

Abstract
The development of multifunctional nanoparticles for medical applications is of growing technological interest. A single formulation containing imaging and/or drug moieties that is also capable of preferential uptake in specific cells would greatly enhance diagnostics and treatments. There is growing interest in plant-derived viral nanoparticles (VNPs) and establishing new platform technologies based on these nanoparticles inspired by nature. Cowpea mosaic virus (CPMV) serves as the standard model for VNPs. Although exterior surface modification is well known and has been comprehensively studied, little is known of interior modification. Additional functionality conferred by the capability for interior engineering would be of great benefit toward the ultimate goal of targeted drug delivery. Here, we examined the capacity of empty CPMV (eCPMV) particles devoid of RNA to encapsulate a wide variety of molecules. We systematically investigated the conjugation of fluorophores, biotin affinity tags, large molecular weight polymers such as polyethylene glycol (PEG), and various peptides through targeting reactive cysteines displayed selectively on the interior surface. Several methods are described that mutually confirm specific functionalization of the interior. Finally, CPMV and eCPMV were labeled with near-infrared fluorophores and studied side-by-side in vitro and in vivo. Passive tumor targeting via the enhanced permeability and retention effect and optical imaging were confirmed using a preclinical mouse model of colon cancer. The results of our studies lay the foundation for the development of the eCPMV platform in a range of biomedical applications.
INTRODUCTION
Nanomaterials are currently under investigation for platform development for applications in nanomedicine. They have favorable properties for the detection, imaging, and treatment of diseases such as cancer and cardiovascular disease as they can carry large payloads of imaging reagents and/or drugs and can be engineered to direct the payload specifically to target cells. The fact that multifunctional units can be designed and developed makes nanoparticles attractive candidates for the development of novel therapeutics and diagnostics.
Many different platforms have been developed, including synthetic man-made nanomaterials and naturally occurring bionanomaterials, such as protein cages and viral nanoparticles (VNPs) 1. Although viral nanotechnology is considered a novel and emerging field, recombinant virus-based materials have been used as vaccines and gene delivery vectors since the 1970s. Several recombinant virus-like particle-based vaccines are now used in the clinic, e.g. the Human papillomavirus (HPV) vaccine Gardasil (Merck & Co Inc.). Several gene therapies based on Adenovirus, Adeno-associated virus, and Lentivirus are undergoing clinical trials 2-4. For the past 20 years chemists, materials scientists, and engineers have developed a range of methodologies that can be applied to fine-tune and engineer VNPs for desired applications. VNPs are genetically encoded biomaterials and can therefore be genetically modified. In addition, chemical engineering procedures including chemical bioconjugation, mineralization, infusion, and encapsulation techniques have been widely developed and applied 5-6. VNPs offer several sites for modification: the exterior surface, the interior surface, the coat protein interface, and the interior cavity, all of which have been utilized for modification. The application of a combination of techniques facilitates the development of highly sophisticated multifunctional nanoprobes 7.
In this work we turned toward the development of the plant virus Cowpea mosaic virus (CPMV). CPMV is a 30 nm-sized icosahedron; it has been used as a model system for various applications ranging from electronic materials 8-10 and sensors 11-12 to imaging probes 13-14. CPMV has been studied extensively because of its biocompatibility, high stress tolerance, low toxicity, and diverse possibilities for surface modification by conjugation and genetic engineering. CPMV nanoparticles consist of 60 copies each of a large (L) and small (S) coat protein. Wild-type CPMV can be produced with ease and in high yields in black-eyed pea plants. Recently a RNA-free version of the capsid, referred to as empty CPMV (eCPMV), has been developed. These are produced by agroinfiltrating Nicotiana benthamiana leaves with a construct expressing the precursor of the L and S coat proteins (VP60) and the virus-derived proteinase (24K) required for its processing 15. The particles produced in this way are completely devoid of RNA of either viral or host origin though structurally identical to wild-type particles in terms of their proteins.
Several bioconjugation chemistries have been successfully applied to modify the exterior surface of CPMV. These include exterior lysine modification using N-hydroxysuccinimide esters 16-17, carbodiimide-mediated amine coupling to solvent-exposed carboxylates 18, and maleimide coupling to genetically or chemically introduced thiols 19-20. Further, advanced bio-orthogonal reactions such as Cu(I)-catalyzed azide-alkyne cycloaddition 21 and hydrazone ligation procedures 22 have been applied with great success. Exterior surface modification of CPMV has been extensively studied and is well understood. In stark contrast, only few studies have been reported that address the chemical engineering of the interior CPMV surface 23-24.
With growing interest to develop CPMV-based nanomaterials for applications in materials and medicine, the generation of multifunctional materials is a requirement and thus interior engineering in addition to exterior labeling is becoming more and more important. For example, CPMV nanoparticles have a natural affinity to cancer cells such as HeLa (cervical cancer), HT-29 (colon cancer), and PC-3 (prostate cancer); surface domains of CPMV specifically interact with cell surface-expressed vimentin and promote cell internalization 25-27. This property can be utilized to image cancer neovasculature or to target CPMV to cancer cells in vitro and in vivo 14, 25, 28. To utilize CPMV probes for the study or targeting of surface vimentin-expressing cells, it is desired to develop chemistries on the interior particle surface, e.g. to install imaging moieties; this will ensure conservation of the particle surface and CPMV-vimentin interaction. CPMV can also be re-directed to other receptors, such as folic acid receptors 29, vascular endothelial growth factor receptor-1 29, and gastrin-releasing peptide receptors 13, by decorating the CPMV particle surface with the appropriate targeting ligands. With the long-term application of targeted drug delivery or optical imaging in mind, it is of interest to establish chemistries that allow interior cargo loading in addition to exterior surface modifications.
The recent development of the eCPMV formulation now opens a new avenue to further advance the CPMV platform technology. In this study, we systematically investigated the interior labeling capacity of RNA-free eCPMV and RNA-containing wild-type CPMV nanoparticles using negatively and positively charged fluorophores, small molecule biotin tags, large polymers such as poly(ethylene glycol), and negatively and positively charged peptide sequences: penta(arginine), hexa(histidine), and FLAG tag peptide. The in vitro and in vivo properties of fluorescently labeled CPMV and eCPMV were investigated in tissue culture and a preclinical mouse model of colon cancer.
