

Abstract
The genetic basis for most of the rare lipid monogenic disorders have been elucidated, but the challenge remains in determining the combination of genes that contribute to the genetic variability in lipid levels in the general population; this has been estimated to be in the range of 40-60 per cent of the total variability. Therefore, the effect of common polymorphisms on lipid phenotypes will be greatly modulated by gene-gene and gene-environment interactions. This approach can also be used to characterise the individuality of the response to lipid-lowering therapies, whether using drugs (pharmacogenetics) or dietary interventions (nutrigenetics). In this regard, multiple studies have already described significant interactions between candidate genes for lipid and drug metabolism that modulate therapeutic response--although the outcomes of these studies have been controversial and call for more rigorous experimental design and analytical approaches. Once solid evidence about the predictive value of genetic panels is obtained, risk and therapeutic algorithms can begin to be generated that should provide an accurate measure of genetic predisposition, as well as targeted behavioural modifications or drugs of choice and personalised dosages of these drugs.
Keywords: lipids, lipoproteins, cholesterol, cholesterol therapy, cardiovascular diseases, atherosclerosis, pharmacogenetics
Introduction
Current knowledge about the genetic basis of lipid diseases has its roots in the 1950s, with the development of techniques to separate and quantitate different plasma lipoprotein classes. The lipid field experienced a definite boost during the 1960s and 1970s with the enhanced capacity to carry out more extensive phenotyping. This contributed to the first classification of hyperlipidaemias by Fredrickson. The work also led to the recognition of the familial basis for some other rare disorders such as abetalipoproteinaemia, which is characterised by the absence in plasma of the 'B' protein of beta-lipoproteins, and its mirror image, Tangier disease, characterised by the almost complete absence of 'A' apo-protein from alpha-lipoproteins in the plasma [1]. During the ensuing years, the molecular bases for some of the rare, monogenic disorders have been uncovered. It is apparent, for example, that hyperlipoproteinaemia type IIa is, for the most part, the result of mutations at the low-density lipo-protein receptor (LDLR) gene; in fact, it is now known that there are over 700 such mutations [2]. Similarly, type III hyperlipoproteinaemia was found to be associated with specific apolipoprotein E (APOE) alleles plus some other poorly characterised trigger(s). Hyperlipoproteinaemia type I was found to be the result of mutations at either of two genes: lipoprotein lipase (LPL) or apolipoprotein C-II (APOC2). One of the most interesting stories about the genetics of lipids is represented by the race to understand the molecular basis of Tangier disease. This was a marathon pursuit that ended up with three groups reporting simultaneously the finding of the ATP-Binding Cassette, Subfamily A, Member 1 (ABCA1) as the gene responsible for Tangier disease [3-5]. These findings were immediately verified by other investigators [6-8]. It turned out to be a very tight finish to a 40-year race. Thus, slowly but surely, many of the phenotypes associated with lipid disorders have been assigned to specific genes; a search of the Online Mendelian Inheritance in Man website (http://www.ncbi.nlm.nih.gov/omim/) using the term 'lipid disorders' reveals that most of the 124 hits already have genes assigned and mutations identified, explaining some of the molecular bases for the clinical/biochemical phenotypes. These disorders tend to be rare, however, and they do not account for the most common dislipidaemias present in the general population. The truth is that it is not known how many genes and single nucleotide polymorphisms (SNPs) will be needed in order to classify people precisely according to their genetic predisposition to cardiovascular disease (CVD), but it is likely that one should gear up for a long and difficult race [9].
At this time, most of the obvious 'candidate' genes have already been identified and characterised, including those coding for the known apolipoproteins, lipolytic enzymes, cellular receptors, lipid transporters and some transcription factors, although the picture provided is neither complete nor clear. Researchers need to identify many more new genes, and this endeavour is being accomplished using multiple approaches, ranging from bioinformatics[10] to whole genome scans [11]. More powerful tools are now needed to harness the complexity of the problem at hand. The necessary power will come from the more potent statistical and bioinformatics tools that are being developed, and by taking into consideration the significant contribution of scores of environmental factors to the phenotypes being considered.
Lipid genetics: Is this a worthy cause?
Diseases of the circulatory system, including CVD, are the major cause of morbidity and mortality in the world, and current trends indicate that this is not going to change in the near future. The good news is that CVD can be, and is, pre-vented [12]. CVD risk is determined by a combination of modifiable and non-modifiable (including genetic) factors, therefore, the current thinking is that an accurate prediction of an individual's genetic predisposition will lead to those who are at higher risk being targetted more efficaciously. Thus, one of the most important outcomes from the search for the genetic factors involved in lipid metabolism will be an increased ability to identify early and accurately 'who is who' in CVD risk. It would be frustrating, however, if all that one could do was to 'label' people without providing proper guidance towards a healthier and longer CVD-free life. This is where the new areas of nutrigenetics, pharmacogenetics and other areas that could fall within the general term of 'envirogenetics' come into play.
Many polymorphisms, many promises, few realisations
Many polymorphisms, at scores of gene loci, have been examined to uncover associations with lipid-related variables; however, very few have shown consistent results, and in many cases they do not provide independent information over that elicited from the currently used biochemical markers. If this is the case, has all this effort been a huge waste of time and resources? The answer is probably 'no'. One should keep in mind that the lifetime tracking of biochemical markers tends to be poor. Therefore, genetic markers could still be used as early predictors, well before the phenotypes are expressed (see Figure 1). Moreover, they have the potential to guide the choice of effective preventive/therapeutic approaches (see Figure 2).
Figure 1.
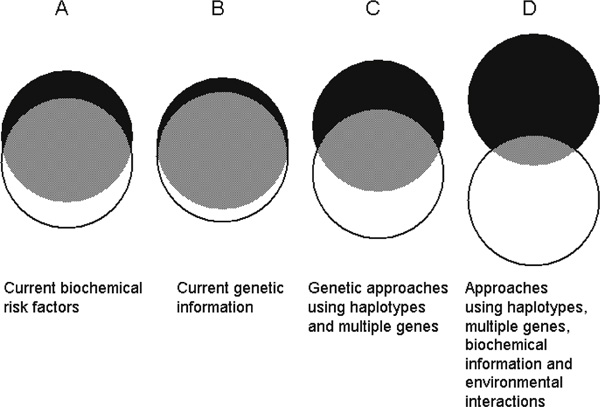
'Reducing the eclipse': The classical biochemical predictors of coronary heart disease still have a significant overlap (grey area) between cases (black circles) and controls (white circles). Current information based on isolated markers does not provide much better separation. The notion is that merging genetic, biochemical and behavioural information will provide the tools to get an early and close-to-complete separation between those individuals who could become 'cases' and those who will remain free of disease.
Figure 2.
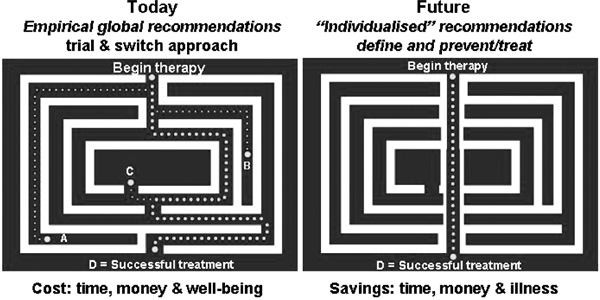
Therapeutic effects of dietary, behavioural and pharmaceutical treatments can be predicted for the 'population', but when it comes to the individual, we are still limited to trial and error. The fields of pharmacogenetics and nutrigenetics aim to cut through the maze and provide more effective personalised recommendations.
The gene that has provided most excitement by far to the genetic epidemiology of lipids is the APOE gene. The potential impact on public health of genetic variation at this locus may have even exceeded the wildest dreams of Utermann et al. when they reported for the first time common variants in the population in 1975 [13]. Other genes are falling far behind in terms of consistency of the results being obtained and impact on the population at large. The reasons for the poor outcome or lack of consistency for this research are many and some of them have been recently analysed and discussed (see Table 1) [14-16].
Table 1.
Most plausible causes of discrepancies among different genetic association studies
| Random errors | Confounders | Interactions |
|---|---|---|
| • Bogus association due to multiple testing | • Phenotype is affected by variants at several genes (locus heterogeneity) | • Lack of consideration of gene-gene interactions |
| • Small sample size | • Phenotype is affected by several variants at each locus (allele heterogeneity) | • Lack of consideration of gene-environment interactions |
| • Inadequate choice of genetic variant | • Population stratification, especially on case-control studies | |
| • Genotyping errors | • Use of single markers instead of haplotypes |
Pharmacogenetics of lipid disorders
It has been rigorously demonstrated that lowering serum lipid levels with pharmacological agents results in significant reductions in CVD events and total mortality in both primary and secondary prevention trials;[17] however, the magnitude of plasma lipid responses to hypolipaemic drug therapies varies considerably among individuals [18]. These differences may be due to the multiple genetic factors that affect pharmaco-kinetics (eg polymorphisms in the genes coding for drug metabolic enzymes) or pharmacodynamics (polymorphisms in drug target genes), as well as a myriad of modifiable factors that may account for as much of the variability as the genetic components themselves. Increasing knowledge of the network of genes that govern drug response should allow for the possibility of optimising drug therapy based on the individual's molecular diagnosis and response [19,20].
