponding β-lactams 59 as a mixture of E and Z isomers (59-E:59-Z = 25:75–11:89) in 96–99% yield with 83–96% ee for 59-Z (major) and 48–83% ee for 59-E (minor) (Scheme 14).64
Scheme 13.

Scheme 14.

The β-lactam synthesis through Rh-catalyzed carbene insertion into a C-H bond is another catalytic process that is applicable to catalytic asymmetric synthesis. The construction of the β-lactam ring based on the intramolecular C-H insertion of carbene species generated from α-diazo amides, initially by photoirradiation and then by Rh2(OAc)4 catalysis, was used for the synthesis of carbapenems, especially thienamycin.65,66 Then, the Rh2(OCOR)4-catalyzed process was successfully employed in the intramolecular C-H insertion reaction of α-diazoacetamides to yield the corresponding β-lactams. For example, the reaction of N-t-butyl-N-arylmethyl-α-diazoacetamides 60 catalyzed by Rh2(OAc)4 gave the trans-β-lactams 62 exclusively via a Rh-carbenoid species 61 in 90–98% yield (Scheme 15).67,68
Scheme 15.

The catalytic asymmetric synthesis of β-lactams through Rh-catalyzed intramolecular C-H insertion reactions of α-diazoacetamides has been investigated using dimeric Rh(II) complexes with chiral carboxylate ligands. For example, the reaction of α-methoxycarbonyl-α-diazoacetamides 63 catalyzed by Rh2(S-PTPA)4 (S-PTPA = N-naphthoylphenyl-(S)-phenylalaninate) afforded cis-β-lactams 64 in excellent yield and 56–74% ee (Scheme 16).69 This reaction system was successfully applied to the asymmetric synthesis of bicyclic β-lactams, which served as key intermediates for the synthesis of enantiopure carbapenems.70 Thus, the reaction of α-methoxycarbonyl-α-diazoacetamides 65 catalyzed by Rh2(N-naphthoyl-(S)-AA)4 (AA = Phe, Ala, Val, PhGly, t-BuGly) afforded β-lactams 66 in 85–94% yield and 83–96% ee (Scheme 16). The best result (94% yield and 96% ee) was obtained with α-diazoacetamide 65a (R, R =-(CH2)5-) and Rh2(PTA)4 (PTA = N-naphthoyl-Ala). In a similar manner, β-lactam 68 (88% ee, 83% yield), a key intermediate in the synthesis of 1β-methylthienamycin, was obtained by the reaction of diazoacetamide 67 catalyzed by Rh2(PTA)4, which was successfully converted into the enantiopure advanced key intermediate 69 in several steps (Scheme 16).70
Scheme 16.

Regio- and stereospecific carbonylation of chiral aziridines catalyzed by [Rh(CO)2Cl]2 gives the corresponding β-lactams in high yields. For example, the reactions of 70 and 72 afforded 71 and 73 in 81 and 89% yield, respectively (Scheme 17).71 This carbonylation process was found to proceed stereospecifically at the carbon bearing a phenyl substituent with complete retention of configuration. This process was successfully applied to the kinetic resolution of racemic 2-phenylaziridine 74 in the presence of (−)-menthol (3 equiv) and [Rh(COD)Cl]2 (5 mol%) as catalyst at 90 °C and 20 atm CO for 24 h to give (S)-β-lactam 75 with 99.5% ee (25% yield) and (R)-74 with 85% ee (56% yield) (Scheme 17).71 It is of interest to note that Co2(CO)8 was found to catalyze the regio- and stereospecific carbonylation of chiral aziridines as well, but with complete inversion of configuration at the more substituted carbon with alkyl or phenyl substituents.72 This process exhibited its exceptional stereochemical integrity in the formation of extremely strained trans-bicyclic β-lactams, 8-aza-7-oxo-[4.2.0]bicyclooctanes, from cis-cyclohexeneaziridines. 72 However, this process has not been applied to asymmetric synthesis.
Scheme 17.

2.2 Asymmetric synthesis of β-lactams through ester enolate-imine cyclocondensation
The chiral lithium enolates generated in situ from N,N-bis(silyl)glycinates 76 reacted with N-PMP-arylaldimines 77 to give trans-3-amino-β-lactams 78 exclusively in fairly good yields and excellent enantiopurity.73 Among the various chiral ester auxiliaries screened, (−)-menthyl and (−)-2-phenylcyclohexyl were found to be highly effective for this reaction. For example, the reaction of 76a (R* = (−)-menthyl) and 76b (R* = (−)-2-phenylcyclohexyl) with N-PMP-benzaldimine (77a) in the presence of LDA in THF at −78 °C afforded (3R,4R)-β-lactam 78a with >99% ee in 65 and 58% yield, respectively (Scheme 18).73 Under the same conditions, the reactions of 76a with other N-PMP-arylaldimines 77b-e (Ar = 4-FC6H4, 4-CF3C6H4, 4-MeOC6H4, 3,4-(MeO)2C6H3) gave the corresponding β-lactams 78b-e with >99% ee, albeit with small amounts of cis-β-lactams 78d,e being obtained in the case of 77d and 77e.73
Scheme 18.

It has also been shown that the chiral ester enolate-imine cyclcocondensation is a highly efficient method for the asymmetric synthesis of 3-hydroxy-β-lactams with excellent enantiopurity7,8. In contrast to the rigid bissilylamino moiety in glycinates 76, the O-protected hydroxyl group is very flexible in hydroxyacetates 79. Thus, both the chiral auxiliaries and the O-protecting groups were screened to find the optimal chiral ester enolate 80 for this process. Then, the combination of TIPS and (−)- or (+)-2-phenylcyclohexyl groups was found to achieve the best results. It should be noted that, in contrast to the reactions of 76, cis-β-lactams 82 were formed exclusively in this process. Chiral enolate 80 was generated from (−)-2-phenylcyclohexyl TIPSO-acetate 79 with LDA in THF at −78 °C for 2 h. N-TMS-arylaldimines 81 (R2 = TMS) were added to the enolate solution and reacted at −78 °C and then gradually warmed to room temperature to give (3R,4S)-3-TIPSO-4-aryl-β-lactams 82 with 96–98% ee in 80–85% yields and 96–98% ee (Scheme 19).74,75 It is worthy of note that (3R,4S)-3-TIPSO-4-phenyl-β-lactam 82a (Table 1) serves as a key intermediate for the highly efficient semisynthesis of paclitaxel and docetaxel (vide infra).74,76
Scheme 19.

Table 1.
Asymmetric synthesis of 3-TIPSO-β-lactams 82 through chiral ester enolate-imine cyclocondensation.
| Entry | R1 | R2 | R3 | β-Lactam | Yield (%) | ee (%) |
|---|---|---|---|---|---|---|
| 1 | Ph | TMS | H | 82a | 85 | 96 |
| 2 | 4-MeOC6H4 | TMS | H | 82b | 80 | 96 |
| 3 | 3,4-(MeO)2C6H3 | TMS | H | 82c | 80 | 98 |
| 4 | Ph | PMP | PMP | 82d | 89 | 98 |
| 5 | 4-FC6H4 | PMP | PMP | 82e | 81 | 98 |
| 6 | 4-CF3C6H4 | PMP | PMP | 82f | 84 | 99 |
| 7 | 2-furyl | PMP | PMP | 82g | 78 | 92 |
| 8 | (E)-PhCH=CH2 | PMP | PMP | 82h | 85 | 96 |
| 9 | 2-furyl-CH=CH2 | PMP | PMP | 82i | 72 | 94 |
| 10 | c-C6H11CH2 | PMP | PMP | 82j | 85 | 90 |
| 11 | Me2CHCH2 | PMP | PMP | 82k | 85 | 92 |
| 12 | Me2C=CH | PMP | PMP | 82l | 60 | 94 |
Although N-TMS-imines 81 (R2 = TMS) are efficient to produce β-lactams 82 (R3 = H) with free NH directly, those imines are limited to arylaldimines due to the instability of N-TMS-alkylaldimines. Thus, N-PMP-imines 81 (R2 = PMP) were employed to successfully expand the scope of this reaction. Table 1 summarizes representative results, including those for N-TMS-imines 81 (R2 = TMS).74,75,77
An asymmetric ester-enolate cyclocondensation process mediated by a ternary chiral ether ligand–lithium amide complex was developed. The reaction of lithium enolate 83, generated from the corresponding 3-pentyl alkanoate (2 equiv) and LDA (2.2 equiv), with N-PMP-aldimine 84 in the presence of (1S,2S)-1,2-dimethoxy-1,2-diphenylethane (86) (2.6 equiv) and additional lithium amide, e.g. LiN(Pr-i)Cy and LiNCy2, (2.2 equiv) gave β-lactam 85 in 12–85% yield and 26–90% ee (Scheme 20).78 The yield and enantioselectivity depend on the structure of the lithium amide used. For example, the reaction of 83a (R1 = Me) with 84a (R2 = Ph) using LiN(Pr-i)Cy afforded 85a in 85% yield and 88% ee, while the same reaction using LiN(Bu-t)Cy gave 85a in 22% yield and 55% ee.
Scheme 20.

3. Synthesis of amino acids, dipeptides and their derivatives by means of β-lactam synthon method
3.1 Asymmetric synthesis of non-protein amino acids
Non-protein amino acids are amino acids which are not produced from protein amino acids by post-translational modification and these amino acids do not have a specific transfer-RNA and codon triplet, and thus the main chains of proteins do not have these amino acid residues.79 Since various non-protein amino acids are important components of medicinally important compounds, the efficient synthesis of non-protein amino acids with high enantiopurity has significance in medicinal chemistry and organic synthesis.
3.1.1 Synthesis of α,β-diamino acids, azetidines and polyamines
α,β-Diamino acids are often components of peptidic antibiotics such as lavendomycin and glumamycin.80 It has been shown that α,β-diamino acids are readily obtained through the hydrolysis of enantiopure 3-amino-β-lactams, which were prepared by chiral ester-enolate cyclocondensation or asymmetric [2+2] ketene-imine cycloaddition reactions (vide supra).7,9 Thus, the hydrolysis of 3-amino-β-lactam (3S,4R)-87 and its epimer, (3R,4R)-87, using 6 M hydrochloric acid gave the corresponding α,β-diamino acids (2S,3R)-88 and (2R,3R)-88, respectively, as hydrochlorides in quantitative yields (Scheme 21). The epimerization of (3S,4R)-87 to (3R,4R)-87 was readily realized through imine formation, deprotonation and protonation.7 Moreover, (3R,4S)-87 can be prepared just by switching the chiral auxiliary to the other enantiomer in the asymmetric ketene–imine [2 + 2] cycloaddition or from the other enantiomer obtained in the enzymatic optical resolution (vide supra). Thus, (3S,4S)-87, (2R,2S)-88 and (2S,2S)-88 can be prepared in the same manner. Furthermore, α,β-diamino acids 88 were reduced to the corresponding α,β-diamino alcohols 89 in high yields using LiAlH4 (Scheme 21). Thus, this protocol provides facile access to all four diastereoisomers of α,β-diamino acids 88 and amino alcohols 89.7,8
Scheme 21.