MATERIALS AND METHODS
Materials
Oregon Green 488 (OG488) maleimide, OG488 succinimidyl ester, and Rhodamine Red C2 (RR) maleimide were purchased from Invitrogen. DyLight 488 (DL488) maleimide, sulfo-NHS-LC-biotin, and maleimide-PEG2-biotin were purchased from Pierce. Polyethylene glycol maleimide (MW 2000 Da) and polyethylene glycol succinimidyl ester (MW 2000 and 5000 Da) were purchased from Nanocs. Maleimido trioxa-6-formyl benzamide (MTFB), succinimidyl-4-formyl benzoate (S-4FB), His6 Tag-HyNic, and FLAG Tag-HyNic were purchased from Solulink. A biotinylated 6-hydrazinopyridyl-polyarginine peptide (bio-R5-HyNic) was synthesized as described elsewhere 30. Aniline and dimethylsulfoxide (DMSO) were purchased from Fisher.
Propagation and isolation of wild-type and empty CPMV particles
CPMV production: Black-eyed peas (Vigna unguiculata) were inoculated with 100 ng/μL CPMV in 0.1 M potassium phosphate buffer (pH 7.0) and propagated for 18-20 days using established procedures 31. Virus concentration in plant extracts was determined by UV/vis spectroscopy (ε260 nm = 8.1 mg−1 mL cm−1), and virus integrity was determined by size exclusion chromatography (see below). eCPMV production: Agrobacterium LBA4404 cultures harboring the binary plasmid pEAQexpress-VP60-24K, which encodes the coat protein precursor VP60 and viral proteinase 24K 32, were introduced into N. benthamiana leaves using syringe-infiltration. Infiltrated tissue was harvested 6 days post infiltration and homogenized in 0.1 M sodium phosphate buffer (pH 7.0). eCPMV was purified further using a protocol adapted from established procedures for wild-type CPMV31 and the particle concentration was determined by UV/vis spectroscopy (ε280 nm = 1.28 mg−1 mL cm−1). eCPMV integrity was examined using transmission electron microscopy on FEI Technai20.
Bioconjugation using CPMV and eCPMV
For all reactions using maleimide and NHS chemistries, CPMV and eCPMV particles were used at a final concentration of 2 mg/mL in 0.1 M potassium phosphate buffer (pH 7.0) and incubated with the chemical label (e.g. dye, biotin) at room temperature overnight, with agitation. For all hydrazone ligation reactions, CPMV and eCPMV particles modified with MTFB (final concentration 0.5 mg/mL) were incubated with the peptide (e.g. FLAG-HyNic) in 0.1 M potassium phosphate buffer (pH 7.0) containing 10 mM aniline catalyst overnight at room temperature, with agitation. The final DMSO concentration was adjusted to 10% of the reaction volume. Particles were purified with 10 kDa molecular weight cut-off centrifugal filter units (Millipore) and analyzed with UV/vis spectroscopy, native and denaturing gel electrophoresis, and size exclusion chromatography. For initial studies, labeling with dyes, S-4FB, and MTFB were performed using 6000 molar excess of the chemical per particle, with biotin and PEG using 2000 molar excess, and with peptides using 360 molar excess. For comparisons between externally and internally dye-labeled CPMV and eCPMV, the molar excess was adjusted to match the number of dyes, as confirmed by UV/vis spectroscopy. For in vivo studies, 4500 molar excess of mPEG5000-NHS was used. Western blotting was performed for biotinylated particles. The 4FB labeling efficiency of CPMV-S4FBE and CPMV-MTFBI was determined using the Solulink 4FB molar substitution ratio protocol (MSR), in which 10 μg of 4FB-modified CPMV particles were mixed with a 0.5 mM solution of 2-hydrazinopyridine-2-HCl (2-HP) prepared in 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5.5). The reaction was incubated at 37°C for 30 min and analyzed by UV/vis spectroscopy. The number of 4FB labels per particle was calculated using the bond-specific extinction coefficient at 350 nm (ε = 18,000 M−1 cm−1). Similarly, peptide attachment was quantified using the bond-specific extinction coefficient at 354 nm (ε = 29,000 M−1 cm−1) for the hydrazone bond.
Size exclusion chromatography (SEC)
All labeled particles were analyzed by SEC using a Superose6 column on the ÄKTA Explorer chromatography system (GE Healthcare). Samples (100 Hg/100 μL) were analyzed at a flow rate of 0.4 mL/min using 0.1 M potassium phosphate buffer (pH 7.0).
Transmission electron microscopy (TEM)
Drops of labeled particles were placed on carbon-coated copper TEM grids (5 μL, 0.1 mg/mL), allowed to adsorb for 5 minutes, washed with DI water, then negatively stained with 2% (w/v) uranyl acetate for 1 minute. Samples were examined using a Zeiss Libra 200FE transmission electron microscope operated at 200 kV.
Gel electrophoresis
Native gel electrophoresis was performed using 1.2% agarose gels in 1x TBE buffer (45 mM Tris, 45 mM boric acid, 1.25 mM EDTA in MilliQ water) with 1x TBE running buffer and 10 μg of sample. Protein subunits were analyzed on denaturing 4-12% NuPAGE gels (Invitrogen) using 1x MOPS running buffer (Invitrogen) and 10 Hg of sample. After separation, the gel was photographed using an AlphaImager (Biosciences) imaging system before and after staining with Coomassie Blue, or further processed for Western blotting.
Western blotting
To detect biotinylated particles, CPMV, CPMV-bioE, eCPMV-bioI, and eCPMV-R5-bioI were analyzed by Western blotting. 1 μg samples were separated on a 4-12% NuPAGE Bis-Tris gel using MOPS buffer (see above). After separation, the proteins were transferred onto a nitrocellulose membrane (Thermo Scientific) using NuPAGE Transfer Buffer (Invitrogen). The membrane was blocked at room temperature for 1 h using 0.1 M TBS (pH 7.6) containing 5% w/v skimmed milk powder and 0.05% w/v Tween 20. Detection was carried out using alkaline phosphatase-conjugated streptavidin (Sigma Aldrich) (1:1000) in blocking buffer solution. Alkaline phosphatase activity was detected using the BCIP/NBT liquid substrate system (Sigma Aldrich).
Avidin agarose affinity binding assay
CPMV, CPMV-bioE, and eCPMV-bioI were tested for binding to avidin agarose resin (Pierce) to confirm interior eCPMV modification with biotin maleimide. The batch method provided by the supplier was used, with some modifications: 50 μg of the samples in 50 μL of 0.1 M potassium phosphate buffer (pH 7.0) were added to 50 μL of the resin (100 μL of slurry) and mixed for 1 h at room temperature, with agitation. The supernatant was then recovered and the resin washed twice using 50 μL of 0.1 M potassium phosphate buffer (pH 7.0). Any bound sample was eluted with 50 μL of 0.5 M glycine-HCl buffer (pH 2.8) and the pH immediately adjusted with 5 μL of 1 M Tris buffer (pH 7.5). The samples were analyzed by running 30 μL of the first recovered and eluted fractions on a denaturing gel.