There is much potential in the use of pharmacogenetics to improve the success of achieving individual target lipid levels, saving lives, time and money (see Figure 2). While current information is sufficiently enticing to prove the concept of the genetic basis of drug responses however, it does not provide a strong incentive for the pharmaceutical industry to adopt the concept, or for the clinician to use these tools in their daily practice.
The initial studies addressing gene-lipid-lowering drug interactions date back to the late 1980s, although this concept did not generate much general attention until the report from the Regression Growth Evaluation Statin Study (REGRESS), related the cholesterol ester transfer protein gene (CETP) locus to differential changes in the progression of atherosclerosis following pravastatin therapy [21]. The common CETP TaqIB polymorphism appeared to predict whether men with coronary artery disease (CAD) would benefit from treatment with pravastatin to delay the progression of coronary atherosclerosis independently of effects on plasma lipids. The puzzling finding was that the people who benefited less were those who carried the protective B2 allele [22]. The solution to this paradox may be scattered through other reports demonstrating that the response of high-density lipoprotein (HDL)-C levels to atorvastatin treatment was associated with the TaqIB polymorphism in the direction that one would have predicted based on the REGRESS results [23,24], and this was especially found to be true in diabetic subjects [25]. Unsurprisingly, however, the locus that has been examined by a larger number of investigators is the APOE gene.
Current pharmacogenetic knowledge is summarised in Table 2. Several of the large lipid-lowering trials and several smaller studies have been contributing with some limited, and frequently controversial, information; this is also happening in other areas of pharmacogenetics [16].
Table 2.
Summary of lipid-related pharmacogenetic studies by loci
| Gene locus | Clinical trial/population | Medication | Statistically significant gene-therapy interaction with biochemical (B) or clinical (C) outcomes | Reference |
|---|---|---|---|---|
| ABCA1 | LCAS | FLUVA | Y (B) | [66] |
| ACE | LCAS | FLUVA | Y (C) | [75] |
| CARE | PRAVA | N | [76] | |
| Canada | GEMF | Y (B) | [77] | |
| APOA4 | PLAC1 | PRAVA | N | [35] |
| APOB | Finland | LOVA | N | [31] |
| Brazil | FLUVA | Y (B) | [67] | |
| APOE | Japan | HRT | Y (B) | [45] |
| KORFPS | HRT | Y (B) | [46] | |
| Japan | HRT | Y (B) | [47] | |
| NMAPS | HRT | N | [48] | |
| Canada | PROB | Y (B) | [28] | |
| Netherlands (FH) | SIMVA | N | [29] | |
| USA (FH) | LOVA | N | [30] | |
| Finland | LOVA | N | [31] | |
| Sweden (FH) | CHOLY/PRAVA | N | [32] | |
| Japan | PRAVA | Y (B) | [33] | |
| Canada | LOVA | Y (B) | [34] | |
| PLAC1 | PRAVA | Y (B) | [35] | |
| USA | ATORVA | Y (B) | [36] | |
| Japan | BEZA | Y (B) | [37] | |
| 4S | SIMVA | Y (C) | [38] | |
| LCAS | FLUVA | Y (B) | [39] | |
| Japan | PRAVA | Y (B) | [40] | |
| Spain | ATORVA+BEZA | Y (B) | [41] | |
| Canada | FENO | Y (B) | [42] | |
| Spain | PRAVA | N | [43] | |
| AT2R1 | REGRESS | PRAVA | N | [101] |
| B1AR | WOSCOPS | PRAVA | N | [78] |
| CD14 | LCAS | FLUVA | N | [79] |
| CETP | REGRESS | PRAVA | Y (C) | [21] |
| WOSCOPS | PRAVA | Y (C) | [23] | |
| Japan | Statin | Y (B) | [24] | |
| Korea | HRT | N | [102] | |
| DALI | ATORVA | Y (B) | [25] | |
| CYP2D6 | Netherlands | SIMVA | Y (B) | [80] |
| Germany | SIMVA | N | [81] | |
| ESR1 | ERA | HRT | Y (B) | [68] |
| Factor XII | WOSCOPS | PRAVA | Y (C) | [82] |
| FGA | REGRESS | PRAVA | Y (C) | [83] |
| LIPC | FATS | LOVA/COLEP/NIA | Y (C) | [65] |
| Japan | HRT | N | [103] | |
| IL-6 | LCAS | FLUVA | N | [79] |
| WOSCOPS | PRAVA | Y (C) | [84] | |
| LDLR | South Africa (FH) | SIMVA | Y (B) | [50] |
| Norway (FH) | LOVA | N | [51] | |
| FHRT (UK) | SIMVA (+) | N | [52] | |
| Japan (FH) | PRAVA+CHOLY | Y (B) | [53] | |
| Canada (FH) | SIMVA | Y (B) | [54] | |
| Netherlands (FH) | SIMVA | N | [55] | |
| UK (FH) | SIMVA | Y (B) | [56] | |
| Brazil (FH) | FLUVA | Y (B) | [57] | |
| Spain (FH) | SIMVA | Y (B) | [58] | |
| FH | SIMVA | Y (B) | [59] | |
| Denmark (FH) | FLUVA | N | [60] | |
| LPL | REGRESS | PRAVA | N | [69] |
| LCAS | FLUVA | Y* (B) | [70] | |
| Canada | FENO | Y (B) | [42] | |
| MMP3 | REGRESS | PRAVA | Y (C) | [85] |
| PON1 | Spain (FH) | SIMVA | N | [72] |
| LCAS | FLUVA | N | [73] | |
| PPARA | Canada | GEMF | Y (B) | [71] |
| Canada | FENO | Y (B) | [42] | |
| PPARG | Canada | Feno | N | [42] |
| TNFA | LCAS | FLUVA | N | [79] |
Studies/populations
4S: Scandinavian Simvastatin Survival Study; CARE: Cholesterol and Recurrent Events; DALI: Diabetes Atorvastatin Lipid Interaction; ERA: Estrogen Replacement and Atherosclerosis trial; FATS: Familial Atherosclerosis Treatment Study; FH: Familial Hypercholesterolaemia; FHRT: Familial Hypercholesterolaemia Regression Trial; KORFPS: Kuo-pio Osteoporosis Risk Factor and Prevention Study; LCAS: Lipoprotein and Coronary Atherosclerosis Study; NMAPS: New Mexico Aging Process Study; PLAC1 Pravastatin Limitation of Atherosclerosis in Coronary Arteries Study-1; REGRESS: Regression Growth Evaluation Statin Study; WOSCOPS: West Of Scotland Coronary Prevention Study.
Medications
ATORVA: atorvastatin; BEZA: bezafibrate; CHOLY: cholestyramine; COLEP: colestipol; FENO: fenofibrate; FLUVA: fluvastatin; GEMF: gemfibrozil; HRT: hormone replacement therapy; LOVA: lovastatin; NIA: niacin; PRAVA: pravastatin; PROB: probucol; SIMVA: simvastatin.
Genes
ABCA1: ATP-binding cassette, subfamily A, member 1; ACE: angiotensin I-converting enzyme; APOB: apolipoprotein B; APOE: apolipoprotein E; APOA4: apolipoprotein A-IV; AT2R1: angiotensin II receptor, vascular type 1; B1AR: beta-1-adrenergic receptor; CD14: monocyte differentiation antigen CD14; CETP: cholesteryl ester transfer protein; CYP2D6: cytochrome P450, subfamily IID; ESR1: estrogen receptor 1; Factor XII: coagulation factor XII; FGA: fibrinogen, A alpha polypeptide; IL6: interleukin 6; LDLR: low-density lipoprotein receptor; LIPC: lipase, hepatic; LPL: lipoprotein lipase; MMP3: matrix metalloproteinase 3; PON1: paraoxonase 1; PPARA: peroxisome proliferator-activated receptor-alpha; PPARG: peroxisome proliferator-activated receptor-gamma; TNFA: tumour necrosis factor, alpha.
*borderline significance (P ≈ 0:05):
APOE: A multifaceted protein
As previously mentioned, the APOE locus has been the most studied locus in lipid pharmacogenetics. Common genetic variability at the APOE gene, as defined by the e2, e3 and e4 alleles, was initially associated with plasma lipoprotein levels, CVD risk and a long list of other non-lipid related traits, such as neurological disorders, eye diseases and cancer [26]. Among all the many loci investigated in terms of variation in plasma lipid levels, the APOE locus has provided the most consistent associations for plasma LDL-C concentrations in the general population. Moreover, it appears that variation at the APOE gene may also be a significant indicator of response to both dietary[27] and pharmacological interventions [20,28-48].