Azetidine is a very challenging amine to synthesize, but the selective reduction of β-lactams using monochloroalane (AlH2Cl) or dichloroalane (AlHCl2) provides one of the most straightforward and efficient routes to enantiopure azetidines.75,81 The reduction of enantiopure 4-aryl-β-lactam 90 with monochloroalane (AlH2Cl) in ether gave the corresponding 2-arylaziridine 91 in high yield without epimerization. Then, the hydrogenolysis of 91 at the N-C2 bond afforded the enantiopure diamine or amino alcohol 92 in quantitative yield (Scheme 22).75 In the same manner, the monochloroalane reduction of tetrapeptide synthon bis-β-lactam 93 gave bisazetidine 94 in moderate yield, which was converted into polyamino ether 95 in quantitative yield via hydrogenolysis. It should be noted that the reductive cleavage of the two azetidine moieties of 94 were much faster than debenzylation in this case (Scheme 22).9,82
Scheme 22.

3.1.2 Synthesis of α-alkyl-α-amino acids and their dipeptides
Various α-alkyl-α-amino acids are known to act as potent substrate-based inhibitors of decarboxylases and aminotransferases.80 In addition, the strategic incorporation of α-alkyl-α-amino acid residues into physiologically active peptides can introduce conformational constraints to stabilize 3D peptide structures.80 The access to enantiopure α-alkyl-α-amino acids is not easy because conventional enzymatic resolution cannot be applied effectively. Fortunately, the β-lactam synthon method provides highly efficient routes to α-alkyl-α-amino acids with high enantiopurity through extremely stereoselective alkylation of 3-amino-β-lactams.
Two protocols have been developed for asymmetric alkylations, i.e. (i) the alkylation of the C-3 position of a β-lactam (Type 1) and (ii) the alkylation of the side chain ester enolate (Type 2) (Figure 2).7,9 As Figure 2 illustrates, in the Type 1 alkylation, an electrophile attacks the β-lactam enolate at the C-3 position from the back side of the C-4 aryl group, while, in the Type 2 alkylation, an electrophile attacks the C1′ position of the lithium enolate, forming a chelate with the β-lactam oxygen, from the opposite side of the C-4 aryl group.
Figure 2.

Type 1 and Type 2 asymmetric alkylations
A typical example of the Type 1 alkylation is shown in Scheme 23. The stereoselective methylation of β-lactam 96 (X = H or MeO) with MeI and LiHMDS gave 3-methyl-β-lactam 97 with >99.5% de in excellent yield. Birch reduction of 97 gave the corresponding amide of α-methyl-α-amino acid 98 (>99.5% ee) in high yield.7,83 Acidic hydrolysis of 98a (X = H) and 98b (X = MeO) should afford (S)-α-methylphenylalanine and (S)-O,O-dimethyl-α-methyldopa with > 99.5% ee, respectively. It should be noted, however, that similar Type 1 alkylations of a bicyclic trans-β-lactam 99, which did not have a 4-aryl group, gave 3-alkyl-β-lactams 100 with >97% de. in 51–80% yields, wherein the electrophiles attacked the C3 position of the β-lactam moiety of 99 from the front face with retention of configuration (Scheme 23).84
Scheme 23.

The Type 2 alkylation was first examined with enantiopure β-lactam ester 101 with 1.0 equivalent of LDA and alkyl halides (BnBr and EtBr). As Scheme 24 shows, deprotonation took place exclusively at C1′ of the ester moiety, leaving C3 of the β-lactam moiety intact. Thus, the alkylation proceeded at C1′ exclusively to give 102 (>98% de), which was subjected to hydrogenolysis on Pd/C to give N-acyl-α-alkyl-α-amino acid ester 103 and subsequent acidic hydrolysis afforded α-alkyl-(R)-alanine 104 (>98% ee) in excellent yield (Scheme 24).7,9,83 The results indicated that the Type 2 alkylation process should be applicable for the asymmetric synthesis of dipeptides or depsipeptide fragments bearing an α-alkyl-α-amino acid residue at the C-terminus.
Scheme 24.

The Type 2 alkylation was successfully applied to the sequential asymmetric double alkylation of chiral β-lactam ester 105 (>99% de, >99% ee) to give phenylalanyl-α-alkylalanines, (S,S)-108 (via (R)-106 and (S)-107) and (S,R)-108 (via (S)-106 and (R)-107) in good overall yields and >99% de, and . In this process, the double alkylation took place exclusively at C1′ and the absolute configuration of C1′ was controlled by changing the order of addition of the two alkyl halides (Scheme 25).9,85
Scheme 25.

The highly efficient triple alkylation of 105 was also achieved through sequential Type 2 double alkylation and Type 1 alkylations (Scheme 26)9,85 For example, sequential double alkylation of β-lactam 105 with methyl iodide and allyl bromide proceeded in the same manner as that in Scheme 25, giving 107b (>99% de), which was followed by the third (Type 1) alkylation with methyl iodide to afford 109 (>99% de) uneventfully in 80% yield for 3 steps. Then, triply alkylated β-lactam 109 was converted into (S)-α-methylphenylalanyl-(R)-α-allylalanine, 110 in 62% yield through deprotection, Birch reduction and separation on a Dowex column (Scheme 26).
Scheme 26.

3.1.3 Asymmetric synthesis of α-hydroxy-β-amino acids and their dipeptides
α-Hydroxy-β-amino acids (isoserines) are key structural motifs in numerous therapeutically important substances. (2R,3S)-3-Amino-2-hydroxy-5-methylhexanoic acid (norstatine) and its analogues, for example, are readily found in peptide-based inhibitors of enzymes such as renin,86,87 HIV-I protease88 and angiotensin-converting enzyme (ACE),89 as well as in a variety of natural products.90 (2R,3S)-3-Phenylisoserines are indispensable constituents of paclitaxel and docetaxel, which are two of the most widely used anticancer drugs in chemotherapy.91,92 Therefore, extensive efforts have been made to develop efficient methods for the syntheses of isoserines with excellent enantiopurity. The β-lactam synthon method has made a significant contribution to solve this synthetic challenge.
Acidic hydrolysis of 3-TIPSO-β-lactams 112 with excellent enantiopurity, obtained from 1-PMP-β-lactam 111, provides a ready access to norstatine (113a, R = isobutyl) and isoserines 113 (Scheme 27).7,93 The ring opening of β-lactams at the N1-C2 bond is substantially accelerated when an electron-withdrawing group, e.g. acyl, carbalkoxy, carbamoyl, sulfonyl, etc., is introduced into the N1 position. For example, the ring-opening coupling of N-Boc-3-hydroxy-β-lactams 114 with various amino acid esters 115 proceeded smoothly to give the corresponding dipeptides 116, bearing an isoserine residue at the N terminus, in high yields without epimerization or racemization under mild conditions without any coupling agent (Scheme 28). Proline methyl ester as well as the Wang resin-bound amino acids were also successfully employed for this coupling.7,94 The high efficiency and atom-economy in this ring-opening coupling process is one of the most salient features of the β-lactam synthon method. It is also worthy of note that enantiopure N-Boc-3-siloxy-β-lactams were readily converted into the corresponding hydroxyethylene isosteres and dihydroxyethylene isosteres, which are key components in various enzyme inhibitors, mimicking tetrahedral intermediates.95,96
Scheme 27.

Scheme 28.

It has been shown that the incorporation of fluorine(s) into a medicinally active compound often improves its pharmacological properties, such as increased membrane permeability, enhanced hydrophobic binding and stability against metabolic oxidation among other merits.43,97,98 As fluorine is absent in living tissue, the incorporation of fluorine(s) into biologically relevant molecules as marker(s) provides a valuable means to monitor protein structures and dynamics as well as drug-protein interactions in vitro and in vivo by 19F NMR. Accordingly, efficient and convenient methods for the synthesis of fluorine-containing compounds of biological and medical interest are in high demand.
The β-lactam synthon method has provided convenient and efficient routes to fluorine-containing α-hydroxy-β-amino acid derivatives and congeners. Enantiopure 3-TIPSO-4-Rf-β-lactams (Rf = fluorine-containing substituent) were prepared through (i) functional-group transformations of 4-isobutenyl-β-lactams, (3R,4S)-82l or (3S,4R)-82l, and (ii) ketene-imine [2+2] cycloaddition followed by enzymatic resolution using PS Amano lipase.13 For instance, the ozonolysis of (3R,4S)-82l afforded the corresponding 4-formyl-β-lactam (3R,4R)-11f (R1 = PMP; R2 = TIPSO) in high yield, which was reacted with diethylaminosulfur trifluoride (DAST) and CBr2F2/Zn to give 4-difluoromethyl-β-lactam (3R,4R)-117 and 4-difluorovinyl-β-lactam (3R,4R)-118, respectively, with >99.9% ee in high yields (Scheme 29).13,44,99 (3R,4R)- and (3S,4S)-1-PMP-4-trifluoromethyl-β-lactams with >99% ee were obtained through enzymatic resolution of racemic cis-3-acetyl-4-CF3-β-lactam prepared through ketene-imine [2+2] cycloaddition.13,43 Enantiopure 1-PMP-3-TIPSO-4-Rf-β-lactams 119, thus obtained, were deprotected by cerium ammonium nitrate (CAN) to give NH-free β-lactams 120, which were converted into the corresponding 1-acyl-, 1-carbalkoxy-, and 1-arenesulfonyl-β-lactams 121 in good yields through reactions with acyl chlorides, chloroformates, and arenesulfonyl chlorides in the presence of an appropriate base (Scheme 30; only one enantiomer is shown for simplicity).13
Scheme 29.