Ni-NTA affinity binding assay
CPMV, eCPMV, CPMV-His6E, and eCPMV-His6I were tested for binding to HisPur Ni-NTA resin (Pierce) to confirm interior eCPMV modification with His6. The batch method provided by the supplier was used, with some modifications: 50 μg of the samples in 100 μL of equilibration buffer (20 mM sodium phosphate, 300 mM sodium chloride, 10 mM imidazole, pH 7.4) were added to 50 μL of the resin (150 μL of slurry) and mixed for 30 min at room temperature, with agitation. The supernatant was then recovered and the resin washed twice using 100 μL of wash buffer (20 mM sodium phosphate, 300 mM sodium chloride, 25 mM imidazole, pH 7.4). Any bound sample was eluted with 50 μL of elution buffer (20 mM sodium phosphate, 300 mM sodium chloride, 250 mM imidazole, pH 7.4). The samples were analyzed by running 30 μL of the first recovered and eluted fractions on a denaturing gel.
Cell cultures
HeLa cells (ATCC) were grown and maintained in minimal essential medium (MEM), while HT-29 cells (ATCC) were cultured in RPMI medium at 37°C in a 5% CO2 humidified atmosphere. The media were supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS), 1% (v/v) L-glutamine, and 1% (v/v) penicillin-streptomycin. All reagents were obtained from Gibco.
Confocal microscopy imaging
HeLa cells (20,000 cells/750 μL MEM/well) were grown for 24 h on glass coverslips placed in an untreated 24-well plate. The media was then replaced with 250 μL of fresh MEM containing 10 μg of CPMV-OG488E or eCPMV-OG488I (~5 × 107 VNPs/cell) and incubated at 37°C, 5% CO2 for 2 h. Post incubation, cells were washed thoroughly with sterile saline and incubated for a further 24 h in fresh medium. The cells were then fixed using 4% v/v paraformaldehyde and 0.3% v/v glutaraldehyde in DPBS (pH 7.2) for 5 min. Cell membranes were stained using wheat germ agglutinin (WGA) conjugated with Alexa Fluor 555 (WGA-A555) (Invitrogen) at 1 μg/mL in 5% goat serum (GS) (Invitrogen) for 45 min. Cell nuclei were stained using 4′,6-diamidino-2-phenylindole (DAPI) (MP Biomedicals) at 0.13 μg/mL in DPBS for 5 min. All steps were carried out in the dark at room temperature; in between each step the coverslips were washed 3x with DPBS. The coverslips were then mounted using Permount (Fisher) on glass slides and sealed using nail polish. Confocal images were obtained using Olympus FluoView™ FV1000 LSCM and data processed using Image J 1.44o (http://imagej.nih.gov/ij).
Fluorescence measurements
50 μL of dye-labeled CPMV and eCPMV were added to a black 384-well plate at a concentration of 50 μM. 0.1 M potassium phosphate buffer was used for pH 7.0 measurements, and 0.1 M MES buffer was used for pH 5.0 measurements. Particles were incubated in their respective buffers for 3 hours. Fluorescence intensity was measured using a Tecan Infinite 200 plate reader (Ex/Em wavelengths 600/665 for A647 and 435/495 for OG488).
Flow cytometry
HeLa cells (750,000 cells/200 μL MEM/well) were added to an untreated 96-well v-bottom plate. CPMV, CPMV-A647E, eCPMV, and eCPMV-A647I particles were added at a concentration of 100,000 particles/cell in triplicates and incubated for 3 hours at 37°C and 5% CO2. Following incubation, cells were spun down at 500 g for 4 minutes. The supernatant was removed, and the cells were resuspended in FACS buffer (0.1 mL 0.5 M EDTA, 0.5 mL FBS, and 1.25 mL 1 M HEPES, pH 7.0 in 50 mL Ca2+ and Mg2+ free PBS). This washing step was repeated twice. The cells were then fixed in 2% (v/v) paraformaldehyde in FACS buffer for 10 minutes at room temperature and washed another three times. Analysis was carried out using the BD LSR II flow cytometer, and a total of 10,000 events per sample were collected.
Tumor homing with HT-29 xenografts
All animal procedures were performed in accordance with approved protocols from the Institutional Animal Care and Use Committee at Case Western Reserve University. Tumor xenografts were established by injecting 5 × 106 HT-29 cells/100 μL of a 1:1 preparation of RPMI medium and Matrigel (Fisher) subcutaneously in the flanks of six week old NCr-nu/nu mice. The mice were maintained on an alfalfa free diet (Teklad) to reduce tissue autofluorescence. Animals were observed closely, and tumor size was measured using calipers. After the tumors reached an average volume of 20 mm3 (10-12 days), the mice were randomly divided into three groups: PBS, CPMV, and eCPMV (n=5). PEG5000E-eCPMV-A647I was administered intravenously at a dose of 200 μg/100 μL sterile PBS and PEG5000E-CPMV-A647E at a dose of 284.3 μg/100 μL sterile PBS to match the number of particles. Both formulations had 50 dyes/particle. Animals were sacrificed 24 h post administration, and the tumors on the flanks were excised and imaged using a Maestro™ fluorescence imaging instrument (yellow excitation and emission filters with an exposure time of 800 ms). After background subtraction in Maestro, the average fluorescence intensity over the tumor area was analyzed using ImageJ.
RESULTS AND DISCUSSION
Production of CPMV and eCPMV
CPMV particles were purified from infected black-eyed pea plants yielding 0.5-1 mg CPMV per 1 g infected leaves. The purity of the virus was confirmed based on the A260:A280 ratio (a ratio of 1.7-1.8 indicates pure and intact particles) and size-exclusion chromatography (SEC). eCPMV particles were produced by co-expression of the precursor to the L and S coat proteins (VP60) and the viral proteinase (24K) in N. benthamiana leaves, and the particles were purified using a modified CPMV extraction procedure. Yields of up to 0.5 mg/g leaf tissue were achieved, somewhat lower than the yields achieved for wild-type CPMV via infection.