Several studies have shown that, following statin therapy, male subjects carrying the APOE2 allele tend to have the greatest LDL-C decreases, whereas APOE4 carriers have the smallest. Based on those observations, one would be tempted to conclude that male APOE4 carriers might benefit the least from statins; however, when clinical events have been examined in relation to statin treatment and APOE genotype, the outcome was paradoxical [38]. Data from the Scandinavian Simvastatin Survival Study (4S) showed that among myocardial infarction survivors, carriers of the APOE4 allele had a nearly twofold increased risk of dying compared with other patients; however, the good news was that the genetic predisposition towards excess mortality was abolished by statin treatment. This was true despite the fact that the effectiveness of simvastatin in reducing LDL-C levels in subjects with different APOE genotypes was not significantly different. Likewise, the Lipoprotein and Coronary Atherosclerosis Study (LCAS) showed that, in agreement with the emerging trend, subjects with the APOE4 allele had a smaller reduction in LDL-C levels with fluvastatin; however, they had similar benefit in terms of CAD progression/regression [39].
The available data support the following notions:
• The APOE genotype may be responsible for a statistically significant but clinically modest variation in the LDL-C response to statins;
• This gene-statin interaction appears to be present mainly in men;
• Despite the slightly lower LDL-C response associated with the presence of APOE4, the increased genetic predisposition towards CVD risk present in carriers of this allele seems to be abolished following statin therapy.
• It should be kept in mind, however, that the data are fragmented and inconsistent, leaving several relevant questions unexplained:
• Why is the gene for a statin interaction observed for the LDL-C response in men not present in women?
• Are male APOE2 carriers likely to have a greater benefit, in terms of cardiovascular mortality and morbidity, than APOE4 or APOE3/3 subjects? This should be expected based on their significantly greater LDL-C reductions; however, this has not been addressed in previous studies, due to the small numbers of subjects involved.
• Which pleiotropic effect is responsible for the fact that APOE4 carriers are less responsive to statins in terms of LDL-C lowering, even though they get similar, or even greater, clinical benefit?
It is interesting to highlight that the pattern of LDL-C responses, according to APOE genotype observed for statins in men, parallels that seen with hormone replacement therapy (HRT) in women. Statins, like HRT, upregulate the number of LDL receptors (LDLRs). Therefore, the APOE2 allele and statins may have additive effects and this may also be the case for HRT and APOE2. Conversely, the apoE4 isoform induces a decrease in LDL receptor numbers, which counteracts the statin-induced increase.
The LDLR gene: Too many mutations, too few subjects
For many years, investigators have considered the LDLR gene an obvious focus of interest to examine the impact of a monogenic disorder (familial hypercholesterolaemia [FH]) on biochemical, clinical and disease phenotypes. The number of mutations in the LDLR gene is growing continuously. Most of these are very rare and associated with the clinical manifestation of FH; however, some of these mutations are relatively common in the population, although there is no clear evidence that they play a major role in the population attributable risk for high cholesterol levels [49]. The LDLR locus has also been an obvious target for the study of the pharmacogenetics of statin therapy. In general, these studies have examined different types of FH mutations in relation to LDL-C lowering by statins in FH heterozygotes [50-60]. The data presented in Table 2 show a balance between those who reported a modulating effect of the LDLR gene and those who did not find any significant interaction. The major caveats for most of these studies are the small sample sizes and the heterogeneity of the populations studied (Table 3).
Table 3.
Summary of lipid-related pharmacogenetic studies according to biochemical/clinical phenotypes
| Phenotype(s) | Clinical trial/population | Medication | Statistically significant gene-therapy interaction with biochemical or clinical outcomes (locus involved) | Reference |
|---|---|---|---|---|
| TC/LDL-C/apoB | LCAS | FLUVA | Y (ACE) | [75] |
| PLAC1 | PRAVA | N (APOA4) | [35] | |
| Brazil | FLUVA | Y (APOB) | [67] | |
| Finland | LOVA | N (APOB) | [31] | |
| USA | ATORVA | Y (APOE) | [36] | |
| Spain | ATORVA+BEZA | Y (APOE) | [41] | |
| Japan | BEZA | Y (APOE) | [37] | |
| LCAS | FLUVA | Y (APOE) | [39] | |
| KORFPS | HRT | Y (APOE) | [46] | |
| Japan | HRT | Y (APOE) | [47] | |
| NMAPS | HRT | N (APOE) | [48] | |
| Canada | LOVA | Y (APOE) | [34] | |
| USA (FH) | LOVA | N (APOE) | [30] | |
| Finland | LOVA | N (APOE) | [32] | |
| PLAC1 | PRAVA | Y (APOE) | [35] | |
| Japan | PRAVA | Y (APOE) | [40] | |
| Japan | PRAVA | Y (APOE) | [33] | |
| Spain | PRAVA | N (APOE) | [43] | |
| Sweden (FH) | PRAVA/CHOLY | N (APOE) | [32] | |
| Canada | PROB | Y (APOE) | [28] | |
| Netherlands (FH) | SIMVA | N (APOE) | [29] | |
| FH | SIMVA | Y (APOE) | [59] | |
| Canada (FH) | SIMVA | Y (APOE) | [54] | |
| Netherlands | SIMVA | Y (CYP2D6) | [80] | |
| Germany | SIMVA | N (CYP2D6) | [81] | |
| Brazil (FH) | FLUA | Y (LDLR) | [57] | |
| Denmark (FH) | FLUVA | N (LDLR) | [60] | |
| Norway (FH) | LOVA | N (LDLR) | [51] | |
| Japan (FH) | PRAVA+CHOLY | Y (LDLR) | [53] | |
| South Africa (FH) | SIMVA | Y (LDLR) | [50] | |
| FH | SIMVA | N (LDLR) | [59] | |
| UK (FH) | SIMVA | Y (LDLR) | [56] | |
| Netherlands (FH) | SIMVA | N (LDLR) | [55] | |
| Canada (FH) | SIMVA | Y (LDLR) | [54] | |
| UK (FHRT) | SIMVA (+) | N (LDLR) | [52] | |
| Spain (FH) | SIMVA | Y (LDLR) | [58] | |
| Japan | HRT | N (LIPC) | [103] | |
| Spain (FH) | SIMVA | N (PON1) | [72] | |
| LCAS | FLUVA | N (PON1) | [73] | |
| HDL-C/APOA1 | LCAS | FLUVA | Y (ABCA1) | [66] |
| Canada | GEMF | Y (ACE) | [77] | |
| Canada | LOVA | Y (APOE) | [34] | |
| Canada | FENO | Y (APOExPPARAxLPL) | [42] | |
| Japan | HRT | [45] | ||
| DALI | ATORVA | [25] | ||
| Korea | HRT | Y (APOE)** | [102] | |
| Japan | Statin | (LDL/HDL) | [24] | |
| ERA | HRT | Y (CETP) | [68] | |
| Japan | HRT | N (CETP) | [103] | |
| Canada | FENO | Y (CETP) | [42] | |
| LCAS | FLUVA | Y (ESR1) | [70] | |
| LCAS | FLUVA | N (LIPC) | [73] | |
| Spain (FH) | SIMVA | N (LPL) | [72] | |
| Canada | GEMF | Y* (LPL) | [71] | |
| Canada | FENO | N (PON1) | [42] | |
| Canada | FENO | N (PON1) | ||
| Y (PPARA) | ||||
| N (PPARG) | ||||
| TG | Spain | ATORVA+BEZA | Y (APOE) | [41] |
| USA | Y (APOE) | [36] | ||
| Canada | ATORVA | Y | [42] | |
| DALI | FENO | (APOExPPARAxLPL) | [25] | |
| Canada | ATORVA | [42] | ||
| Canada | FENO | Y (CETP) | [42] | |
| FENO | N (LPL) | |||
| N (PPARG) | ||||
| Disease end points/disease assessment | LCAS | FLUVA | Y (ACE) | [75] |
| CARE | PRAVA | Y (ACExGP3A) | [76] | |
| 4S | SIMVA | Y (APOE) | [38] | |
| REGRESS | PRAVA | N [AT2R1] | [101] | |
| WOSCOPS | PRAVA | N (B1AR) | [78] | |
| LCAS | FLUVA | N (CD14) | [79] | |
| REGRESS | PRAVA | Y (CETP) | [21] | |
| WOSCOPS | PRAVA | Y (CETP) | [23] | |
| WOSCOPS | PRAVA | Y (Factor XII) | [82] | |
| REGRESS | PRAVA | Y (FGA) | [83] | |
| LCAS | FLUVA | N (IL6) | [79] | |
| WOSCOPS | PRAVA | Y (IL6) | [84] | |
| FATS | LOVA/COLEP/ | Y (LIPC) | [65] | |
| REGRESS | NIA | N (LPL) | [69] | |
| REGRESS | PRAVA | Y (MMP3) | [85] | |
| LCAS | PRAVA | Y* (LPL) | [70] | |
| LCAS | FLUVA | N (PON1) | [73] | |
| Spain (FH) | FLUVA | N (PON1) | [72] | |
| LCAS | SIMVA | N (TNFA) | [79] | |
| FLUVA |
Phenotypes
apoB: Apolipoprotein B; APOA1: apoliprotein AI; HDL: High-density lipoprotein cholesterol; LDL-C: low-density lipoprotein-C; TC: total cholesterol; TG: triglyceride.