Scheme 30.

Methanolysis of β-lactams 121 in the presence of triethylamine and 4-dimethylaminopyridine (DMAP) at room temperature afforded the corresponding β-Rf-α-siloxy-β-amino acid methyl esters 122 in modest-to-quantitative yields (Scheme 31).13 Furthermore, the ring-opening coupling of β-lactams 121 with various α- and β-amino acid esters gave the corresponding dipeptides, 123 and 124, bearing fluoroisoserine residues, in 50–100% yields (Scheme 31).13 [Note: Scheme 31 only shows the reactions of (3R,4R)-121 for simplicity, but (3S,4S)-121 reacted in a similar manner.]
Scheme 31.

The Type 1 alkylation of 3-siloxy-β-lactams 125 proceeded with excellent diastereoselectivity to give 3-alkyl-3-siloxy-β-lactams 126 with >99% de, which were converted into NH-β-lactams via CAN deprotection, followed by acidic hydrolysis to afford the corresponding α-alkyl-α-hydroxy-β-amino acids 127 in excellent yields (Scheme 32).13,100 β-Lactams 126 were also converted into 1-Boc-β-lactams 128 via NH-β-lactams via CAN deprotection and subsequent reaction with Boc-anhydride.13,100 The facile methanolysis of 1-Boc-β-lactams 128 proceeded at room temperature to give N-Boc-α-alkyl-α-hydroxy-β-amino acid methyl esters 129 in excellent yields (Scheme 32).100 1-Boc-3-hydroxy-β-lactams 130 were prepared by deprotection of β-lactams 128 with HF/pyridine and subjected to ring-opening coupling with (S)-Leu-OMe to give the corresponding dipeptides 131 in high yields (Scheme 33).100
Scheme 32.

Scheme 33.

4. Applications of β-lactam synthon method for the synthesis of medicinally active compounds
4.1 Synthesis of paclitaxel, docetaxel and new-generation taxoids
Paclitaxel (Taxol®; Figure 3), a complex diterpene isolated from the bark of Taxus brevifolia (Pacific yew), is a leading FDA-approved drug for the treatment of advanced ovarian cancer (1992), breast cancer (1994), AIDS-related Karposi’s sarcoma (1997), non-small-cell lung cancer (1999) and other cancers (Figure 3).91,101,102 Docetaxel (Figure 3), the first semisynthetic analogue (“taxoid”) of paclitaxel, was also approved by the FDA in 1996 against breast cancer and has been extensively used for the treatment of various cancers such as those of lung, ovarian, prostate and others.91,92,102
Figure 3.

Chemical structures of paclitaxel and docetaxel
Paclitaxel and docetaxel are cytotoxic drugs which act as mitotic spindle poisons by accelerating tubulin polymerization in microtubules, but stabilize them and arrest cell mitosis at the G2/M phase, leading to apoptosis.103–106 Although the total synthesis of paclitaxel has been accomplished as outstanding hallmark of academic research by six research groups via very long steps,107–112 the semisynthesis of paclitaxel from 10-deacetylbaccatin III, 132 (P = R1 = H), a renewable natural product abundantly isolated from the leaves of Taxus baccata (European yew) and Taxus wallichiana (Himalayan yew), has been found to be the practical approach for the manufacturing of this drug.7,74,113–115
The β-lactam synthon method has played a crucial role in the highly efficient semisynthesis of paclitaxel and docetaxel. For instance, paclitaxel was synthesized through ring-opening coupling of (3R,4S)-1-benzoyl-3-EEO-4-phenyl-β-lactam 132a (R = Ph) with 7-TES-baccatin 133a (R1 = Ac, P = Et3Si) at the C13 position, affording 134a, followed by deprotection of the 1-ethoxyethyl (EE) and TES groups with 0.5% HCl in ethanol in high overall yield (Scheme 34).7 β-Lactam 132a and this ring-opening coupling protocol were also employed to complete the total synthesis of paclitaxel.107–110. In a similar manner, docetaxel was obtained in high overall yield via ring-opening coupling of (3R,4S)-1-Boc-3-EEO-4-phenyl-β-lactam 132b (R = t-BuO) with 7,10-diTroc-baccatin 133b (R1 = P = Cl3CCH2OCO), giving 134b, followed by deprotection with Zn/acetic acid/methanol (Scheme 34).7,76 β-Lactams 132a and 132b were derived from (3R,4S)-3-TIPSO-4-phenyl-β-lactams 82a or 82d (see Table 1) prepared by chiral ester enolate-imine cyclocondensation (see Section 2.2)74,75 or (3R,4S)-3-acetoxy-4-phenyl-β-lactam 30 (see Scheme 7) prepared by ketene-imine [2+2] cycloaddition followed by enzymatic optical resolution.54
Scheme 34.

This highly efficient ring-opening coupling process, “Ojima-Holton coupling”, opened an avenue for the synthesis and structure-activity relationship (SAR) studies on a variety of paclitaxel congeners, “taxoids”.76,77,115–119 The standard procedure for the coupling quickly evolved to the use of 1-acyl- or 1-carbalkoxy-3-TIPSO-β-lactams and LiHMDS as the base.7
Extensive SAR studies on taxoids have led to the discovery of highly potent new-generation taxoids, which have modifications at C3′, C10 and/or C2 and possess 2-to-3 orders of magnitude higher potencies than paclitaxel and docetaxel against multidrug resistant (MDR) as well as paclitaxel-resistant cancer cell lines. Selected new-generation taxoids exhibit much better efficacy than paclitaxel and docetaxel against human tumor xenografts in animal models.77,117–119 Selected new-generation taxoids are listed in Table 2 and their in vitro activities against several cancer cell lines in Table 3.
Table 2.
Selected examples of new-generation taxoids.
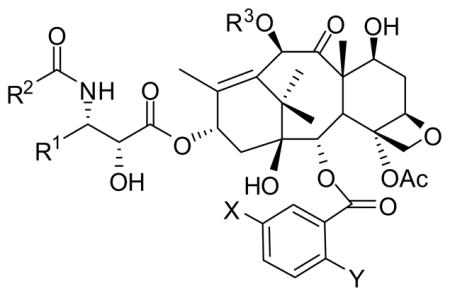
| |||||
|---|---|---|---|---|---|
| Taxoid | R1 | R2 | R3 | X | Y |
| paclitaxel | Ph | Ph | Ac | H | H |
| docetaxel | Ph | t-BuO | H | H | H |
| SBT-1213 | Me2C=CH | t-BuO | EtCO | H | H |
| SBT-1214 | Me2C=CH | t-BuO | c-PrCO | H | H |
| SBT-1216 | Me2C=CH | t-BuO | Me2NCO | H | H |
| SBT-11033 | Me2CHCH2 | t-BuO | EtCO | MeO | H |
| SBT-121303 | Me2C=CH | t-BuO | EtCO | MeO | H |
| SBT-121313 | Me2C=CH | t-BuO | EtCO | MeO | MeO |
| SBT-121602 | Me2C=CH | t-BuO | Me2NCO | Me | H |
| SBT-12823-3 | CF3 | t-BuO | Me2NCO | Cl | H |
| SBT-12854 | F2C=CH | t-BuO | Me2NCO | H | H |
| SBT-12855-1 | F2C=CH | t-BuO | MeOCO | MeO | H |
Table 3.
Cytotoxicity (IC50, nM) of selected new-generation taxoids against human cancer cell lines.
| Taxoid | MCF-7a | HT-29b | CFPAC-1c | DLD-1d | LCC6-MDRe | NCI/ADRf |
|---|---|---|---|---|---|---|
| paclitaxel | 1.7 | 12 | 68 | 300 | 346 | 550 |
| docetaxel | 1.0 | -- | -- | -- | 120 | 723 |
| SBT-1213 | 0.18 | 0.37 | 4.6 | 3.9 | -- | 4.0 |
| SBT-1214 | 0.20 | 0.73 | 0.38 | 3.8 | -- | 3.9 |
| SBT-1216 | 0.13 | 0.052 | 0.66 | 5.4 | -- | 7.4 |
| SBT-11033 | 0.36 | -- | -- | -- | -- | 0.61 |
| SBT-121303 | 0.36 | -- | 0.89 | -- | 0.90 | 0.79 |
| SBT-121313 | 0.30 | 3.6 | 0.025 | 13 | -- | -- |
| SBT-121602 | 0.08 | 0.003 | 0.31 | 0.46 | -- | -- |
| SBT-12823-3 | 0.17 | 0.45 | -- | -- | -- | 1.9 |
| SBT-12854 | 0.18 | 0.46 | 0.35 | 0.25 | -- | 4.3 |
| SBT-12855-1 | 0.11 | -- | -- | -- | -- | 0.92 |
breast cancer cell line (Pgp−).
colon cancer cell line (Pgp−).
pancreatic cancer cell line.
colon cancer cell line (Pgp+).
breast cancer cell line (Pgp+).
ovarian cancer cell line (Pgp+).
In order to investigate the bioactive conformation of paclitaxel, conformationally restricted macrocyclic paclitaxel congeners were designed and synthesized by using a combination of the β-lactam synthon method and an intramolecular Heck reaction or Ru-catalyzed ring-closing metathesis (RCM).119–121
For example, a 3′N-C2-linked novel macrocyclic taxoid 138 was synthesized using an intramolecular Heck reaction in the macrocyclization step (Scheme 35).121 Enantiopure 1-(3-iodobenzoyl)-3-TESO-4-phenyl-β-lactam 135 was prepared from (3R,4S)-3-TIPSO-4-phenyl-β-lactam 82a (see Table 1). The ring-opening coupling of 135 with 2-(2-vinylbenzoyl)-7-TES-baccatin (136) under the standard conditions gave the iodo-vinyl-taxoid 137 in high yield. The intramolecular Heck reaction of 137 catalyzed by a Pd(0)-AsPh3 complex at 55 °C, followed by deprotection afforded the 19-membered exo-methylene-macrocyclic taxoid 138 in 56% yield. The macrocyclic taxoid 138 exhibited a very good IC50 value of 67 nM against LCC6-WT human breast cancer cell line 121.
Scheme 35.