Structural properties of (e)CPMV
One of the advantages of working with bionanoparticles such as VNPs is that their structures are known to atomic resolution. The structure of CPMV has been solved to 2.8 Å resolution and its coordinates are available at the Virus Particle Explorer database (viperdb.scripps.edu). The 30 nm-sized CPMV capsids have icosahedral symmetry and are formed by 60 copies of two different types of coat proteins, the S and L subunits. The S subunit (213 amino acids) folds into one jelly roll β-sandwich, the A domain, and the L subunit (374 amino acids) folds into two jelly roll β-sandwich domains: the B domain that covers the carboxy-terminus and the C domain that covers the amino-terminus. The three domains form the asymmetric unit and are arranged in a similar surface lattice to T = 3 viruses, except they have different polypeptide sequences; therefore the particle structure is described as pseudo T = 3, or P = 3, symmetry. While the B and C domains are clustered around the icosahedral three-fold axis and form hexamers, the A domain is clustered around the five-fold axis, forming pentamers 33 (Figure 1A & B). The interior surface is accessible through 12 pores at the five-fold axis; at its narrowest point the opening of the pore is 0.75 nm (as measured using PyMol 1.4.1 software) (Figure 1C).
Figure 1. The structure of CPMV.

CPMV consists of 60 copies of a small subunit (S, blue) and a large two-domain subunit (L, green and red). A) Exterior view and B) interior view. C) Pore structure at the five-fold axis; the pore diameter was measured to be 0.75 nm. D) CPMV displays 300 reactive lysines on the surface, five per asymmetric unit. E) CPMV also displays cysteines selectively on the interior surface, 8 per asymmetric unit (Cys 4 – yellow, 108 – forest green, 119 – cyan, 132 – purple, 177 – orange, 187 – green, 295 – blue, and 355 – red). Inset shows 90 degree rotation, revealing hidden reactive thiol of Cys 4. F) View of interior with surface-exposed thiols highlighted (Cys 4 – brown, 187 – pale green, 295 – sky blue, and 355 – light orange).
The chemical addressability of the exterior surface has been extensively studied, and it is known that CPMV displays up to 300 reactive solvent-exposed surface lysine side chains (Figure 1D), all of which can be chemically labeled 16-17. CPMV particles do not display any reactive cysteine side chains on their exterior particle surface. However, reactive cysteine residues are located on the solvent-exposed interior particle surface (Figure 1E & F). In earlier studies Wang et al. showed that small chemical modifiers such as ethylmercury phosphate (EMP), 5-maleimidofluorescein 23, and thiol-selective stilbene derivatives 24 could be introduced to CPMV and covalently attached to interior cysteine residues.
Studying the structure of CPMV using PyMol software, we located eight cysteines per asymmetric unit on the solvent-exposed interior surface (Figure 1E & F): Cys 4 on the S protein, and Cys 108, 119, 132, 177, 187, 295, and 355 on the L protein. Thiols from two cysteine side chains, Cys 295 on L and Cys 4 on S, were found to be solvent-exposed (the thiol of Cys 4 appears to be exposed in a small pocket, which can be seen when looking at the asymmetric unit; see inset in Figure 1F). The thiols of Cys 187 and Cys 355 appear to be engaged in a disulfide bond (Figure 1E & F). Previous data from Wang et al. indicated that EMP reacted with Cys 295 on L but also indicated that 5-maleimidofluorescein attached to cysteines on both the S and L subunit; the exact positions could not be identified 23. Based on the structural data, we propose that CPMV nanoparticles display at least 120 reactive cysteine residues, one each on L (Cys 295) and S (Cys 4) subunits. It is important to note that VNPs, although often depicted as rigid closed shells, are highly dynamic structures that can undergo various reversible structural transitions. The structural data generated from crystallography is just a snapshot.
Interior labeling with fluorophores
We studied the chemical reactivity of both eCPMV and CPMV toward several fluorophores. Prior to chemical labeling, eCPMV and CPMV were treated with 10 mM tris(2-carboxyethyl)phosphine (TCEP) in 0.1 M phosphate buffer pH 7.0. TCEP is a reducing reagent; treatment was performed to ensure that thiols were reduced and reactive toward maleimide-containing compounds. The reducing agent was removed using centrifugal spin filters with a cut-off of 10K prior to introduction of maleimide-containing fluorophores: Oregon Green 488 (OG488), DyLight 488 (DL488), Rhodamine Red (RR), and Alexa Fluor 647 (A647) (Figure 2A). Different conditions were tested, i.e. varying incubation times and excess of reagent used (see Materials and Methods), and we found that the reaction efficiency reached a plateau using a 2000-fold excess of reagents. To ensure maximum labeling, the reactions were carried out under forcing conditions using a molar excess of 6000 fluorophores per eCPMV and CPMV particle; the protein concentration was kept at 2 mg/mL (350 nM for CPMV, 500 nM for eCPMV). The reaction was allowed to proceed overnight, eCPMV-dye and CPMV-dye conjugates were purified using centrifugal filters with a size cut-off of 10K, and samples were resuspended in buffer and analyzed by UV/vis spectroscopy, SEC, TEM, and native and denaturing gel electrophoresis (Figures 2, S1, and S2).
Figure 2.

Characterization of CPMV-dye conjugates. A) Schematic of internal functionalization of eCPMV and CPMV with OG488 and RR (DL488 and A647 structures are proprietary). B) Representative data of the labeling efficiency of the various dyes for eCPMV vs. CPMV. eCPMV data are shown as solid bars on the left, while CPMV data are shown as striped bars on the right. Asterisks denote statistical significance between eCPMV and CPMV formulations (** p < 0.01). C) eCPMV- and CPMV-OG488 conjugates on a 1.2% agarose gel. The gel was visualized under UV light (left) and after Coomassie staining (right). 1=CPMV; 2=eCPMV; 3=CPMV-OG488; 4=eCPMV-OG488. D) Same particles on a denaturing 4-12% SDS-PAGE gel visualized under UV light (left) and after Coomassie staining (right). M=SeeBlue Plus2 molecular weight marker. E) eCPMV-RR and CPMV-RR conjugates on a SDS-PAGE gel visualized under UV light (left) and after Coomassie staining (right). 1=CPMV; 5=CPMV-RR; 6=eCPMV-RR.