Studies/populations
4S: Scandinavian Simvastatin Survival Study; CARE: Cholesterol and Recurrent Events; DALI: Diabetes Atorvastatin Lipid Intervention; ERA: Estrogen Replacement and Atherosclerosis trial; FATS: Familial Atherosclerosis Treatment Study; HRT: hormone replacement therapy; KORFPS: Kuopio Osteoporosis Risk Factor and Prevention Study; LCAS: Lipoprotein and Coronary Atherosclerosis Study; NMAPS: New Mexico Aging Process Study; PLAC1: Pravastatin Limitation of Atherosclerosis in Coronary Arteries Study-1; REGRESS: Regression Growth Evaluation Statin Study; WOSCOPS: West Of Scotland Coronary Prevention Study.
Medications
ATORVA: atorvastatin; BEZA: bezafibrate; CHOLY: cholestyramine; COLEP: colestipol; FENO: fenofibrate; FLUVA: fluvastatin; GEMF: gemfibrozil; LOVA: lovastatin; NIA: niacin; PRAVA: pravastatin; PROB: probucol; SIMVA: simvastatin.
Genes
ABCA1: atp-binding cassette, subfamily a, member 1; ACE: Angiotensin I-converting enzyme; APOB: Apolipoprotein B; APOE: Apolipoprotein E; APOA4: Apolipoprotein A-IV; AT2R1: Angiotensin II receptor, vascular type 1; B1AR: Beta-1-adrenergic receptor; CD14: Monocyte differentiation antigen CD14; CETP: Cholesteryl ester transfer protein; CYP2D6: Cytochrome P450, subfamily IID; ESR1: Estrogen receptor 1; Factor XII: Coagulation factor XII; FGA: Fibrinogen, A alpha polypeptide; GP3A: Platelet-specific antigen system PL(A1); IL6: Interleukin 6; LDLR: Low-density lipoprotein receptor; LIPC: Lipase, hepatic; LPL: Lipoprotein lipase; MMP3: Matrix metalloproteinase 3; PON1: Paraoxonase 1; PPARA: Peroxisome proliferator-activated receptor-alpha; PPARG: Peroxisome proliferator-activated receptor-gamma; TNFA: Tumour necrosis factor, alpha.
*borderline significance (p ≈ 0.05); **the phenotype reported is low-density lipoprotein-C/high-density lipoprotein-C.
CETP: a 'rising' gene
The CETP gene and the product of its expression are emerging as important players in the future of risk assessment, pharmacogenetics and new drug targets. The association between common variants and HDL-C concentrations is physiologically relevant and fairly consistent among populations [61]. The consistency of the associations between CETP polymorphisms and HDL-C levels can be compared with those observed for the APOE gene in relation to LDL-C levels. The same cannot be said, however, about its association with CVD risk [62], which remains controversial. With regard to the interaction between common variants and drug treatment, there is growing evidence for a significant role for the CETP locus in determining the efficacy of statin therapy [21,23-25].
The hepatic lipase (LIPC) gene: Protective or not?
Hepatic lipase (HL) is a plasma lipolytic enzyme that participates in the metabolism of intermediate-density lipoprotein and large LDL into smaller, denser LDL particles, and in the conversion of HDL2 to HDL3 during reverse cholesterol transport. It has also been suggested that HL acts as a ligand for cell-surface proteoglycans in the uptake of lipoproteins by cell-surface receptors. HL deficiency is characterised by mildly elevated concentrations of triglyceride-rich LDL and HDL particles, as well as impaired metabolism of postprandial triglyceride-rich lipoproteins, which may result in premature atherosclerosis. Conversely, increased HL activity has been associated with increased small, dense LDL particles and decreased HDL2 concentrations, which may also result in increased CAD risk.
Four common SNPs on the 5'-flanking region of the HL gene (LIPC) [-763(A/G); -710(T/C); -514(C/T) and -250(G/A)] appear to be in total linkage disequilibrium and define a unique haplotype which has been associated with variation in HL activity and HDL-C levels [63]. The less common A allele of the SNP at position -250 was associated with lower HL activity and buoyant LDL particles. Normal and CAD subjects heterozygous for the A allele had lower HL activity and significantly more buoyant LDL particles. Homozygosity for this allele (AA) was associated with an even lower HL activity. The A allele was associated with higher HDL2-C [64].
Given the wide spectrum of effects that HL exerts on lipoprotein metabolism, and the significance of the promoter variant(s), it is reasonable to hypothesise that genetic variation at this locus may also be involved in variability in the response to hypocholesterolaemic treatment. This notion was recently explored by Zambon et al. in the Familial Atherosclerosis Treatment Study (FATS) trial [65]. Following intensive lipid-lowering therapy, subjects with the CC genotype at the -514(C/T) polymorphism, which is equivalent to the A allele previously described above for the -250(G/A) polymorphism, had a greater decrease in HL activity compared with carriers of the T allele. Consistent with this effect on HL, these subjects also experienced a greater improvement in their LDL-subclass distribution, as well as increases in HDL2-C concentrations. In addition to the greater improvement in their lipid profile, CC subjects also demonstrated the greatest coronary stenosis regression, as determined by quantitative angiography. This is the first report of a pharmacogenetic interaction for the LIPC locus and, although promising, needs replication by other investigators.
Other lipid genes (ABCA 1 [66], APOA4 [35], APOB [31,67], ERA [68], LPL [42,69,70], and PPARA[42,71]) and PON1 [72,73], despite their interest as candidate genes for lipid disorders and the number of publications showing different degrees of associations, have been less studied and it is too soon to make a verdict about their contribution to the individual variability in response. Some of these genes are beginning to emerge as serious players, especially when it comes to therapies involving fibric acid derivatives. In this case, it comes as little surprise that the PPARA gene is already showing significant effects [42,71]. There are, however, some notorious absences, such asthe 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase (HMGCR) gene, for which no data have been yet reported [74].
Considering the multifactorial nature of CVD and the potential pleiotropic effects of the statins, other genes related mainly to hypertension, coagulation, inflammation, vascular structure and drug metabolism have been examined. Again, the outcome has been mixed and based on too few studies and subjects (see Table 2) [68,75-85].
Nutrigenetics
Nutrigenomics and nutrigenetics are emerging as promising multidisciplinary systems that focus on studying the interactions between nutrition, genetic factors and health outcomes, using similar technical and conceptual developments to those described for pharmacogenomics and pharmacogenetics. The terms 'nutrigenomics' and 'nutrigenetics' have largely been used interchangeably;[86-89] however, there are significant conceptual differences in their approaches and aims.
Nutrigenomics applies to the comprehensive, genome-wide assessment of the effects of dietary factors or interventions, and is concerned with the systematic assessment of how nutrients modify the overall expression pattern in cells and tissues of interest. By contrast with nutrigenetics, nutrigenomics does not focus on interindividual differences with regard to the effects of nutrients, but rather focuses on differences among several dietary conditions or factors in quantitative measures of expression and their association with specific phenotype characteristics.
Nutrigenetics, however, describes the interactions between nutrients (or dietary habits) and the characteristics of individuals, which, to some extent, will be determined by their genetic makeup. Thus, nutrigenetics is based on observations of dietary responses in individuals and tests the hypothesis that interindividual differences in the observed response may be associated with the presence or absence of individual-specific biological markers, in most cases SNPs, which may allow prediction of individual dietary responses [27,90,91].
Although nutrigenetics and nutrigenomics share many of the concepts of pharmacogenetics and pharmacogenomics, there also important differences: 1) the latter apply to the segment of the population needing drug therapy, whereas the former apply to everyone; and 2) in general, the magnitude of the differences resulting from nutritional effects is much smaller than that expected from pharmacological agents, making the characterisation of interactions more difficult.
Nutrigenetics is moving forward through similar convoluted paths as those described for pharmacogenetics [27,91]. The findings of this author's group, using well-characterised population studies[92-95] as well as dietary intervention trials [96-98], support statistically significant interactions between several candidate genes, plasma lipid concentrations and dietary factors.