3′NBz-C14β-linked novel macrocyclic taxoids, SB-T-2053 and SB-T-2054, were synthesized using Ru-catalyzed intramolecular coupling reactions of taxoid-dienes 141. Taxoid-dienes 141a and 141b were synthesized though ring-opening coupling of (3R,4S)-1-(2-allylbenzoyl)-3-TESO-4-phenyl-β-lactam 139a and (3R,4S)-1-(2-vinyl)-4-phenyl-3-TESO-β-lactam 139b with 7-TES-14β-allyloxybaccatin III (140), respectively, in high yields under the standard conditions (Scheme 36).120
Scheme 36.

The RCM of 141a using the first-generation Grubbs Ru catalyst at room temperature, followed by deprotection of the silyl groups, gave the expected 15-membered macrocyclic taxoid SB-T-2053 in high yield (Scheme 36).120,122 However, the attempted RCM of 141b did not occur and an unanticipated novel coupling took place to give the 15-membered macrocyclic taxoid SB-T-2054, in which the double bond of the linkage was conjugated to the benzoyl moiety, in good yield after deprotection (Scheme 36).120 This novel coupling reaction is mediated by the Ru complex, but not catalytic, and likely to involve Ru-allyl complexes as key intermediates.120 These macrocyclic taxoids were examined for their activities in tubulin polymerization and microtubule stabilization as well as their potencies against six human cancer cell lines. It was found that both SBT-2053 and SBT-2054 possessed virtually the same activity as that of paclitaxel in the tubulin polymerization/depolymerization assay. Moreover, SBT-2054 was found to be as potent as paclitaxel in the cytotoxicity assay, which is the closest conformationally restricted paclitaxel congener to date, mimicking the binding structure of paclitaxel in microtubules.120,123
4.2 Synthesis of cryptophycins by means of N-acyl-β-lactam macrolactonization
Cryptophycins are macrocyclic cytotoxins produced by cyanobacteria, which exhibit potent tumor-selective antitumor activities by binding microtubules and disrupting cellular mitosis (Figure 4). These compounds are particularly active against MDR cancer cell lines. Synthetic cryptophycin-52 (Cr-52) is currently under clinical development for treatment against solid tumors and has shown an exceptionally potent cytotoxicity against the MCF-7 breast cancer cell line (IC50 = 0.037 nM).124 Dechlorocryptophycin-52 also exhibits good activity in the tubulin assembly assay (IC50 = 3 μM; for Cr-1, IC50 = 2.5 μM) and excellent cytotoxicity against the MCF-7 cell line (IC50 = 0.3 nM).124
Figure 4.

Cryptophycins
An efficient synthesis of the macrolide core of cryptophycins has been achieved by using the N-acyl-β-lactam macrolactonization protocol.125,126 The key steps in the total synthesis of cryptophycin-24 (arenastatin A) are illustrated in Scheme 37, which represents the synthetic strategy for this class of compounds.126 The key β-lactam 144 was prepared in 53% yield for 3 steps through the coupling of the Northern/Western fragment 142 with N-(p-MeO-Phe)-β-lactam 143, the Eastern fragment. The macrolactonization of 144 proceeded through cyanide-initiated β-lactam ring opening using Bu4NCN and subsequent lactonization to give the 16-membered macrocycle 145 in 68% yield. The attempted direct ring-opening coupling using NaH and NaHMDS as base was unsuccessful, presumably due to the instability of 144 under basic conditions. The introduction of the C3′-phenyl moiety through the Heck reaction gave the desired product, but only in modest yield. Epoxidation of the resulting styryl double bond with dimethyldioxirane (DMD) gave cryptophycin-24 as a 2:1 (β:α) diastereomeric mixture, which was separated by reverse-phase HPLC. This strategy was also employed for the total synthesis of dechlorocryptophycin-52.126
Scheme 37.

4.3 Synthesis of β-turn mimetics of enkephalin via β-lactam macrocyclization
In connection with the investigations into the binding conformation of enkephalins,127 endogenous opioid pentapeptides, to specific opiate receptors, conformationally constrained non-peptide β-turn mimetics 146 (Figure 5) were designed and synthesized using a β-lactam macrocyclization protocol.128
Figure 5.

Conformationally constrained non-peptide b-turn mimics
The synthesis of the novel 4→1 β-turn mimic 146a (X = NH) is illustrated in Scheme 38 as a typical example.128 β-Lactam 147, an (S)-Tyr-Gly-Gly mimic synthon, was prepared from (3S,4S)-1-TBS-3-but-3-enyl-4-(4-TBSO-benzyl)-β-lactam, which was obtained through highly stereoselective alkylation of (4S)-1-TBS-4-(4-TBSO-benzyl)-β-lactam, via NaIO4 oxidation and peptide coupling with a glycinate. (S)-Tyr-(S)-Leu-OMe hydrazide 148 was prepared by coupling Leu-OMe with (S)-BocNHNH-Tyr-OH, which was derived from (R)-Tyr and BocNHNH2 via diazonium species. The coupling of 147 with 148 was carried out by converting 147 into the corresponding acid chloride, followed by reaction with 148 in the presence of AgCN to give the β-lactam key intermediate 149a in 49% yield. Then, the Boc group was removed from 149a with trifluoracetic acid (TFA) and the subsequent β-lactam macrocyclization, followed by silicon group deprotection, gave Leu-enkephalin mimic 146a in 94% yield for 3 steps.128
Scheme 38.

4.4 Stereoselective synthesis of indolizidine alkaloids via β-lacatm ring expansion
Indolizidine alkaloids are among the most abundant naturally occurring alkaloid natural products and possess various biological activities of medicinal interest. For example, indolizidine (-)-205A, an alkaloid from poison dart frogs extracts, as well as castanospermine and swainsonine, extracted from plant sources have been extensively studied for their syntheses and biological activities.129–131
The β-lactam synthon method was applied to the construction of the indolizidine skeleton through ring expansion of piperidinyl-β-lactams 153.132 The preparation of β-lactams 153 is illustrated in Scheme 39. An aza-Diels-Alder reaction of β-lactam 150 with Danishefsky’s diene 151 gave a 3:1 mixture of cycloadducts 152a and 152b, which were separated by column chromatography.132 β-Lactam 152a was reduced successively with L-selectride and NaBH4 to give a 3:2 mixture of epimeric alcohols, which were protected with TBS and separated by chromatography to afford piperidinyl-β-lactams 153a and 153b. It is interesting to note that the same reduction-protection sequence for β-lactam 152b gave 153c exclusively.133
Scheme 39.

The ring-expansion reaction of three piperidinyl-β-lactams, 153a, 153b and 153c, catalyzed by sodium methoxide proceeded smoothly at 0 °C to afford the corresponding epimeric indolizidines, 154a, 154b and 154c, respectively, in quantitative yields (Scheme 40).133 Although the preparation of piperidinyl-β-lactams needs improvements, the ring-expansion process is highly efficient and this methodology can be applied to the construction of other izidine alkaloid skeletons.
Scheme 40.

5. Synthesis of 3- to 8-membered nitrogen-heterocycles from β-lactams
As any of the four bonds in the β-lactam ring can be selectively cleaved by varying either the reaction conditions and/or the substrate, the β-lactam ring system can be used for the construction of N-containing heterocycles of different ring sizes. Various examples of intramolecular ring expansions leading to functionalized enaminones, γ-lactams and lactones as well as peptide- and non-peptide based macrocycles through intra- or intermolecular nucleophilic attack have been reported in literature.11,134–136
5.1 Formation of aziridines through ring contraction of β-lactams
The reductive cleavage of the N1-C2 bond of 4-(2-chloroisopropyl)-β-lactam 155 with LiAlH4 gave β-hydroxyethylaziridine 156 in modest-to-fairly good yield through a tandem reduction-cyclization process (Scheme 41).137
Scheme 41.

Treatment of 2-aryl-3,3-dichloroazetidines 158, derived from β-lactams 157 by monochloroalane reduction, with sodium methoxide in methanol gave aziridine ketals 159 in 60–90% yield for two steps (Scheme 42). This reaction is likely to involve 2-azetines 160 and highly strained azabicyclo[1.1.0]cyclobutanes 161, as key intermediates. The ring contraction of 161 through C-N bond cleavage generates aziridine oxonium salts 162, which react with methoxide ion to give 159 (Scheme 42).138
Scheme 42.

5.2 Formation of pyrrolidines through one-carbon ring expansion of β-lactams
The reaction of (2R,3R)-3-alkoxy-4-formyl-β-lactams 11 (see Scheme 3) with t-BuMe2SiCN (TBSCN) catalyzed by molecular iodine gave 5-cyanopyrrolidin-2-ones 163 in moderate-to-high yield with high syn selectivity (86–100%) (Scheme 43).139,140 The syn/anti selectivity depends mostly on the N-substituents and PMP gave the best results, i.e. 100% syn selectivity. This skeletal rearrangement involves the C3-C4 bond cleavage, acyliminium ion formation and cyanide addition. This mechanism was supported by a DFT calculation using 4-formyl-β-lactam 11 (R1 = Me, R2 = Ph) and TMSCN. Other nucleophiles such as allyltrimethylsilanes and propargylsilane can be used in this unique reaction.139
Scheme 43.

The ring-expansion reaction of enantiopure 4-(arylimino)-β-lactams 164, which were readily derived from 4-formyl-β-lactams 11 with arylamines, in the presence of a catalytic amount of tetrabutylammonium cyanide (Bu4NCN) proceeded smoothly in acetonitrile at room temperature to give the corresponding 5-aryliminopyrrolidin-2-ones 165 without racemization in 44–70% yields via N1-C4 bond breakage (Scheme 43).141 The acidic hydrolysis of 165a (R1 = Me; R2 = PMP; R3 = PMP) gave succinimide 166 in 64% yield. It is worth mentioning that the same reaction sequence in one pot from 4-formyl-β-lactam 11 afforded succinimide 166 in 50–55% yields, which were better than the overall yields in the original three-step process (Scheme 41).141
5.3 Formation of 6-membered nitrogen-heterocycles via two-carbon ring expansion of β-lactams
Reduction of 4-(2-TBSO-ethyl)-β-lactams 167 with monochloroalane, followed by TBS deprotection and mesylation, gave 2-(2-TBSO-ethyl)azetidines 168 in 3 steps. The subsequent reaction of 168 with sodium acetate in acetonitrile, followed by basic hydrolysis, gave cis-3-alkoxy/phenoxy-4-hydroxypiperidines 170 in moderate overall yields through two-carbon ring expansion (Scheme 44).142 This ring expansion process involves the ammonium ion of 1-azabicyclo[2.2.0]hexane 169 as the key intermediate, which can explain the 3,4-cis stereochemistry of 170 as the result of exo-attack of acetate ion to cleave the 1–4 bond.142 Closely related reactions using 3-alkoxyl-4-(2-bromo-1,1-dimethylethyl)-β-lactams afforded 3,3-dimethyl-4-X-5-alkoxypiperidines (X = Br, OAc, OH, CN, N3) in high-to-excellent yields.143,144 The reaction of 168 (R1 = i-Pr) with LiBr in acetonitrile gave cis-3-alkoxy-4-bromopiperidine 171, which was converted into piperidin-3-one 172 via the enol-ether in fairly good yield for 2 steps (Scheme 44).142
Scheme 44.