In all cases, SEC and TEM confirmed that the particles remained structurally sound (Figures S1 and S2). The degree of conjugation was quantified, with the number of dye moieties per particle calculated based on the UV/vis spectrum using the concentration ratio of dye to eCPMV or CPMV. The concentrations were calculated using the Beer-Lambert law and the respective extinction coefficients: OG488 ε491 nm = 81,000 M−1 491 cm−1, DL488 ε493 nm = 70,000 M−1 493 cm−1, RR ε573 nm = 119,000 M−1 573 cm−1, A647 ε651 nm = 265,000 M−1 cm−1, CPMV ε260 nm = 8.1 mg−1 mL cm−1, MW of CPMV = 5.6 × 106 g mol−1, eCPMV ε280 nm = 1.28 mg−1 cm−1, and MW of eCPMV = 3.94 × 106 g mol−1. The data are summarized in Figure 2B. Overall, the labeling efficiency was significantly higher for each dye tested using eCPMV compared to CPMV. For eCPMV, it was found that approximately 110 OG488, 140 DL488, 60 RR, and 50 A647 were attached, while for CPMV, there were only 30 OG488, 30 DL488, less than 10 RR, and 10 A647 attached. The reproducibility and error lies within 10 dyes per particle. CPMV and eCPMV are structurally identical, but CPMV contains nucleic acids and eCPMV is nucleic acid-free. Labeling studies thus indicate that the presence of nucleic acids reduces labeling efficiency. It is possible the nucleic acids block the pores and thus reduce diffusion of the dyes into the capsid cavity. Electrostatic repulsion may also play a role. It is interesting to note that labeling with the negatively charged OG488 dye was most effective for eCPMV (see Figure 2A). The negative dye mimics the charge of the natural nucleic acid cargo; diffusion into the interior cavity and conjugation might thus be favored.
eCPMV- and CPMV-dye conjugates were analyzed by native and denaturing gel electrophoresis, and the gels were visualized under UV light before and white light after Coomassie staining (Figure 2C-E). In native gels, intact VNPs are analyzed. The appearance of fluorescent bands under UV light confirms that the labels were indeed covalently attached to the particles, with brighter bands corresponding with more dyes attached. We had previously shown that exterior dye conjugation alters the mobility of CPMV in native agarose gels 13. The conjugation of chemical modifiers neutralizes the positive charge from the exterior lysine side chains, leading to increased mobility toward the anode. CPMV and eCPMV particles labeled with negatively or positively charged dyes on interior cysteines do not show an altered mobility in the gel, indicating that the labels are indeed attached to the interior.
Denaturing gels were analyzed to determine whether the dyes were attached to the S or L protein. A greater shift was observed in RR labeling of the L protein compared to OG488 labeling, most likely due to the almost 50% greater molar mass of RR (Figure 2D & E). It was found that in any case, dyes were attached to both the S and L proteins. Selective attachment to S or L was not observed for any formulation tested, indicating that a reactive cysteine is present on both subunits. This was found to be true even for the CPMV-RR formulation in which we quantified less than 10 dyes per CPMV. The fact that the dye distribution follows a random pattern might indicate that the thiols on S and L are both highly reactive. This is consistent with previous data from Wang et al. that indicated 5-maleimidofluorescein was attached to cysteines on both the S and L subunits of CPMV at higher dye-to-subunit ratios 23.
CPMV- and eCPMV-biotin conjugates: labeling (e)CPMV inside and out
Next, we sought to determine whether labels were indeed attached to the interior surface. The employed maleimide chemistry is selective toward thiols; however, cross-reactivity with lysines has been reported 34. Since CPMV and eCPMV display 300 reactive lysine side chains on the exterior we sought to rule out that non-specific conjugation to exterior lysines instead of interior cysteine conjugation occurred. To do this, we chose biotin as a label. Biotin is a small molecule, a vitamin that specifically binds with high affinity to streptavidin and avidin. It is a popular label employed in biochemistry. Two particle conjugates were made: 1) CPMV was labeled at exterior lysines using 2000 molar excess of an N-hydroxysuccinimide reactive biotin probe (referred to as CPMV-bioE) and 2) eCPMV was labeled at interior cysteines using 2000 molar excess of a maleimide-activated biotin derivative (referred to as eCPMV-bioI) (Figure 3A).
Figure 3.

A) Schematic of biotin functionalization of eCPMV-bioI and CPMV-bioE, respectively B) Biotinylated particles on a 1.2% agarose gel visualized after Coomassie staining. 1=CPMV; 2=eCPMV; 3=CPMV-biotin; 4=eCPMV-biotin. C) Same particles on a 4-12% denaturing SDS-PAGE gel stained with Coomassie. M=SeeBlue Plus2 molecular weight marker. D) Western blot probed with streptavidin-alkaline phosphatase confirms biotinylation. E) Schematic of the avidin bead binding assay. F) Flow through and eluted particles from binding assay on a SDS-PAGE gel after staining with Coomassie. 5=CPMV flow through; 6=CPMV-bioE flow through; 7= eCPMV-bioI flow through; 8=bound CPMV; 9=bound CPMV-bioE; 10=bound eCPMV-bioI.
Purified CPMV-bioE and eCPMV-bioI conjugates were analyzed on native and denaturing gels and by Western blotting (Figure 3B-D). Native gel electrophoresis gave a first indication that biotin labels were indeed attached to interior cysteine side chains. Altered mobility is observed comparing CPMV and eCPMV. The absence of negatively charged RNA in eCPMV results in a retardation of movement through the gel toward the anode (Figure 3B), as previously described 19. Exterior labeling of CPMV with biotin results in an increased mobility of CPMV-bioE versus CPMV towards the anode. The increased mobility can be explained by altered exterior surface modification. Biotin is a non-charged chemical modifier; conjugation to the exterior lysines of CPMV results in neutralization of the positive charged derived from lysine side chains, resulting in an overall more negatively charged particle. Labeling the interior of eCPMV does not affect the size or charge of the particle. Despite similar degree of biotin labeling as CPMV-bioE (Figure 3D), eCPMV-bioI appears to have comparable electrophoretic mobility as eCPMV, thus indicating that labels were indeed attached to the interior cysteines (as opposed to non-specific external lysine conjugation) (Figure 3B).
To analyze the S and L proteins and to determine whether labels introduced were selective to just one or both coat proteins, denaturing gel electrophoresis and Western blotting were conducted. Membranes were probed with an alkaline phosphatase-labeled streptavidin. Data confirm successful biotinylation. Further, data indicate that biotin was attached to both the S and L proteins for both formulations, CPMV-bioE and eCPMV-bioI. This is in agreement with fluorescent dye attachment and indicates that reactive thiols are present on each coat protein unit (Figure 3D).