Individuals who respond better to one type of recommendation than to another can now begin to be characterised. For example, a low fat, low cholesterol strategy may be especially beneficial in terms of lowering plasma cholesterol levels in those subjects carrying the APOE4 allele at the APOE gene [99]. This also applies to other CVD risk factors, such as HDL concentrations. The levels of HDL are also modulated by dietary, behavioural and genetic factors. It has recently been reported that the effect of dietary intake of polyunsaturated fatty acids (PUFAs) on HDL-cholesterol concentrations is modulated by a common genetic polymorphism in the promoter region of the APOA1 gene. Thus, subjects carrying the A allele at the -75 G/A polymorphism show an increase in HDL-C concentrations with increased intakes of PUFAs, whereas those homozygotes for the more common G allele show the expected lowering of HDL-C levels as the intake of PUFAs goes up [93]. Most interestingly, significant interactions have also been reported between the intake of fat, specifically of animal origin, and variability at the HL gene [94]. These data could identify a segment of the population especially susceptible to diet-induced atherosclerosis. Moreover, these data could shed some light on the impaired ability of certain ethnic groups to adapt to new nutritional environments, as is clearly seen for Native Americans and Asian Indians [95].
This knowledge should pave the way for more successful dietary recommendations based on genetic factors; this may help to reduce CVD risk more efficiently than the current universal recommendations. The same concept is clearly applicable to other age-related disorders.
Summary
The data presented here suggest that at some time in the future it will be possible to make preventive recommendations and medical decisions based on an individual's specific genetic makeup; however, this is a process that will slowly permeate into clinical practice and public health systems. Before this occurs, there is much that needs to be done [9]. First, the level of consistency among scientific reports needs to be increased before researchers can move to issues of sensitivity and specificity, as has traditionally been the case for other non-genetic assays. It should also be emphasised that most of the findings have been reported primarily in white males, and there is evidence that males and females respond differently to intervention, and that the same genetic determinants may not translate equally into phenotypes. Moreover, it is known that different ethnic groups have different haplotypes, and that the findings reported for white populations do not necessarily apply to other ethnic groups, underscoring the need for comprehensive examination of these questions in different ethnic groups and geographical areas.
Once researchers have solid evidence about the predictive value of genetic panels, they can start generating risk and therapeutic algorithms that should provide the measure of genetic predisposition, as well as targeted behavioural modifications or drugs of choice and also personalised dosages. Finally, there is a need to put these developments to the test in 'real life'. The feasibility and success of this concept will need to be demonstrated using clinical trials comparing standard care versus approaches using genetic information.
The criteria for pharmacogenetic studies are very similar to those previously suggested for genetic association studies:[100]
1. The heritability of the phenotype needs to be established. This is already obvious for CVD; however, it is less established for some of the intermediate phenotypes used in association studies, and there have not been studies investigating this heritability for response to pharmacological therapies aimed at decreasing cardiovascular risk;
2. The choice of candidate genes needs to be justified mainly on the basis of functional information;
3. Subjects should be carefully matched to account for potential genetic and environmental confounders;
4. Despite the issue highlighted in point number 2 above, the reality is that some exciting findings may result from fortuitous observations; however, the findings from these exploratory analyses need to be substantiated by replication in an independent sample or by functional studies involving in vitro or in vivo laboratory experimentation;
5. Sometimes, significant findings are reported after subjecting the data to multiple comparisons in several groups and subgroups. Efforts to account for errors due to multiple comparisons need to be clearly specified. Associations should be reported using odds ratios and confidence limits, as well as P-values;
6. Selective publication is almost unavoidable. It is more rewarding and easier to report significant associations than to report a lack of association, whether this is a new finding or refutes previous reports. It should be remembered that well-designed and ably carried out 'negative' research can be very valuable. Again, in this case, demonstration of the statistical power of the study to show lack of association is of paramount importance;
7. Replication of findings is important, both internally within a report and also by other researchers. Therefore, such reports are also important to the field; however, in order to avoid cluttering the current literature, confirmatory studies should also provide some novel information beyond mere replication.
Acknowledgements
Supported by NIH/NHLBI grant no. HL54776, NIH/NHLBI contract no. 1-38038 and contracts 53-K06-5-10 and 58-1950-9-001 from the US Department of Agriculture Research Service, the author would like to express his deepest gratitude to the late Richard H. Ward, who was a continual source of learning and inspiration during the initial stages of the author's genetic research -- he feels very privileged to have had him as collaborator and friend.
References
- Fredrickson DS. 'Phenotyping. On reaching base camp (1950-1975)'. Circulation. 1993;87:III1–III15. [PubMed] [Google Scholar]
- Heath KE, Gahan M, Whittall RA. et al. 'Low-density lipo-protein receptor gene (LDLR) world wide website in familial hypercholesterolaemia: Update, new features and mutation analysis'. Atherosclerosis. 2001;154:243–246. doi: 10.1016/S0021-9150(00)00647-X. [DOI] [PubMed] [Google Scholar]
- Bodzioch M, Orso E, Klucken J. et al. 'The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease'. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- Rust S, Rosier M, Funke H. et al. 'Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1'. Nat Genet. 1999;22:352–355. doi: 10.1038/11921. [DOI] [PubMed] [Google Scholar]
- Brooks-Wilson A, Marcil M, Clee SM. et al. 'Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency'. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- Brousseau ME, Schaefer EJ, Dupuis J. et al. 'Novel mutations in the gene encoding ATP-binding cassette 1 in four Tangier disease kindreds'. J Lipid Res. 2000;41:433–441. [PubMed] [Google Scholar]
- Lawn RM, Wade DP, Garvin MR. et al. 'The Tangier disease gene product ABC1 controls the cellular apolipoprotein-mediated lipid removal pathway'. J Clin Invest. 1999;104:R25–R31. doi: 10.1172/JCI8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remaley AT, Rust S, Rosier M. et al. 'Human ATP-binding cassette transporter 1 (ABC1): Genomic organization and identification of the genetic defect in the original Tangier disease kindred'. Proc Natl Acad Sci USA. 1999;96:12685–12690. doi: 10.1073/pnas.96.22.12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordovas JM. 'Cardiovascular disease genetics: A long and winding road'. Curr Opin Lipidol. 2003;14:47–54. doi: 10.1097/00041433-200302000-00009. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Olivier M, Hubacek JA. et al. 'An apolipo-protein influencing triglycerides in humans and mice revealed by comparative sequencing'. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- Ordovas JM. 'HDL genetics: Candidate genes, genome wide scans and gene-environment interactions'. Cardiovasc Drugs Ther. 2002;16:273–281. doi: 10.1023/A:1021769523568. [DOI] [PubMed] [Google Scholar]