Two-carbon ring expansion of β-lactams 173 through the N1-C4 bond cleavage via 175 to form 176, followed by 1,6-conjugate addition of a malonate to 176, lactamization via 177, and deprotection of the resulting 178, gave glutarimides 174 (Scheme 45). Trans-4-hydroxyphenyl-β-lactams 173 were reacted with t-butyl methyl malonate in the presence of potassium t-butoxide to give 1,3,4,5-substituted glutarimides 174 in 54–77% yields, as illustrated in Scheme 45.145 The relative stereochemistry of the 3, 4 and 5 positions depends on the nature of R1 and R2. Thus, the reactions of trans-β-lactams 173a (R1 = benzyl; R2 = Ph) and 173b (R1 = cyclohexylmethyl; R2 = Ph) exclusively gave trans-trans glutarimides 174a and 174b, respectively, while that of 173c (R1 = Me; R2 = CH2CO2Me) afforded 174c as a 2:1 mixture of trans-trans and cis-trans isomers.145
Scheme 45.

The reaction of 4-acyloxy-β-lactams 179 with an acid chloride and DBU afforded 1,3-oxazin-6-ones 180 in modest-to-good yields (Scheme 46).146 The mechanism of this unique process is likely to involve the formation of highly strained N-acylazetone 181, which rapidly undergoes electrocyclic ring opening to generate N-acylimidoylketene 182. Then, the cyclization of 182 gave 1,3-oxazin-6-one 180.147 A theoretical study on this reaction based on ab initio and DFT analysis has confirmed that two pseudopericyclic reactions are involved. The first key reaction is a retro-[4-exo-dig] cyclization to generate 183 instead of a thermal conrotatory electrocyclic ring opening, followed by another exothermic pseudopericyclic reaction to form 180 in place of a six-electron disrotatory electrocyclization.146
Scheme 46.

The C3-C4 bond cleavage of 4-formyl-β-lactam 183a (X = O) and 4-arylimino-β-lactam 183b (X = ArN) was mediated by stannous chloride and the subsequent 6-membered ring formation gave 1,4-dihydrooxane 184a (X = O) and pyrazine-2,3-dione 184b (X = ArN), respectively, in good-to-excellent yield (Scheme 47).22
Scheme 47.

5.4 Formation of 8-membered rings through [3,3] sigmatropic rearrangement of β-lactams
The [3,3] sigmatropic (Cope) rearrangement of 3,4-dialkenyl-β-lactams 185 proceeded at 120 °C in a sealed tube using toluene as solvent to give the 8-membered lactams, tetrahydroazocinones 186, in 60–100% yield (Scheme 48).148 This rearrangement involves the cleavage of the C3-C4 bond of the β-lactam.
Scheme 48.

This sigmatropic rearrangement was found to be completely stereospecific. Thus, the reactions of enantiopure (3S,4R)-β-lactam 185a and (3R,4S)-β-lactam 185a, bearing a (R)-1-phenylethyl group at N1, gave (6S)-186a and (6R)-186a in 75 and 83% yield, respectively, as a single product in each case. The observed excellent stereospecificity was attributed to the boat-like conformations of these diastereomeric β-lactams, as illustrated in Scheme 49.148
Scheme 49.

6. Use of β-lactam skeleton as a uniquely rigid scaffold for drug discovery
6.1 β-Lactam cholesterol absorption inhibitors
Atherosclerotic coronary heart disease (CHD) is the major cause of death in the United States, and it has been shown that CHD is associated with elevated serum cholesterol levels, to which cholesterol from dietary or intestinal sources is the major contributor.15 Accordingly, the serum cholesterol level should be effectively controlled by blocking the intestinal sources of cholesterol. Then, the cholesterol absorption in the intestine can be blocked by inhibiting acyl-CoA:cholesterol O-acyltransferase (ACAT).15 Thus, ACAT inhibitors have been extensively studied, including β-lactams, which led to the discovery of ezetimibe, a trans-β-lactam, as a potent and highly efficacious cholesterol absorption inhibitor.15 Ezetimibe was approved by the FDA for the treatment of hypercholesterolemia in 2002 and combination therapies with statins have been extensively studied as well.
In a patented asymmetric synthesis of ezetimibe (Scheme 50), a key intermediate, (S)-hydroxypentanoyl-(S)-oxazolidinone 189 (99:1 dr), was prepared by the Corey-Bakshi-Shibata (CBS) asymmetric reduction of 188 in excellent yield. Oxazolidinone 188 was obtained through coupling of δ-keto acid 187 and (S)-phenyloxazolidinone in excellent yield. The asymmetric enolate-imine cyclocondensation of 189 with imine 190 (via in situ silylation and TMS-enolate formation) afforded β-amino amide 191 with high stereoselectivity. The N-silylation followed by TBAF-catalyzed cyclization of 191 gave ezetimibe in high yield.15,149
Scheme 50.

Another synthesis of ezetimibe includes a one-step highly diastereoselective formation of trans-β-lactam 194 through enolate-imine cyclocondensation of (S)-hydroxy-γ-lactone 192 with 4-BnO-C6H4CH= NC6H4-F-4 (193) (Scheme 51). The sodium periodate oxidation of the diol moiety of 194 to the corresponding 3-formyl-β-lactam, which was subjected to Mukaiyama aldol condensation with (TMSO)(4-F-C6H4)C=CH2 (195), followed by desilylation, to give the β-lactam 196. Hydrogenation of the double bond, followed by asymmetric CBS reduction of the ketone moiety and removal of the benzyl protection by hydrogenolysis, afforded ezetimibe with >99% ee in good overall yield.150
Scheme 51.

6.2. β-Lactam-based combretastatin mimics
Combretastatins are naturally occurring diarylstilbenes, isolated from the stem of Combretum caffrum (South African bushwillow tree).151,152 These compounds exhibit strong cytotoxicity and are known to share the same tubulin-binding site as colchicine.153 Among these, the highly potent combretastatin A-4 was selected for drug development and its phosphate derivative, combretastatin A-4-phosphate, a water-soluble prodrug, is currently in clinical trials for the treatment of thyroid cancer (Figure 6).152,154 As the cis-olefin moiethy of combretastatin A-4 is prone to isomerization, which causes a loss of activity, the rigid β-lactam ring system was used to develop a series of β-lactam-based structurally stable combretastatin mimics, some of which exhibited promising activities (Figure 6).19
Figure 6.

Combretastatin A-4, its derivative and b-lactam-based mimics
Combretastatin A-4 mimic 197a was synthesized via the Reformatsky reaction of phenylbromoacetate 198 and imine 199, followed by desilylation (Scheme 52).19 Mimics 197b and 197c were synthesized through the Staudinger ketene-imine cycloaddition reaction of ketenes generated from 200a and 200b with imine 199, forming β-lactams 201a and 201b, respectively, followed by desilylation and reductive removal of the benzyl or Cbz group (Scheme 52).19 The reported chemical yields for these mimics are modest, at best, for some reason. The IC50 values of these mimics were determined to be 9.6, 0.8, and 4.5 nM, respectively, indicating that they are as potent as, or more potent than, combretastatin A-4 against the MCF-7 human breast cancer cell line.19 Although these mimics have been found to be potent and promising, a substantial improvement in the syntheses is necessary for their further development as drug candidates.
Scheme 52.

6.3 β-Lactam-based novel vasopressin V1a antagonists
Vasopressin and oxytocin are neurohypophysical hormones which bind to specific receptors of the G protein-coupled receptor (GPCR) superfamily.155 Vasopressin V1a, V1b and oxytocin receptors activate phospholipase C, which leads to secretion of inositol 1,4,5-triphosphate and diacylglycerol that mobilize intracellular calcium and activation of protein kinase C.155 Besides their functions in the cardiovascular system, vasopressin antagonists have been recommended for some CNS applications.156 Although numerous vasopression V1 receptor antagonists have been developed, none of them had been reported to penetrate the central nervous system (CNS) efficiently, until novel β-lactam-based antagonists such as 202a (SRX246) were discovered.
Based on its structural similarities to ketoconazole, a known antagonist of the luteinizing hormone releasing hormone (LHRH) receptor, LY307174 (IC50 = 45 nM for human V1a receptor) was identified as the lead structure for extensive SAR studies (Figure 7).14 After systematic SAR studies, two highly potent compounds, 202a (SRX246) and 202b (SRX251) (Figure 7 and Scheme 53) were discovered, which exhibited Ki values of 0.3 and 0.66 nM, respectively. When administered orally, these two compounds reached ca. 100-fold higher brain levels in rats than their in vitro receptor affinities. Thus, these two compounds are currently under drug development for human clinical evaluation.14
Figure 7.

Rational design of b-lactam based LHRH receptor antagonists
Scheme 53.