We developed an assay using avidin agarose beads to determine whether labels were indeed attached on the interior capsid surface using eCPMV (Figure 3E). CPMV, CPMV-bioE, and eCPMV-bioI particles were mixed with avidin agarose beads. CPMV and eCPMV-bioI were not expected to bind to the beads, as neither of these formulations display surface-exposed biotin groups. In contrast, CPMV-bioE displaying multiple biotin labels on its exterior surface was expected to strongly bind to the beads via the biotin-avidin interaction. Each formulation was incubated with the beads, beads were washed and the flow through collected. Then, the beads were treated with 0.5 M glycine-HCl buffer, pH 2.8 to disrupt the biotin-avidin interaction and elute any bound particles from the beads. Both the flow through from the washing steps and the eluent after treatment were collected and analyzed on a denaturing gel (Figure 3F). As expected, CPMV and eCPMV-bioI were detected in the washing steps indicating these formulations did not interact with the avidin agarose beads. CPMV-bioE was not detected in the flow through but was detected in the final eluent as these particles did bind to the beads (Figure 3F). These observations support that maleimide chemistry is indeed selective and that biotin labels in eCPMV-bioI were attached to the interior cysteine residues.
Labeling the eCPMV and CPMV interior with high molecular weight polymers
Next, we sought to test whether labeling with high molecular weight polymers could also be achieved. Polymers present an important building block in medical research; they can be used for covalent or non-covalent drug loading and controlled drug release. For example, the chemotherapeutic doxorubicin was loaded onto the polymer polystyrene sulfonic acid and subsequently encapsulated into VNPs from Hibiscus chlorotic ringspot virus (HCRSV) 35. In a different study, PEG-based polymers were grown on the surface of the phage Qβ; the chemotherapeutic doxorubicin was then covalently introduced into multivalent binding pockets provided by the polymers 36.
Our goal was to test whether high molecular weight polymers could diffuse inside the interior cavity and covalently attach to the interior cysteine side chains; we chose PEG-maleimide with a MW of 2000 Da as a test molecule. We compared the labeling efficiency of eCPMV with CPMV when treated with mPEG2000-maleimide. To rule out cross-reactivity and confirm functionalization of the interior, we carried out the same reactions using eCPMV and CPMV particles with surface lysines labeled with succinimidyl-4-formyl benzoate (S-4FB). Reaction of S-4FB-covered eCPMV and CPMV with an N-hydroxysuccinimide ester-activated PEG with a MW of 2000 Da (mPEG2000-NHS) was used as a control to verify that the lysines are unavailable for modification with PEG (Figure 4A).
Figure 4.
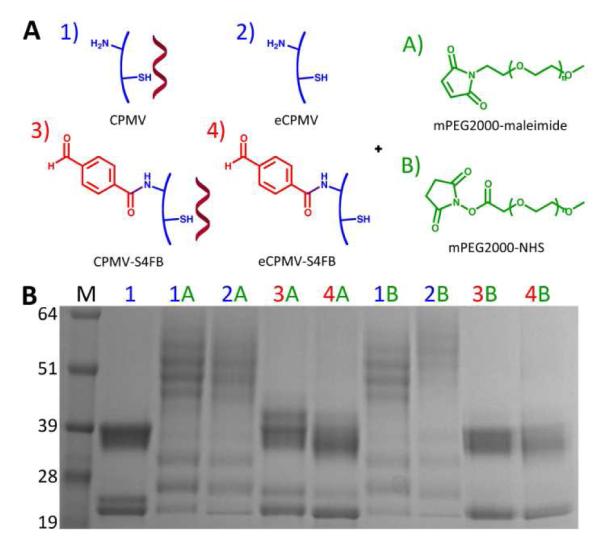
A) Reactions to determine functionalizability of eCPMV and CPMV with maleimide- and NHS-mPEG2000 when surface lysines are either available or unavailable for modification. B) Results of the reactions on a 4-12% denaturing SDS-PAGE gel stained with Coomassie Blue. M=SeeBlue Plus2 molecular weight marker; 1=CPMV; 2=eCPMV; 3=CPMV-S4FB; 4=eCPMV-S4FB; A=mPEG2000-maleimide; B=mPEG2000-NHS.
An electrophoretic mobility shift assay using denaturing gel electrophoresis was carried out to ascertain covalent attachment of PEG. Successful PEG conjugation was verified by the appearance of a laddering effect on the gel (Figure 4B). The additional, lower mobility bands correspond to coat proteins that have been labeled with PEG. With the surface lysines free, PEG labeling was achieved using both maleimide (lanes 1A and 2A) and NHS (lanes 1B and 2B) chemistries. However, while PEG could be attached to eCPMV and CPMV covered with S-4FB using mPEG2000-maleimide (lanes 3A and 4A), as expected no PEG conjugation was observed with mPEG2000-NHS (lanes 3B and 4B). In addition, densitometry analysis of the S protein bands was performed to determine an estimate of the degree of labeling. The L protein bands were not used because they were less distinct, but the degree of covalent modification appears to be comparable if not greater than the S protein. The density ratio of labeled to unlabeled S coat proteins indicate approximately 80% PEG labeling for eCPMV and 75% for CPMV, regardless of S-4FB coverage. These findings indicate that mPEG2000-maleimide is indeed attached to the interior cysteine residues. This is further supported by SEC data, where CPMV particles labeled at the exterior surface with mPEG2000-NHS elute earlier from the column, consistent with an increase in size. In contrast, the elution profile of eCPMV particles labeled with mPEG2000-maleimide at the interior surface resembles that of unlabeled particles, suggesting that PEG chains are presented on the interior surface (Figure S2).
It is interesting that the high molecular weight polymer PEG was able to diffuse into the eCPMV and CPMV formulations and react with interior cysteines. The pore at the five-fold axis (Figure 1C) was measured to be 0.75 nm at its narrowest point. Consistent with these structural measurements, previous experiments showed that rigid gold nanoparticles with a diameter of 1.4 nm could not diffuse inside CPMV particles 23. Now, the size of PEG in solution can be calculated based on the Flory dimension with RF = aN3/5, where a is the persistence length of the PEG monomer (a = 0.35 nm) 37 and N is the number of PEG monomers (N = 45 for PEG2000) 38. This gives a Flory dimension for PEG with a MW of 2000 Da of RF PEG2000 = 3.45 nm. Based on the Flory dimension, it would appear that their size would prevent the polymers from being able to diffuse inside the cavity and react with interior cysteines. However, the Flory radius is only an estimate of the size when PEG is in a mushroom conformation. PEG is a highly flexible polymer, and its conformation in solution is dynamic. The steric hindrance from the small pore size would promote the brush conformation. When the PEG is stretched out in this conformation, it is small enough to diffuse through the pores. In addition, the pore size of CPMV was determined by the crystal structure and based on a snapshot. The combination of the flexible polymer and dynamic CPMV structure provides access for large polymer systems to enter the interior of the nanoparticles. At this point, it is unknown whether the whole molecule is within the particle. It is possible that only the maleimide portion of the molecule entered the cavity and reacted with the Cys 4 residue that is in close proximity to the pore. Nevertheless, these findings of polymer loading may open the door for polymer-mediated drug loading studies.