- Grundy SM. 'United States Cholesterol Guidelines 2001: Expanded scope of intensive low-density lipoprotein-lowering therapy'. Am J Cardiol. 2001. pp. 23J–27J. [DOI] [PubMed]
- Utermann G, Jaeschke M, Menzel J. 'Familial hyper-lipoproteinemia type III: Deficiency of a specific apolipoprotein (apoA-III) in the very low density lipoproteins'. FEBS Lett. 1975;56:352–355. doi: 10.1016/0014-5793(75)81125-2. [DOI] [PubMed] [Google Scholar]
- Lazzeroni LC, Karlovich CA. 'Genotype to phenotype: Associations, errors and complexity'. Trends Genet. 2002;18:283–284. doi: 10.1016/S0168-9525(02)02702-6. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP, Ntzani EE, Trikalinos TA. et al. 'Replication validity of genetic association studies'. Nat Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- Ryan SG. 'Regression to the truth: Replication of association in pharmacogenetic studies'. Pharmacogenomics. 2003;4:201–207. doi: 10.1517/phgs.4.2.201.22631. [DOI] [PubMed] [Google Scholar]
- Auer J, Berent R, Eber B. 'Lessons learned from statin trials'. Clin Cardiol. 2001;24:277–280. doi: 10.1002/clc.4960240404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordovas JM, Shen AH. 'Genetics, the environment and lipid abnormalities'. Curr Cardiol Rep. 2002;4:508–513. doi: 10.1007/s11886-002-0115-4. [DOI] [PubMed] [Google Scholar]
- Meisel C, Gerloff T, Kirchheiner J. et al. 'Implications of pharmacogenetics for individualizing drug treatment and for study design'. J Mol Med. 2003;81:154–167. doi: 10.1007/s00109-002-0417-4. [DOI] [PubMed] [Google Scholar]
- Lindpaintner K. 'Pharmacogenetics and the future of medical practice'. J Mol Med. 2003;81:141–153. doi: 10.1007/s00109-002-0416-5. [DOI] [PubMed] [Google Scholar]
- Kuivenhoven JA, Jukema JW, Zwinderman AH. et al. 'The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The Regression Growth Evaluation Statin Study Group'. N Engl J Med. 1998;338:86–93. doi: 10.1056/NEJM199801083380203. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, Cupples LA, Corella D. et al. 'Association of cholesteryl ester transfer protein-TaqIB polymorphism with variations in lipoprotein subclasses and coronary heart disease risk: The Framingham study'. Arterioscler Thromb Vasc Biol. 2000;20:1323–1329. doi: 10.1161/01.ATV.20.5.1323. [DOI] [PubMed] [Google Scholar]
- Freeman DJ, Wilson V, McMahon AD. et al. 'A polymorphism of the cholesteryl ester transfer protein (CETP) gene predicts cardiovascular (CV) events in the West of Scotland Coronary Prevention Study (WOSCOPS)'. Atherosclerosis. 2000;151:91. doi: 10.1016/j.ehj.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Kotake H, Sekikawa A, Tokita Y. et al. 'Effect of HMG-CoA reductase inhibitor on plasma cholesteryl ester transfer protein activity in primary hypercholesterolemia: Comparison among CETP/TaqIB genotype subgroups'. J Atheroscler Thromb. 2002;9:207–212. doi: 10.5551/jat.9.207. [DOI] [PubMed] [Google Scholar]
- van Venrooij FV, Stolk RP, Banga JD. et al. 'Common cholesteryl ester transfer protein gene polymorphisms and the effect of atorvastatin therapy in type 2 diabetes'. Diabetes Care. 2003;26:1216–1223. doi: 10.2337/diacare.26.4.1216. [DOI] [PubMed] [Google Scholar]
- Eichner JE, Dunn ST, Perveen G. et al. 'Apolipoprotein E polymorphism and cardiovascular disease: A HuGE review'. Am J Epidemiol. 2002;155:487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- Ordovas JM. 'Gene-diet interaction and plasma lipid responses to dietary intervention'. Biochem Soc Trans. 2002;30:68–73. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Nestruck AC, Bouthillier D, Sing CF. et al. 'Apolipoprotein E polymorphism and plasma cholesterol response to probucol'. Metabolism. 1987;36:743–747. doi: 10.1016/0026-0495(87)90110-7. [DOI] [PubMed] [Google Scholar]
- de Knijff P, Stalenhoef AFH, Mol MJTM. et al. 'Influence of apoE polymorphism on the response to simvastatin treatment in patients with heterozygous familial hypercholesterolemia'. Atherosclerosis. 1990;83:89–97. doi: 10.1016/0021-9150(90)90134-5. [DOI] [PubMed] [Google Scholar]
- O'Malley JP, Illingworth DR. 'The influence of apolipo-protein E phenotype on the response to lovastatin therapy in patients with heterozygous familial hypercholesterolemia'. Metabolism. 1990;39:150–154. doi: 10.1016/0026-0495(90)90068-N. [DOI] [PubMed] [Google Scholar]
- Ojala JP, Helve E, Ehnholm C. et al. 'Effect of apolipoprotein E polymorphism and XbaI polymorphism of apolipoprotein B on response to lovastatin treatment in familial and non-familial hypercho-lesterolaemia'. J Intern Med. 1991;230:397–405. doi: 10.1111/j.1365-2796.1991.tb00464.x. [DOI] [PubMed] [Google Scholar]
- Berglund L, Wiklund O, Eggertsen G. et al. 'Apolipoprotein E phenotypes in familial hypercholesterolaemia: Importance for expression of disease and response to therapy'. J Intern Med. 1993;233:173–178. doi: 10.1111/j.1365-2796.1993.tb00670.x. [DOI] [PubMed] [Google Scholar]
- Watanabe J, Kobayashi K, Umeda F. et al. 'Apolipoprotein E polymorphism affects the response to pravastatin on plasma apolipoproteins in diabetic patients'. Diabetes Res Clin Pract. 1993;20:21–27. doi: 10.1016/0168-8227(93)90018-Z. [DOI] [PubMed] [Google Scholar]
- Carmena R, Roederer G, Mailloux H. et al. 'The response to lovastatin treatment in patients with heterozygous familial hypercholes-terolemia is modulated by apolipoprotein E polymorphism'. Metabolism. 1993;42:895–901. doi: 10.1016/0026-0495(93)90066-W. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, Lopez-Miranda J, Perez-Jimenez F. et al. 'Effect of apolipoprotein E and A-IV phenotypes on the low density lipoprotein response to HMG-CoA reductase inhibitor therapy'. Atherosclerosis. 1995;113:157–166. doi: 10.1016/0021-9150(94)05439-P. [DOI] [PubMed] [Google Scholar]
- Pedro-Botet J, Schaefer EJ, Bakker-Arkema RG. et al. 'Apolipoprotein E genotype affects plasma lipid response to atorvastatin in a gender specific manner'. Atherosclerosis. 2001;158:183–193. doi: 10.1016/S0021-9150(01)00410-5. [DOI] [PubMed] [Google Scholar]
- Yamada M. 'Influence of apolipoprotein E polymorphism on bezafibrate treatment response in dyslipidemic patients'. J Atheroscler Thromb. 1997;4:40–44. doi: 10.5551/jat1994.4.40. [DOI] [PubMed] [Google Scholar]
- Gerdes LU, Gerdes C, Kervinen K. et al. 'The apolipoprotein epsilon4 allele determines prognosis and the effect on prognosis of simvastatin in survivors of myocardial infarction: A substudy of the Scandinavian simvastatin survival study'. Circulation. 2000;101:1366–1371. doi: 10.1161/01.CIR.101.12.1366. [DOI] [PubMed] [Google Scholar]
- Ballantyne CM, Herd JA, Stein EA. et al. 'Apolipoprotein E genotypes and response of plasma lipids and progression-regression of coronary atherosclerosis to lipid-lowering drug therapy'. J Am Coll Cardiol. 2000;36:1572–1578. doi: 10.1016/S0735-1097(00)00918-9. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Homma Y. 'Effects of low-dose pravastatin on plasma levels of lipids and apolipoproteins in Japanese type II hyperlipoproteinemic subjects with apolipoprotein E phenotype E3/2, E3/3, and E4/3'. J Clin Pharmacol. 2001;41:1055–1058. doi: 10.1177/00912700122012643. [DOI] [PubMed] [Google Scholar]
- Garcia-Otin AL, Civeira F, Aristegui R. et al. 'Allelic polymorphism -491A/T in apoE gene modulates the lipid-lowering response in combined hyperlipidemia treatment'. Eur J Clin Investig. 2002;32:421–428. doi: 10.1046/j.1365-2362.2002.00996.x. [DOI] [PubMed] [Google Scholar]
- Brisson D, Ledoux K, Bosse Y. et al. 'Effect of apolipoprotein E, peroxisome proliferator-activated receptor alpha and lipoprotein lipase gene mutations on the ability of fenofibrate to improve lipid profiles and reach clinical guideline targets among hypertriglyceridemic patients'. Pharmacogenetics. 2002;12:313–320. doi: 10.1097/00008571-200206000-00007. [DOI] [PubMed] [Google Scholar]
- Pena R, Lahoz C, Mostaza JM. et al. 'Effect of apoE genotype on the hypolipidaemic response to pravastatin in an outpatient setting'. J Intern Med. 2002;251:518–525. doi: 10.1046/j.1365-2796.2002.00991.x. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, Mooser V. 'The APOE locus and the pharmacogenetics of lipid response'. Curr Opin Lipidol. 2002;13:113–117. doi: 10.1097/00041433-200204000-00001. [DOI] [PubMed] [Google Scholar]