The synthetic routes to 202a and 202b are illustrated in Scheme 53.14 A key intermediate β-lactams 206 were synthesized using the asymmetric Staudinger ketene-imine cycloaddition of (R)-phenyloxazolidinylacetyl chloride (18; see Scheme 5) with functionalized imines 205 in the presence of a base, which proceeded with excellent stereoselectivity. Imine 205 was prepared from (R)-N-Cbz-Asp-OBu-t (203) via coupling with an amine [a: (S)-1-phenylethylamine; b: N-methyl-(3-trifluoromethyl)phenylmethylamine] to give amides 204, followed by removal of benzyl protection and imine formation with cinnamaldehyde. The asymmetric Staudinger cycloaddition of 18 with 205 in the presence of triethylamine, followed by treatment with formic acid gave β-lactam 206 in 64–69% yield. The amide coupling of 206a and 206b with 1,4′-dipiperidine afforded 202a (SRX246) and 202b (SRX251), respectively, in >90% yield (Scheme 53).14
7. Conclusions
This Tetrahedron Report has summarized the recent advances in the synthesis of β-lactams with high enantiomeric purity, the chemistry of β-lactams, the β-lactam synthon method and its applications to the synthesis of non-protein amino acids, their derivatives and peptides as well as biologically active natural and unnatural compounds of medicinal interest. In addition, the discovery and development of novel β-lactams with anticancer activity, cholesterol absorption inhibitory activity and CNS activity by exploiting the rigid and unique β-lactam scaffold are described. Traditionally, the organic chemistry and medicinal chemistry of β-lactams have been directly related to β-lactam antibiotics. However, it is obvious that the potential of β-lactams has been explored extensively in an impressive breadth, which has created an entirely new field of chemical and medicinal research. Thus, it is safe to say that this field of research will continue to expand and flourish.
Biographies

Anushree Kamath was born in Mangalore, India. She obtained her B.S. Chem (Hons) from St. Stephen’s College, Delhi in 2005 and M.S. from the Indian Institute of Technology, Roorkee in 2007. In the same year, she moved to France to join Dr. Andrew E. Greene’s group at the Universite de Grenoble for her doctoral thesis, where she worked under Dr. Philippe Delair on the total synthesis of natural products. Since June, 2011, she has been working as a postdoctoral research associate with Prof. Iwao Ojima at the Institute of Chemical Biology and Drug Discovery (ICB&DD), Stony Brook University, New York. Her current research focuses on the development of drug-delivery platforms for tumor-targeted cancer chemotherapy.