Interior peptide loading using CPMV and eCPMV using hydrazone ligations
Recently, it has been shown that it is possible to chemically link peptides to the outer surface of CPMV particles in order to catalyze the deposition of specific minerals around the particles 39. Here we have investigated whether peptides could be introduced to the interior of eCPMV and CPMV in order to promote specific mineralization within particles since mineralized particles could have applications in nanomedicine. Three candidate peptides were chosen: the positively charged penta(arginine) and hexa(histidine) peptides as well as the negatively charged FLAG tag peptide (Figure 5A). To facilitate efficient loading with charged peptides, we turned toward bio-orthogonal chemistries. Standard coupling procedures using maleimide-activated reagents have slow reaction kinetics and large excesses of reagents have to be used to facilitate efficient labeling. Cu(I)-catalyzed azide-alkyne cycloaddition and hydrazone ligation chemistry overcome these limitations 5; these chemistries are highly efficient bioconjugation methods that require low concentrations and excesses of the reagent or ligand of interest. Peptide conjugation was carried out using a two-step hydrazone ligation procedure: first, interior cysteines on eCPMV and CPMV were labeled with benzaldehydes using the maleimide-reactive linker maleimido trioxa-6-formyl benzamide (MTFB); second, peptide coupling was performed using hydrazinopyridine-modified peptide conjugates (Figure 5A & B). MTFB was introduced using a 6000-fold excess to ensure maximum labeling. Quantification of MTFB modification was determined using the Solulink MSR assay and 2-hydrazinopyridine. The resulting hydrazone bond is UV traceable (A at 350 nm, ε = 18,000 M−1 cm−1). We found that reaction with MTFB showed poor reproducibility; between 40 and 80 MTFB linkers were attached (Figure 5C, solid bars). No statistical significant differences between eCPMV and CPMV were observed. This is consistent with observations made using PEG2000 and may be explained by the hydrophilic nature of the ligand; MTFB contains a PEG3 spacer (Figure 5A).
Figure 5.

Hydrazone ligation. A) Functionalization of the exterior of CPMV with S-4FB (top) and the interior of eCPMV and CPMV with MTFB (middle) to provide aldehyde ligation handles. Example of hydrazone ligation using 2-hydrazinopyridine to quantify the molar substitution ratio (bottom). B) Polyarginine, hexahistidine, and FLAG peptides attached using hydrazone ligation. C) Representative data of the labeling efficiency of MTFB (solid bars) and the various peptides (striped bars) to the interior of eCPMV vs. CPMV. D) Western blot probed with streptavidin-alkaline phosphatase confirms successful incorporation of biotinylated R5 peptide into the interior of eCPMV. M=SeeBlue Plus2 molecular weight marker; 1=eCPMV; 2=eCPMV-bioI; 3=eCPMV-R5I. E) Affinity of Ni-NTA to histidines was exploited for testing the binding of CPMV, CPMV-His6E, eCPMV, and eCPMV-His6I to Ni-NTA beads using a similar test as performed for biotinylated particles. F) Schematic of the bead binding assay (top) illustrates the expected results. SDS-PAGE gel stained with Coomassie (bottom) confirms only CPMV-His6E binds to the beads, indicating internal attachment of His6 to eCPMV. M=SeeBlue Plus2 molecular weight marker; 4=washed CPMV; 5=washed CPMV-His6; 6=washed eCPMV; 7=washed eCPMV-His6; 8=eluted CPMV; 9=eluted CPMV-His6; 10=eluted eCPMV; 11=eluted eCPMV-His6.
The aldehyde moiety of eCPMV-MTFBI and CPMV-MTFBI was then coupled with the hydrazine functionality of the penta(arginine), hexa(histidine), and FLAG tag peptides. Hydrazone chemistry has recently been applied to the CPMV platform and was shown to be a versatile strategy allowing the decoration of CPMV with targeting ligands specific for vascular endothelial growth factor receptor-1 22. We optimized the ligation reaction further using the catalyst aniline, which accelerates the rate of hydrazone bond formation by two orders of magnitude, allowing the reaction to proceed rapidly even at neutral and basic pH (the optimum of pH for hydrazone chemistry is 4.5) 40.
eCPMV and CPMV peptide conjugates were purified from excess reagents and characterized using SEC, UV/Vis spectroscopy, denaturing gel electrophoresis, and Western blotting. The R5 peptide used in this study displays a biotin tag (Figure 5B), allowing detection using enzyme-tagged streptavidin probes and Western blotting. Western blotting was performed using alkaline phosphatase-conjugated streptavidin. Alkaline phosphatase activity was detected using the BCIP/NBT liquid substrate system. Data confirmed the covalent decoration of eCPMV and CPMV with R5 peptides. In agreement with dye-, biotin-, and PEG-labeling, labels were found to be introduced to both the S and L proteins (Figure 5D).
Quantification of peptide labeling was based on the UV-traceable hydrazone bond formed (A at 354 nm, ε = 29,000 M−1 cm−1). Overall, eCPMV appeared to show better reactivity. From the data, it was not clear whether the charges of the various peptides impact the success of labeling. It appears that peptide labeling using hydrazone ligation results in high variability. Statistical analysis showed no significant differences between the conjugates. The previous dye-labeling studies indicated that negatively charged molecules may be preferred for entrance through the pores. However, the negatively charged FLAG tag is also larger than the positively charged penta(arginine) and hexa(histidine) peptides, a fact that could explain why this trend was not observed for the peptides. Between 20 and 40 peptides could be introduced using a molar excess of 360 peptides per CPMV/eCPMV. We attempted to increase the labeling efficiency by increasing the molar excess of peptides used or by extending the incubation time. However, in both cases aggregation was observed and recovered yield was less than 10%, indicating that the charged peptides induce electrostatically driven aggregation upon a threshold. We have observed similar trends in exterior peptide conjugation experiments using charged peptides. It has been shown that metals ions can be diffused into the internal cavity and converted to metal or metal oxide 41. Based on previous observations showing that 60 genetically introduced hexa(histidine) peptides were sufficient to serve as nucleation centers to promote external mineralization of eCPMV with cobalt 42, we propose that chemical labeling with up to 40 peptides will achieve similar results.