- Somekawa Y, Wakabayashi A. 'Relationship between apoli-poprotein E polymorphism, menopausal symptoms, and serum lipids during hormone replacement therapy'. Eur J Obstet Gynecol Reprod Biol. 1998;79:185–191. doi: 10.1016/S0301-2115(98)00008-6. [DOI] [PubMed] [Google Scholar]
- Heikkinen AM, Niskanen L, Ryynanen M. et al. 'Is the response of serum lipids and lipoproteins to postmenopausal hormone replacement therapy modified by ApoE genotype?'. Arterioscler Thromb Vasc Biol. 1999;19:402–407. doi: 10.1161/01.ATV.19.2.402. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Sanada M, Nakagawa H. et al. 'Phenotype of apolipoprotein E influences the lipid metabolic response of postmeno-pausal women to hormone replacement therapy'. Maturitas. 2001;38:297–304. doi: 10.1016/S0378-5122(01)00171-2. [DOI] [PubMed] [Google Scholar]
- Garry PJ, Baumgartner RN, Brodie SG. et al. 'Estrogen replacement therapy, serum lipids and polymorphism of the apolipoprotein E gene'. Clin Chem. 1999;45:1214–1223. [PubMed] [Google Scholar]
- Nicholls P, Young IS, Graham CA. 'Genotype/phenotype correlations in familial hypercholesterolaemia'. Curr Opin Lipidol. 1998;9:313–317. doi: 10.1097/00041433-199808000-00005. [DOI] [PubMed] [Google Scholar]
- Jeenah M, September W, Van Roggen FG. et al. 'Influence of specific mutations at the LDL-receptor gene locus on the response to simvastatin therapy in Afrikaner patients with heterozygous familial hypercholesterolaemia'. Atherosclerosis. 1993;98:51–58. doi: 10.1016/0021-9150(93)90222-G. [DOI] [PubMed] [Google Scholar]
- Leren TP, Hjermann I. 'Is responsiveness to lovastatin in familial hypercholesterolaemia heterozygotes influenced by the specific mutation in the low-density lipoprotein receptor gene?'. Eur J Clin Investig. 1995;25:967–973. doi: 10.1111/j.1365-2362.1995.tb01975.x. [DOI] [PubMed] [Google Scholar]
- Sun XM, Patel DD, Knight BL. et al. 'Influence of genotype at the low density lipoprotein (LDL) receptor gene locus on the clinical phenotype and response to lipid-lowering drug therapy in heterozygous familial hypercholesterolaemia. The Familial Hypercholester-olaemia Regression Study Group'. Atherosclerosis. 1998;136:175–185. doi: 10.1016/S0021-9150(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Kajinami K, Yagi K, Higashikata T. et al. 'Low-density lipo-protein receptor genotype-dependent response to cholesterol lowering by combined pravastatin and cholestyramine in familial hypercholesterolemia'. Am J Cardiol. 1998;82:113–117. doi: 10.1016/S0002-9149(98)00230-6. [DOI] [PubMed] [Google Scholar]
- Couture P, Brun LD, Szots F. et al. 'Association of specific LDL receptor gene mutations with differential plasma lipoprotein response to simvastatin in young French Canadians with heterozygous familial hypercholesterolemia'. Arterioscler Thromb Vasc Biol. 1998;18:1007–1012. doi: 10.1161/01.ATV.18.6.1007. [DOI] [PubMed] [Google Scholar]
- Sijbrands EJ, Lombardi MP, Westendorp RG. et al. 'Similar response to simvastatin in patients heterozygous for familial hypercholes-terolemia with mRNA negative and mRNA positive mutations'. Atherosclerosis. 1998;136:247–254. doi: 10.1016/S0021-9150(97)00216-5. [DOI] [PubMed] [Google Scholar]
- Heath KE, Gudnason V, Humphries SE. et al. 'The type of mutation in the low density lipoprotein receptor gene influences the cholesterol-lowering response of the HMG-CoA reductase inhibitor simvastatin in patients with heterozygous familial hypercholesterolaemia'. Atherosclerosis. 1999;143:41–54. doi: 10.1016/S0021-9150(98)00274-3. [DOI] [PubMed] [Google Scholar]
- Salazar LA, Hirata MH, Quintao EC. et al. 'Lipid-lowering response of the HMG-CoA reductase inhibitor fluvastatin is influenced by polymorphisms in the low-density lipoprotein receptor gene in Brazilian patients with primary hypercholesterolemia'. J Clin Lab Anal. 2000;14:125–131. doi: 10.1002/(SICI)1098-2825(2000)14:3<125::AID-JCLA7>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Real JT, Chaves FJ, Civera M. et al. '[Influence of FH Valencia 1 and 2 mutations of the LDL receptor gene on the response to simvastatin in subjects with molecularly defined heterozygous familial hypercholesterolemia in Spain]'. Med Clin (Barc) 2001;116:81–85. doi: 10.1016/s0025-7753(01)71731-9. [DOI] [PubMed] [Google Scholar]
- Vohl MC, Szots F, Lelievre M. et al. 'Influence of LDL receptor gene mutation and apoE polymorphism on lipoprotein response to simvastatin treatment among adolescents with heterozygous familial hypercholesterolemia'. Atherosclerosis. 2002;160:361–368. doi: 10.1016/S0021-9150(01)00584-6. [DOI] [PubMed] [Google Scholar]
- Brorholt-Petersen JU, Jensen HK, Raungaard B. et al. 'LDL-receptor gene mutations and the hypocholesterolemic response to statin therapy'. Clin Genet. 2001;59:397–405. doi: 10.1034/j.1399-0004.2001.590604.x. [DOI] [PubMed] [Google Scholar]
- Miller M, Rhyne J, Hamlette S. et al. 'Genetics of HDL regulation in humans'. Curr Opin Lipidol. 2003;14:273–279. doi: 10.1097/00041433-200306000-00007. [DOI] [PubMed] [Google Scholar]
- Ordovas JM. 'Genetic polymorphisms and activity of cholesterol ester transfer protein (CETP): Should we be measuring them?'. Clin Chem Lab Med. 2000;38:945–949. doi: 10.1515/CCLM.2000.139. [DOI] [PubMed] [Google Scholar]
- Jansen H, Verhoeven AJ, Weeks. et al. 'Common C-to-T substitution at position -480 of the hepatic lipase promoter associated with a lowered lipase activity in coronary artery disease patients'. Arterioscler Thromb Vasc Biol. 1997;17:2837–2842. doi: 10.1161/01.ATV.17.11.2837. [DOI] [PubMed] [Google Scholar]
- Zambon A, Deeb SS, Hokanson JE. et al. 'Common variants in the promoter of the hepatic lipase gene are associated with lower levels of hepatic lipase activity, buoyant LDL, and higher HDL2 cholesterol'. Arterioscler Thromb Vasc Biol. 1998;18:1723–1729. doi: 10.1161/01.ATV.18.11.1723. [DOI] [PubMed] [Google Scholar]
- Zambon A, Deeb SS, Brown BG. et al. 'Common hepatic lipase gene promoter variant determines clinical response to intensive lipid-lowering treatment'. Circulation. 2001;103:792–798. doi: 10.1161/01.CIR.103.6.792. [DOI] [PubMed] [Google Scholar]
- Lutucuta S, Ballantyne CM, Elghannam H. et al. 'Novel polymorphisms in promoter region of ATP binding cassette transporter gene and plasma lipids, severity, progression and regression of coronary atherosclerosis and response to therapy'. Circ Res. 2001;88:969–973. doi: 10.1161/hh0901.090301. [DOI] [PubMed] [Google Scholar]
- Guzman EC, Hirata MH, Quintao EC. et al. 'Association of the apolipoprotein B gene polymorphisms with cholesterol levels and response to fluvastatin in Brazilian individuals with high risk for coronary heart disease'. Clin Chem Lab Med. 2000;38:731–736. doi: 10.1515/CCLM.2000.103. [DOI] [PubMed] [Google Scholar]
- Herrington DM, Howard TD, Hawkins GA. et al. 'Estrogen-receptor polymorphisms and effects of estrogen replacement on high-density lipoprotein cholesterol in women with coronary disease'. N Engl J Med. 2002;346:967–974. doi: 10.1056/NEJMoa012952. [DOI] [PubMed] [Google Scholar]
- Kastelein JJ, Groenemeyer BE, Hallman DM. et al. 'The Asn9 variant of lipoprotein lipase is associated with the -93G promoter mutation and an increased risk of coronary artery disease. The REGRESS Study Group'. Clin Genet. 1998;53:27–33. doi: 10.1034/j.1399-0004.1998.531530106.x. [DOI] [PubMed] [Google Scholar]
- Sing K, Ballantyne CM, Ferlic L. et al. 'Lipoprotein lipase gene mutations, plasma lipid levels, progression/regression of coronary atherosclerosis, response to therapy, and future clinical events. Lipoproteins and Coronary Atherosclerosis Study'. Atherosclerosis. 1999;144:435–442. doi: 10.1016/S0021-9150(99)00004-0. [DOI] [PubMed] [Google Scholar]
- Bosse Y, Pascot A, Dumont M. et al. 'Influences of the PPAR alpha-L162V polymorphism on plasma HDL(2)-cholesterol response of abdominally obese men treated with gemfibrozil'. Genet Med. 2002;4:311–315. doi: 10.1097/00125817-200207000-00010. [DOI] [PubMed] [Google Scholar]