Iwao Ojima received his B.S., M.S., and Ph.D. (1973) from the University of Tokyo, Japan. He joined the Sagami Institute of Chemical Research and held a position of Senior Research Fellow until 1983. He joined the faculty at the Department of Chemistry, State University of New York at Stony Brook (now Stony Brook University) first as Associate Professor (1983), was promoted to Professor (1984), Leading Professor (1991), and then to Distinguished Professor (1995). He served as the Department Chairman (1997–2003). He has been serving as the founding Director for the Institute of Chemical Biology and Drug Discovery (ICB&DD) from 2004. His research program includes the discovery and development of anticancer agents, targeted drug delivery systems and new-generation antimicrobials. His awards and honors include Arthur C. Cope Scholar Award (1994), E. B. Hershberg Award for Important Discoveries of Medicinally Active Substances (2001), the Medicinal Chemistry Hall of Fame (2006) from the American Chemical Society; The Chemical Society of Japan Award (1999); Outstanding Inventor Award (2002) from the Research Foundation of the State University of New York; Elected Fellows of J. S. Guggenheim Memorial Foundation (1995), American Association for the Advancement of Science (1997), The New York Academy of Sciences (2000), and American Chemical Society (2010).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Georg GI. The Organic Chemistry of β-Lactams and references cited therein. VCH; New York: 1992. [Google Scholar]
- 2.Aoki H, Sakai H, Kohsaka M, Konomi T, Hosoda J. J Antibiotics. 1976;29:890–901. doi: 10.7164/antibiotics.29.492. [DOI] [PubMed] [Google Scholar]
- 3.Hashimoto M, Komori T, Kamiya T. J Am Chem Soc. 1976;98:3023–3025. doi: 10.1021/ja00426a063. [DOI] [PubMed] [Google Scholar]
- 4.Imada A, Kitano K, Kintaka K, Muroi M, Asai M. Nature. 1981;289:590–591. doi: 10.1038/289590a0. [DOI] [PubMed] [Google Scholar]
- 5.Ojima I, Shimizu N, Qiu X, Chen HJC, Nakahashi K. Bull Soc Chim France. 1987:649–658. [Google Scholar]
- 6.Hatanaka N, Abe R, Ojima I. Chem Lett. 1981;10:1297–1298. [Google Scholar]
- 7.Ojima I. Acc Chem Res. 1995;28:383–389. [Google Scholar]
- 8.Ojima I. In: Advances in Asymmetric Synthesis. Hassner A, editor. JAI Press, Inc; Greenwich, CT: 1995. pp. 95–146. [Google Scholar]
- 9.Ojima I, Delaloge F. Chem Soc Rev. 1997;26:377–386. [Google Scholar]
- 10.Deshmukh AR, Bhawal BM, Krishnaswamy D, Govande VV, Shinkre BA, Jayanthi A. Curr Med Chem. 2004;11:1889–1920. doi: 10.2174/0929867043364874. [DOI] [PubMed] [Google Scholar]
- 11.Alcaide B, Almendros P. Curr Med Chem. 2004;11:1921–1949. doi: 10.2174/0929867043364856. [DOI] [PubMed] [Google Scholar]
- 12.Palomo C, Aizpurua JM, Ganboa I, Oiardide M. Curr Med Chem. 2004;11:1837–1872. doi: 10.2174/0929867043364900. [DOI] [PubMed] [Google Scholar]
- 13.Ojima I, Kuznetsova L, Ungureanu IM, Pepe A, Zanardi I, Chen J. In: ACS Symp Ser 911: Fluorine-containing Synthons. Soloshonok V, editor. American Chemical Society/Oxford University Press; Washington, D.C: 2005. pp. 544–561. [Google Scholar]
- 14.Guillon CD, Koppel GA, Brownstein MJ, Chaney MO, Ferris CF, Lu SF, Fabio KM, Miller MJ, Heindel ND, Hunden DC, Cooper RDG, Kaldox SW, Skelton JJ, Dressman BA, Clay MP, Steinberg MI, Bruns RF. Bioorg Med Chem. 2007;15:2054–2080. doi: 10.1016/j.bmc.2006.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burnett D. Curr Med Chem. 2004;11:1873–1887. doi: 10.2174/0929867043364865. [DOI] [PubMed] [Google Scholar]
- 16.Banik I, Becker FF, Banik BK. J Med Chem. 2003;46:12–15. doi: 10.1021/jm0255825. [DOI] [PubMed] [Google Scholar]
- 17.Banik BK, Becker FF, Banik I. Bioorg Med Chem. 2004;12:2523–2528. doi: 10.1016/j.bmc.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 18.Banik BK, Banik I, Becker FF. Euro J Med Chem. 2010;45:846–848. doi: 10.1016/j.ejmech.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 19.O’Boyle N, Carr M, Greene L, Bergin O, Nathwani S, McCabe T, Lloyd D, Zisterer D, Meegan M. J Med Chem. 2010;53:8569–8584. doi: 10.1021/jm101115u. [DOI] [PubMed] [Google Scholar]
- 20.Del Buttero PMG, Roncoroni M. Tetrahedron Lett. 2006;47:2209–2211. [Google Scholar]
- 21.Palomo C, Aizpurua JM, Ganboa I, Oiardide M. Amino Acids. 1999;16:321–343. doi: 10.1007/BF01388175. [DOI] [PubMed] [Google Scholar]
- 22.Alcaide B, Martin-Cantalejo Y, Rodriguez-Lopez J, Sierra M. J Org Chem. 1993;58:4767–4770. [Google Scholar]
- 23.Staudinger H. Justus Liebigs Ann Chem. 1907;356:51–123. doi: 10.1002/jlac.19717540118. [DOI] [PubMed] [Google Scholar]
- 24.Venturini A, Gonzalez J. Mini Rev Med Chem. 2006;3:185–194. [Google Scholar]
- 25.Cossio FP, Arrieta A, Sierra MA. Acc Chem Res. 2008;41:925–936. doi: 10.1021/ar800033j. [DOI] [PubMed] [Google Scholar]
- 26.Dumas S, Hegedus LS. J Org Chem. 1994;59:4967–4971. [Google Scholar]
- 27.Georg GI, Ravikumar VT. In: Organic Chemistry β-lactams. GIG, editor. VCH Publishing; New York, NY: 1993. pp. 295–368. [Google Scholar]
- 28.Hegedus LS, Montgomery J, Narukawa Y, Snustad DC. J Am Chem Soc. 1991;113:5784–5791. [Google Scholar]
- 29.Cossio FP, Ugalde JM, Lopez X, Lecea B, Palomo C. J Am Chem Soc. 1993;115:995–1004. [Google Scholar]
- 30.Lopez R, Sordo TL, Sordo JA, Gonzalez J. J Org Chem. 1993;58:7036–7037. [Google Scholar]
- 31.Cossio FP, Arrieta A, Lecea B, Ugalde JM. J Am Chem Soc. 1994;116:2085–2093. [Google Scholar]
- 32.Wang Y, Liang Y, Jiao L, Du DM, Xu J. J Org Chem. 2006;71:6983–6990. doi: 10.1021/jo0611521. [DOI] [PubMed] [Google Scholar]
- 33.Li B, Wang Y, Du DM, Xu J. J Org Chem. 2007;72:990–997. doi: 10.1021/jo0622734. [DOI] [PubMed] [Google Scholar]
- 34.Linder MR, Podlech J. Org Lett. 2001;3:1849–1851. doi: 10.1021/ol015891+. [DOI] [PubMed] [Google Scholar]
- 35.Palomo C, Oiarbide M, Esnal A, Landa A, Miranda J, Linden A. J Org Chem. 1998;63:5838–5846. doi: 10.1021/jo980354x. [DOI] [PubMed] [Google Scholar]
- 36.Aszodi J, Bonnet A, Teusch G. Tetrahedron. 1990;46:1579–1586. [Google Scholar]
- 37.Bose A, Manhas M, van der Veen J, Bari S, Wagle D. Tetrahedron. 1992;48:4831–4844. [Google Scholar]
- 38.Hubschwerlen C, Schmid G. Helv Chim Acta. 1983;66:2206–2209. [Google Scholar]
- 39.Banik B, Manhas M, Kaluza Z, Barakat J, Bose A. Tetrahedron Lett. 1992;33:3603–3606. [Google Scholar]
- 40.Chincholkar P, Puranik VG, Deshmukh ARAS. Tetrahedron. 2007;63:9179–9187. [Google Scholar]
- 41.Wagle DR, Garai C, Chiang J, Monteleone MG, Kurys BE, Strohmeyer TW, Hegde VR, Manhas MS, Bose AK. J Org Chem. 1988;53:4227–4236. [Google Scholar]
- 42.Li L, Thomas SA, Klein LL, Yeung CM, Maring CJ, Grampovnik DJ, Lartey PA, Plattner JJ. J Med Chem. 1994;37:2655–2663. doi: 10.1021/jm00043a005. [DOI] [PubMed] [Google Scholar]
- 43.Kuznetsova L, Ungureanu I, Pepe A, Zanardi I, Wu X, Ojima I. J Fluor Chem. 2004;125:487–500. [Google Scholar]
- 44.Ojima IV, KL, Sun L. In: ACS Sym Ser 949: Current Fluoroorganic Chemistry New Synthetic Directions, Technologies, Materials and Biological Applications. Soloshonok V, Yamazaki KMT, Welch JT, Honek J, editors. American Chemical Society/Oxford University Press; Washington, D.C: 2007. pp. 288–304. [Google Scholar]
- 45.Niu C, Miller MJ. Tetrahedron Lett. 1995;36:497–500. [Google Scholar]
- 46.Evans DA, Sjogren EB. Tetrahedron Lett. 1985;26:3783–3786. [Google Scholar]
- 47.Ruhland B, Gordon ABE, Gallop M. J Am Chem Soc. 1996;118:253–254. [Google Scholar]
- 48.Delpiccolo C, Mata E. Tetrahedron: Asymm. 2002;13:905–910. [Google Scholar]
- 49.Alcaide B, Rodriguez-Vicente A. Tetrahedron Lett. 1999;40:2005–2006. [Google Scholar]
- 50.Chen J, Kuznetsova L, Ungureanu I, Ojima I. In: Enantioselective Synthesis of Amino Acids. 2. Juaristi E, Soloshonok V, editors. John Wiley; New York: 2005. pp. 447–476. [Google Scholar]
- 51.Brieva R, Crich JZ, Sih CJ. J Org Chem. 1993;58:1068–1075. [Google Scholar]
- 52.Banik BK, Manhas MS, Bose AK. J Org Chem. 1994;59:4714–4716. [Google Scholar]
- 53.Patel RN. Eur. Pat. Appl. 634492 A1. Bristol-Myers Squibb Co; USA: 1995
- 54.Holton RA, Vu P. PCT Int. Appl. WO 2001/029245. Florida State University; USA: 2001
- 55.Kuznetsova LV, Pepe A, Ungureanu IM, Pera P, Bernacki RJ, Ojima I. J Fluor Chem. 2008;129:817–828. doi: 10.1016/j.jfluchem.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.France S, Weatherwax A, Taggi AE, Lectka T. Acc Chem Res. 2004;37:592–600. doi: 10.1021/ar030055g. [DOI] [PubMed] [Google Scholar]
- 57.Wack H, Drury W, Taggi A, Ferraris D, Lectka T. Org Lett. 1999;1:1985–1988. doi: 10.1021/ol9903234. [DOI] [PubMed] [Google Scholar]
- 58.Taggi A, Hafez A, Wack H, Young B, Drury W, Lectka T. J Am Chem Soc. 2000;122:7831–7832. [Google Scholar]
- 59.France S, Wack H, Hafez A, Taggi A, Witsil D, Lectka T. Org Lett. 2002;4:1603–1605. doi: 10.1021/ol025805l. [DOI] [PubMed] [Google Scholar]
- 60.Hodous BL, Fu GC. J Am Chem Soc. 2002;124:1578–1579. doi: 10.1021/ja012427r. [DOI] [PubMed] [Google Scholar]
- 61.Zhang YR, He L, Wu X, Shao PL, Ye S. Org Lett. 2008;10:277–280. doi: 10.1021/ol702759b. [DOI] [PubMed] [Google Scholar]
- 62.Huang X, Chen X, Ye S. J Org Chem. 2009;74:7585–7587. doi: 10.1021/jo901656q. [DOI] [PubMed] [Google Scholar]
- 63.Duguet N, Campbell CD, Slawin AMZ, Smith DAD. Org Biomol Chem. 2008;6:1108–1113. doi: 10.1039/b800857b. [DOI] [PubMed] [Google Scholar]
- 64.Sereda O, Blanrue A, Wilhelm R. Chem Commun. 2009;45:1040–1042. doi: 10.1039/b817991c. [DOI] [PubMed] [Google Scholar]
- 65.Ponsford RJaSR. JCS CHEM COMM. 1979:846–847. [Google Scholar]
- 66.Brown P, Southgate R. Tetrahedron Lett. 1986;27:247. [Google Scholar]
- 67.Doyle MP, Shanklin MS, Oon SM, Pho HQ, van der Heidet FR, Veal WR. J Org Chem. 1988;53:3384–3386. [Google Scholar]
- 68.Doyle MP, Oon SM, van der Heide FR, Brown CB. Bioorg Med Chem Lett. 1993;3:2409–2414. [Google Scholar]
- 69.Watanabe N, Anada M, Hashimoto S, Ikegami S. Synlett. 1994:1031–1033. [Google Scholar]
- 70.Anada M, Watanabe N, Hashimoto S. Chem Commun. 1998:1517–1518. [Google Scholar]
- 71.Calet S, Urso F, Alper H. j Am Chem Soc. 1989;111:931–934. [Google Scholar]
- 72.Piotti MEaAH. J Am Chem Soc. 1996;118:111–116. [Google Scholar]
- 73.Ojima I, Habus I. Tetrahedron Lett. 1990;31:4289–4292. [Google Scholar]
- 74.Ojima I, Habus I, Zhao M, Zucco M, Park Y, Sun C, Brigaud T. Tetrahedron. 1992;48:6985–7012. [Google Scholar]
- 75.Ojima I, Habus I, Zhao M. J Org Chem. 1991;56:1681–1683. [Google Scholar]
- 76.Ojima I, Sun CM, Zucco M, Park YH, Duclos O, Kuduk SD. Tetrahedron Lett. 1993;34:4149–4152. [Google Scholar]
- 77.Ojima I, Slater JS, Kuduk SD, Takeuchi CS, Gimi RH, Sun CM, Park YH, Pera P, Veith JM, Bernacki RJ. J Med Chem. 1997;40:267–278. doi: 10.1021/jm960563e. [DOI] [PubMed] [Google Scholar]
- 78.Fujieda H, Kanai M, Kambara T, Iida A, Tamioka K. J Am Chem Soc. 1997;119:2060–2061. [Google Scholar]
- 79.Dunnill P. Nature. 1966;210:1265–167. doi: 10.1038/2101267a0. [DOI] [PubMed] [Google Scholar]
- 80.Jung M. Chemistry and Biochemistry of Amino Acids. Chapman and Hall; New York: 1985. [Google Scholar]
- 81.Brandi A, Cicchi S, Cordero FM. Chem Rev. 2008;108:3988–4035. doi: 10.1021/cr800325e. [DOI] [PubMed] [Google Scholar]
- 82.Ojima I, Zhao M, Yamato T, Nakahashi K, Abe R. J Org Chem. 1991;56:5263–5277. [Google Scholar]
- 83.Ojima I, Chen HJ, Qiu X. Tetrahedron. 1988;44:5307–5318. [Google Scholar]
- 84.Colson P-JaHLS. J Org Chem. 1993;58:5918–5924. [Google Scholar]
- 85.Ojima I, Chen HJ, Nakahashi K. J Am Chem Soc. 1988;110:278–281. [Google Scholar]
- 86.Thaisrivongs S, Pals DT, Kroll LT, Turner SR, Han FS. J Med Chem. 1987;30:976–982. doi: 10.1021/jm00389a004. [DOI] [PubMed] [Google Scholar]
- 87.Iizuka K, Kamijo T, Kubota T, Akahane K, Umeyama Y, Kiso Y. J Med Chem. 1988;31:701–704. doi: 10.1021/jm00399a001. [DOI] [PubMed] [Google Scholar]
- 88.Huff JR. J Med Chem. 1991;34:2305–2314. doi: 10.1021/jm00112a001. [DOI] [PubMed] [Google Scholar]
- 89.Okino TM, Matsuda H, Murakami M, Yamaguchi K. Tetrahedron Lett. 1993;34:501–504. [Google Scholar]
- 90.Cardillo G, Tomasini C. Chem Soc Rev. 1996;25:117–128. [Google Scholar]
- 91.Rowinsky EK. Ann Rev Med. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- 92.Kingston DGI. J Nat Prod. 2009;72:507–515. doi: 10.1021/np800568j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ojima I, Park YH, Sun CM, Zhao M, Brigaud T. Tetrahedron Lett. 1992;33:5739–5742. [Google Scholar]
- 94.Ojima I, Sun CM, Park YH. J Org Chem. 1994;59:1249–1250. [Google Scholar]
- 95.Ojima I, Wang T, Ng EW. Tetrahedron Lett. 1998;39:923–926. [Google Scholar]
- 96.Palomo C, Aizpurua JM, Cuevas C. J Chem Soc Chem Commun. 1994;1994:1957–1958. [Google Scholar]
- 97.Kukhar VP, Soloshonok VA. Fluorine Containing Amino Acids: Synthesis and Properties. Wiley; Chichester, UK: 1994. [Google Scholar]
- 98.Ojima I. Biomedical Frontiers of Fluorine Chemistry. American Chemical Society; Washington, DC: 1996. [Google Scholar]
- 99.Ojima I, Lin S, Slater JC, Wang T, Pera P, Bernacki RJ, Ferlini CGS. Bioorg Med Chem. 2000;8:1576–1585. doi: 10.1016/s0968-0896(00)00093-6. [DOI] [PubMed] [Google Scholar]
- 100.Ojima I, Wang T, Delaloge F. Tetrahedron Lett. 1998;39:3663–3666. [Google Scholar]
- 101.Suffness M. Taxol: Science and Applications. CRC Press; New York: 1995. [Google Scholar]
- 102.Rowinsky EK, Onetto N, Canetta RM, Arbuck SG. Semin Oncol. 1992;19:646–662. [PubMed] [Google Scholar]
- 103.Schiff PB, Fant J, Horwitz SB. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 104.Schiff PB, Horwitz SB. Proc Natl Acad Sci, U S A. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jordan M, Wilson L. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 106.Jordan MA. Curr Med Chem: Anti-Cancer Agents. 2002;2:1–17. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]
- 107.Holton RA, Kim HB, Somoza C, Liang F, Biediger RJ, Boatman PD, Shindo M, Smith CC, Kim S, Nadizadeh H, Suzuki Y, Tao C, Vu P, Tang S, Zhang P, Murthi KK, Gentile LN, Liu JH. J Am Chem Soc. 1994;116:1599–1600. [Google Scholar]
- 108.Nicolaou KC, Yang Z, Liu JJ, Ueno H, Nantermet PG, Guy RK, Claiborne CF, Renaud J, Couladouros EA, Paulvannan K, Sorensen EJ. Nature. 1994;367:630–634. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]
- 109.Danishefsky S, Masters J, Young W, Link J, Snyder L, Magee T, Jung D, Isaacs R, Bornmann W, Alaimo C, Coburn C, Di Grandi M. J Am Chem Soc. 1996;118:2843–2859. [Google Scholar]
- 110.Wender PA, Badham NF, Conway SP, Floreancig PE, Glass TE, Houze JB, Krauss NE, Lee D, Marquess DG, McGrane PL, Meng W, Natchus MG, Shuker AJ, Sutton JC, Taylor RE. J Am Chem Soc. 1997;119:2757–2758. [Google Scholar]
- 111.Mukaiyama T, Shiina I, Iwadare H, Saitoh M, Nishimura T, Ohkawa N, Sakoh H, Nishimura K, Tani Y, Hasegawa M, Yamada K, Saitoh K. Chem-Eur J. 1999;5:121–161. [Google Scholar]
- 112.Morihira K, Hara R, Kawahara S, Nishimori T, Nakamura N, Kusama H, Kuwajima I. J Am Chem Soc. 1998;120:12980–12981. [Google Scholar]
- 113.Denis JN, Greene AE, Guénard D, Guéritte-Voegelein F, Mangatal L, Potier PA. J Am Chem Soc. 1988;110:5917–5919. [Google Scholar]
- 114.Guénard D, Guéritte-Vogelein F, Potier P. Acc Chem Res. 1993;26:160–167. [Google Scholar]
- 115.Holton RA, Biediger RJ, Boatman PD. In: Taxol®: Science and Applications. Suffness M, editor. CRC Press; New York: 1995. pp. 97–121. [Google Scholar]
- 116.Georg GI, Boge TC, Cheruvallath ZS, Clowers JS, Harriman GCB, Hepperle M, Park H. In: Taxol®: Science and Applications. Suffness M, editor. CRC Press; New York: 1995. pp. 317–375. [Google Scholar]
- 117.Ojima I, Slater JC, Michaud E, Kuduk SD, Bounaud PY, Vrignaud P, Bissery MC, Veith J, Pera P, Bernacki RJ. J Med Chem. 1996;39:3889–3896. doi: 10.1021/jm9604080. [DOI] [PubMed] [Google Scholar]
- 118.Ojima I, Chen J, Sun L, Borella CP, Wang T, Miller ML, Lin S, Geng X, Kuznetsova L, Qu C, Gallager D, Zhao X, Zanardi I, Xia S, Horwitz SB, Mallen-StClair J, Guerriero JL, Bar-Sagi D, Veith JM, Pera P, Bernacki RJ. J Med Chem. 2008;51:3203–3221. doi: 10.1021/jm800086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ojima I, Das M. J Nat Prod. 2009;72:554–565. doi: 10.1021/np8006556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun L, Geng X, Geney R, Li Y, Simmerling C, Li Z, Lauher JW, Xia S, Horwitz SB, Veith JM, Pera P, Bernacki RJ, Ojima I. J Org Chem. 2008;73:9584–9593. doi: 10.1021/jo801713q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Geng X, Miller M, Lin S, Ojima I. Org Lett. 2003;5:3733–3736. doi: 10.1021/ol0354627. [DOI] [PubMed] [Google Scholar]
- 122.Geney R, Sun L, Pera P, Bernacki RJ, Xia S, Horwitz SB, Simmerling CL, Ojima I. Chem & Biol. 2005;12:339–348. doi: 10.1016/j.chembiol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 123.Sun L, Simmerling C, Ojima I. ChemMedChem. 2009;4:719–731. doi: 10.1002/cmdc.200900044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eggen MJ, Georg GI. Med Res Rev. 2002;22:85–101. doi: 10.1002/med.10002. [DOI] [PubMed] [Google Scholar]
- 125.Eggen M, Nair S, Georg G. Org Lett. 2001;3:1813–1815. doi: 10.1021/ol010044s. [DOI] [PubMed] [Google Scholar]
- 126.Vidya R, Eggen MJ, Nair SK, Georg GI, Himes RH. J Org Chem. 2003;68:9687–9693. doi: 10.1021/jo0302197. [DOI] [PubMed] [Google Scholar]
- 127.Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, Morris HR. Nature. 1975;258:577–579. doi: 10.1038/258577a0. [DOI] [PubMed] [Google Scholar]
- 128.Gardner B, Nakanishi H, Kahn M. Tetrahedron. 1993;49:3433–3448. [Google Scholar]
- 129.Michael JP. Nat Prod Rep. 2004;21:625–649. doi: 10.1039/b310689f. [DOI] [PubMed] [Google Scholar]
- 130.Cenci di Bello I, Fleet G, Namgoong SK, Tadano K, Winchester B. Biochem J. 1989;259:855–861. doi: 10.1042/bj2590855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Olden K, Breton P, Grzegorzewski K, Yasuda Y, Gause BL, Oladipa A, Oredipe A, Newton SA, White SL. Pharmacology and Therapeutics. 1991;50:285–290. doi: 10.1016/0163-7258(91)90046-o. [DOI] [PubMed] [Google Scholar]
- 132.Alcaide B, Almendros P, Alonso JM, Moustafa FA, Torres MR. SynLett. 2001;10:1531–1534. [Google Scholar]
- 133.Alcaide B, Almendros P, Alonso JM, Aly MF. Chem Eur J. 2003;9:3415–3426. doi: 10.1002/chem.200304712. [DOI] [PubMed] [Google Scholar]
- 134.Alcaide B, Almendros P, Aragoncillo C. Chem Eur J. 2002;8:3646–3652. doi: 10.1002/1521-3765(20020816)8:16<3646::AID-CHEM3646>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 135.Alcaide B, Aly M, Rodriguez C, Rodriguez-Vicente A. J Org Chem. 2000;65:3453–3459. doi: 10.1021/jo991984h. [DOI] [PubMed] [Google Scholar]
- 136.Banfi L, Guanti G, Rasparini M. Eur J Org Chem. 2003;2003:1319–1336. [Google Scholar]
- 137.Van Brabandt W, Dejaegher Y, Van Landeghem R, De Kimpe N. Org Lett. 2006;8:1101–1104. doi: 10.1021/ol053015g. [DOI] [PubMed] [Google Scholar]
- 138.Dejaegher Y, Mangelinckx S, De Kimpe N. J Org Chem. 2002;67:2075–2081. doi: 10.1021/jo010914j. [DOI] [PubMed] [Google Scholar]
- 139.Alcaide B, Almendros P, Cabrero G, Callejo R, Ruiz M, Arno M, Domingo L. Adv Synth Catal. 2010;352:1688–1700. [Google Scholar]
- 140.Alcaide B, Almendros P, Cabrere G, Ruiz MP. Chem Comm. 2008:615–618. doi: 10.1039/b715661h. [DOI] [PubMed] [Google Scholar]
- 141.Alcaide B, Almendros P, Cabrero G, Ruiz M. Org Lett. 2005;7:3981–3984. doi: 10.1021/ol051504a. [DOI] [PubMed] [Google Scholar]
- 142.Mollet K, Catak S, Waroquier M, van Speybroeck V, D’hooghe M, De Kimpe N. J Org Chem. 2011;76:8361–8375. doi: 10.1021/jo201556t. [DOI] [PubMed] [Google Scholar]
- 143.Van Brabandt W, Van Landeghem R, De Kimpe N. Org Lett. 2006;8:1105–1108. doi: 10.1021/ol0530676. [DOI] [PubMed] [Google Scholar]
- 144.Mollet K, Broeckx L, D’hooghe M, De Kimpe N. Heterocycles. 2011;84:431–447. [Google Scholar]
- 145.Cabell LA, McMurray JS. Tetrahedron Lett. 2002;43:2491–2493. [Google Scholar]
- 146.Alajarin M, Sanchez-Andrada P. J Org Chem. 2001;66:8470–8477. doi: 10.1021/jo015922e. [DOI] [PubMed] [Google Scholar]
- 147.Alajarin M, Vidal A, Sanchez-Andrada P, Tovar F, Ochoa Gines. Org Lett. 2000;2:965–968. doi: 10.1021/ol0056168. [DOI] [PubMed] [Google Scholar]
- 148.Almendros P, Aragoncillo C, Cabrero G, Callejo R, Carrascosa R, Luna A, del Campo TM, Pardo MC, Quiros MT, Redondo MC, Rodriguez-Ranera C, Rodriguez-Vicente A, Ruiz MP. Arkivoc. 2010;2010:74–92. [Google Scholar]
- 149.Tiruvettipuram KT, Fu X, Tann CH, McAllister TL, Chiu JS, Colon C. US Pat. 6,207,822 B1. Schering Corporation; 2001
- 150.Wu G, Wong YS, Chen X, Ding Z. J Org Chem. 1999;64:3714–3718. doi: 10.1021/jo990428k. [DOI] [PubMed] [Google Scholar]
- 151.Watt JM, Gerdina M. The Medicinal and Poisonous Plants of Southern and Eastern Africa. E. & S. Livingstone Ltd; Edinburgh and London, U.K: 1962. [Google Scholar]
- 152.Cooney M, Ortiz J, Bukowski R, Remick S. Curr Oncol Rep. 2005;7:90–95. doi: 10.1007/s11912-005-0033-x. [DOI] [PubMed] [Google Scholar]
- 153.Cragg G, Kingston D, Newman D. Anticancer Agents from Natural Products. CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- 154.Young SL, Chaplin DJ. Expert Opin Invest Drugs. 2004;13:1171–1182. doi: 10.1517/13543784.13.9.1171. [DOI] [PubMed] [Google Scholar]
- 155.Neumann IDaLR. Advances in Vaspressin and Oxytocin: from genes to behaviour to disease. Elsevier; 2008. [DOI] [PubMed] [Google Scholar]
- 156.Tribollet E, Barberis C, Jard S, Dubois-DAuphin M, Dreifuss JJ. Brain Research. 1988;442:105–118. doi: 10.1016/0006-8993(88)91437-0. [DOI] [PubMed] [Google Scholar]