- Tomas M, Senti M, Garcia-Faria F. et al. 'Effect of simvastatin therapy on paraoxonase activity and related lipoproteins in familial hypercholesterolemic patients'. Arterioscler Thromb Vasc Biol. 2000;20:2113–2119. doi: 10.1161/01.ATV.20.9.2113. [DOI] [PubMed] [Google Scholar]
- Turban S, Fuentes F, Ferlic L. et al. 'A prospective study of paraoxonase gene Q/R192 polymorphism and severity, progression and regression of coronary atherosclerosis, plasma lipid levels, clinical events and response to fluvastatin'. Atherosclerosis. 2001;154:633–640. doi: 10.1016/S0021-9150(00)00495-0. [DOI] [PubMed] [Google Scholar]
- Maitland-van der Zee AH, Klungel OH, Stricker BH. et al. 'Genetic polymorphisms: Importance for response to HMG-CoA reductase inhibitors'. Atherosclerosis. 2002;163:213–222. doi: 10.1016/S0021-9150(01)00725-0. [DOI] [PubMed] [Google Scholar]
- Marian AJ, Safavi F, Ferlic L. et al. 'Interactions between angiotensin-I converting enzyme insertion/deletion polymorphism and response of plasma lipids and coronary atherosclerosis to treatment with fluvastatin: The lipoprotein and coronary atherosclerosis study'. J Am Coll Cardiol. 2000;35:89–95. doi: 10.1016/S0735-1097(99)00535-5. [DOI] [PubMed] [Google Scholar]
- Bray PF, Cannon CP, Goldschmidt-Clermont P. et al. 'The platelet Pl(A2) and angiotensin-converting enzyme (ACE) D allele polymorphisms and the risk of recurrent events after acute myocardial infarction'. Am J Cardiol. 2001;88:347–352. doi: 10.1016/S0002-9149(01)01677-0. [DOI] [PubMed] [Google Scholar]
- Bosse Y, Vohl MC, Dumont M. et al. 'Influence of the angiotensin-converting enzyme gene insertion/deletion polymorphism on lipoprotein/lipid response to gemfibrozil'. Clin Genet. 2002;62:45–52. doi: 10.1034/j.1399-0004.2002.620106.x. [DOI] [PubMed] [Google Scholar]
- White HL, Maqbool A, McMahon AD. et al. 'An evaluation of the beta-1 adrenergic receptor Arg389Gly polymorphism in individuals at risk of coronary events. A WOSCOPS substudy'. Eur Heart J. 2002;23:1087–1092. doi: 10.1053/euhj.2001.3037. [DOI] [PubMed] [Google Scholar]
- Elghannam H, Tavackoli S, Ferlic L. et al. 'A prospective study of genetic markers of susceptibility to infection and inflammation, and the severity, progression, and regression of coronary atherosclerosis and its response to therapy'. J Mol Med. 2000;78:562–568. doi: 10.1007/s001090000154. [DOI] [PubMed] [Google Scholar]
- Mulder AB, van Lijf HJ, Bon MA. et al. 'Association of polymorphism in the cytochrome CYP2D6 and the efficacy and toler-ability of simvastatin'. Clin Pharmacol Ther. 2001;70:546–551. doi: 10.1067/mcp.2001.120251. [DOI] [PubMed] [Google Scholar]
- Geisel J, Kivisto KT, Griese EU. et al. 'The efficacy of simvastatin is not influenced by CYP2D6 polymorphism'. Clin Pharmacol Ther. 2002;72:595–596. [PubMed] [Google Scholar]
- Zito F, Lowe GD, Rumley A. et al. 'Association of the factor XII 46C.T polymorphism with risk of coronary heart disease (CHD) in the WOSCOPS study'. Atherosclerosis. 2002;165:153–158. doi: 10.1016/S0021-9150(02)00196-X. [DOI] [PubMed] [Google Scholar]
- de Maat MP, Kastelein JJ, Jukema JW. et al. '-455G/A polymorphism of the beta-fibrinogen gene is associated with the progression of coronary atherosclerosis in symptomatic men: proposed role for an acute-phase reaction pattern of fibrinogen. REGRESS group'. Arterioscler Thromb Vasc Biol. 1998;18:265–271. doi: 10.1161/01.ATV.18.2.265. [DOI] [PubMed] [Google Scholar]
- Basso F, Lowe GD, Rumley A. et al. 'Interleukin-6-174 > C polymorphism and risk of coronary heart disease in West of Scotland coronary prevention study (WOSCOPS)'. Arterioscler Thromb Vasc Biol. 2002;22:599–604. doi: 10.1161/01.ATV.0000013283.84306.1A. [DOI] [PubMed] [Google Scholar]
- de Maat MP, Jukema JW, Ye S. et al. 'Effect of the stromelysin-1 promoter on efficacy of pravastatin in coronary atherosclerosis and restenosis'. Am J Cardiol. 1999;83:852–856. doi: 10.1016/S0002-9149(98)01073-X. [DOI] [PubMed] [Google Scholar]
- Peregrin T. 'The new frontier of nutrition science: nutrige-nomics'. J Am Diet Assoc. 2001;101:1306. doi: 10.1016/S0002-8223(01)00309-1. [DOI] [PubMed] [Google Scholar]
- van Ommen B, Stierum R. 'Nutrigenomics: Exploiting systems biology in the nutrition and health arena'. Curr Opin Biotechnol. 2002;13:517–521. doi: 10.1016/S0958-1669(02)00349-X. [DOI] [PubMed] [Google Scholar]
- Trayhurn P. 'Nutritional genomics -- 'Nutrigenomics''. Br J Nutr. 2003;89:1–2. doi: 10.1079/BJN2002780. [DOI] [PubMed] [Google Scholar]
- Muller M, Kersten S. 'Opinion. Nutrigenomics: Goals and strategies'. Nat Rev Genet. 2003;4:315–322. doi: 10.1038/nrg1047. [DOI] [PubMed] [Google Scholar]
- Krauss RM. 'Atherogenic lipoprotein phenotype and diet-gene interactions'. J Nutr. 2001;131:340S–343S. doi: 10.1093/jn/131.2.340S. [DOI] [PubMed] [Google Scholar]
- Vincent S, Planells R, Defoort C. et al. 'Genetic polymorphisms and lipoprotein responses to diets'. Proc Nutr Soc. 2002;61:427–434. doi: 10.1079/PNS2002177. [DOI] [PubMed] [Google Scholar]
- Corella D, Tucker K, Lahoz C. et al. 'Alcohol drinking determines the effect of the APOE locus on LDL-cholesterol concentrations in men: The Framingham Offspring Study'. Am J Clin Nutr. 2001;73:736–745. doi: 10.1093/ajcn/73.4.736. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, Corella D, Cupples LA. et al. 'Polyunsaturated fatty acids modulate the effects of the APOA1 G-A polymorphism on HDL-cholesterol concentrations in a sex-specific manner: The Framingham Study'. Am J Clin Nutr. 2002;75:38–46. doi: 10.1093/ajcn/75.1.38. [DOI] [PubMed] [Google Scholar]
- Ordovas JM, Corella D, Demissie S. et al. 'Dietary fat intake determines the effect of a common polymorphism in the hepatic lipase gene promoter on HDL metabolism: Evidence of a strong dose-effect in this gene-nutrient interaction in the Framingham Study'. Circulation. 2002;106:2315–2321. doi: 10.1161/01.CIR.0000036597.52291.C9. [DOI] [PubMed] [Google Scholar]
- Tai ES, Ordovas JM, Corella D. et al. 'The TaqIB and -629C > A polymorphisms at the cholesteryl ester transfer protein locus: associations with lipid levels in a multiethnic population. The 1998 Singapore National Health Survey'. Clin Genet. 2003;63:19–30. doi: 10.1034/j.1399-0004.2003.630104.x. [DOI] [PubMed] [Google Scholar]
- Weggemans RM, Zock PL, Ordovas JM. et al. 'Apoprotein E genotype and the response of serum cholesterol to dietary fat, cholesterol and cafestol'. Atherosclerosis. 2001;154:547–555. doi: 10.1016/S0021-9150(00)00720-6. [DOI] [PubMed] [Google Scholar]
- Weggemans RM, Zock PL, Ordovas JM. et al. 'Genetic polymorphisms and lipid response to dietary changes in humans'. Eur J Clin Invest. 2001;31:950–957. doi: 10.1046/j.1365-2362.2001.00906.x. [DOI] [PubMed] [Google Scholar]
- Perez-Martinez P, Ordovas JM, Lopez-Miranda J. et al. 'Polymorphism exon 1 variant at the locus of the scavenger receptor class B type I gene: influence on plasma LDL cholesterol in healthy subjects during the consumption of diets with different fat contents'. Am J Clin Nutr. 2003;77:809–813. doi: 10.1093/ajcn/77.4.809. [DOI] [PubMed] [Google Scholar]
- Ordovas JM. 'The genetics of serum lipid responsiveness to dietary interventions'. Proc Nutr Soc. 1999;58:171–187. doi: 10.1079/PNS19990023. [DOI] [PubMed] [Google Scholar]
- Ordovas JM. 'Cardiovascular disease genetics: A long and winding road'. Curr Opin Lipidol. 2003;14:47–54. doi: 10.1097/00041433-200302000-00009. [DOI] [PubMed] [Google Scholar]
- van Geel PP, Pinto YM, Zwinderman AH. et al. 'Increased risk for ischaemic events is related to combined RAS polymorphism'. Heart. 2001;85:458–462. doi: 10.1136/heart.85.4.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Park JB, Han KO. et al. 'A common mutation in cholesteryl ester transfer protein gene and plasma HDL cholesterol level before and after hormone replacement therapy in Korean postmeno-pausal women'. Korean J Intern Med. 2002;17:83–87. doi: 10.3904/kjim.2002.17.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somekawa Y, Umeki H, Kobayashi K. et al. 'Effects of hormone replacement therapy and hepatic lipase polymorphism on serum lipid profiles in postmenopausal Japanese women'. J Clin Endocrinol Metab. 2002;87:4766–4770. doi: 10.1210/jc.2002-020245. [DOI] [PubMed] [Google Scholar]