To verify that peptide labels were indeed conjugated to the interior particle surface, we adapted the agarose bead assay aforementioned for use in determining the spatial location of introduced biotin labels (see above). Agarose beads labeled with nickel-nitrilotriacetic acid (Ni-NTA) were used. Hexa(histidine) sequences have a high affinity to Ni-NTA (Figure 5E). We tested four particle formulations: eCPMV, CPMV, eCPMV-His6I and CPMV-His6E. The latter formulation was generated by decorating the exterior CPMV surface with benzaldehydes using the NHS derivative S-4FB, followed by hydrazone ligation using the HyNic-hexa(histidine) peptide. eCPMV, CPMV, eCPMV-His6I, and CPMV-His6E were all found in the flow through. However, only CPMV-His6E displaying exterior hexa(histidine) tags was detected in the eluent after treatment with imidazole, a chemical known to disrupt the Ni-NTA - hexa(histidine) interaction. Thus, out of all the formulations, only CPMV-His6E had any affinity to the Ni-NTA beads. This is as expected and indicates that the peptide sequences in eCPMV-His6I were indeed attached to the interior surface; there was no indication of eCPMV-His6I being bound to the beads. Since CPMV-His6E was also found in the initial flow through, binding to the beads was incomplete (Figure 5F). The difference in binding between this assay and the previous assay for biotinylated particles could be due to the 9 orders of magnitude lower affinity of the interaction between His6 and Ni-NTA (Kd = 10−6 M) 43 compared to the interaction between biotin and avidin (Kd = 10−15 M) 44. Overall, these data support that peptide labels were selectively attached to the interior surface. This opens a new avenue for peptide-mediated internal mineralization of CPMV.
Toward medical applications: cellular imaging and tumor homing of internally and externally labeled (e)CPMV
Fluorescent dyes are widely employed in optical imaging, and the versatility of fluorescently labeled CPMV nanoparticles has been demonstrated for several applications: CPMV-dye conjugates have been used i) to target and image cancer cells in vitro and in vivo 13, 25, 29, ii) for intravital vascular imaging, including tumor neovasculature mapping 14, 45, and iii) to study its biodistribution in vivo 46. In each case, the fluorophores were attached to the exterior CPMV surface. The conjugation of the imaging labels to the interior surface would offer a clear advantage. For example, it would allow modification of exterior residues with other biomedically relevant moieties such as targeting ligands to re-direct and target specific cells and tissues and PEG, a hydrophilic polymer used to shield nanomaterials and increase their pharmacokinetics while reducing undesired side effects such as immunogenicity. Here, we evaluated the use of internally labeled eCPMV versus externally labeled CPMV for optical imaging applications in tissue culture and preclinical tumor mouse models.
First, we evaluated the optical stability and molecular quenching of dye-labeled CPMV and eCPMV (Figure 6). We examined CPMV-OG488E particles labeled with 80 OG488 at exterior surface lysine side chains and eCPMV-OG488I labeled with 70 OG488 at interior cysteines for potential use in confocal microscopy studies. In addition, we looked at non-PEGylated and PEGylated formulations of CPMV-A647E and eCPMV-A647I labeled with 50 dyes each for use in flow cytometry and tumor homing studies, respectively. Fluorescence was measured for 50 μM solutions of the particles, maintaining equal amounts of dyes and particles for comparison. Measurements indicated that some quenching occurred when labels were conjugated to the interior eCPMV surface, and the fluorescence intensity reached only about half of the fluorescence intensity measured for CPMV decorated with dyes on its exterior surface. This phenomenon was independent of the dye used, i.e. OG488 or A647, and was also pH-independent, i.e. pH 7.0 versus 5.0 (Figure 6). Despite the weakened fluorescence signal of the eCPMV formulation compared to CPMV, the fluorescence is still suitable for optical imaging applications, and the potential for additional functionalities on the exterior of eCPMV remains promising. As the interior labeled eCPMV particles appeared strongly fluorescent in gels even though they are less fluorescent when measured in bulk in aqueous buffers, environmental factors clearly play a role in observed fluorescence. A difference may also be observed when imaging the particles within cells. Consequently, we went on to evaluate the in vitro and in vivo properties of eCPMV.
Figure 6.
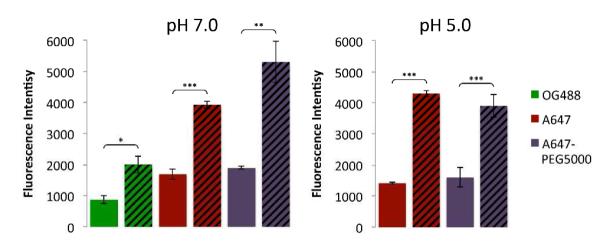
Fluorescence intensity data of interior labeled eCPMV compared to exterior labeled CPMV in pH 7.0 potassium phosphate buffer (left) and of A647-labeled particles in pH 5.0 MES buffer (right). eCPMV data are shown as solid bars on the left, while CPMV data are shown as striped bars on the right. Asterisks denote statistical significance between fluorescence intensities of eCPMV and CPMV formulations (* p < 0.05, ** p < 0.01, *** p < 0.001).
Cell imaging was studied using HeLa cells, a cervical cancer cell line. HeLa cells are an ideal model for this analysis because previous studies have shown that CPMV binds to vimentin displayed on the surface of HeLa cells and is then taken up by endocytosis 26. OG488- and A647-labeled constructs from the fluorescence measurements were used for confocal microscopy imaging and flow cytometry analysis, respectively. For confocal microscopy studies, live cells were incubated with CPMV-OG488E and eCPMV-OG488I, washed, and subsequently fixed. Cell membranes were stained with wheat germ agglutinin, and nuclei were stained with DAPI (Figure 7A-D). There were no apparent differences between the CPMV-OG488E and eCPMV-OG488I formulations, and fluorescent signals were comparable. From z-stacks analyzed using ImageJ software, CPMV and eCPMV nanoparticles were found to be internalized. Flow cytometry measurements were performed to gain quantitative data (Figure 7E & F). Signals obtained from eCPMV-A647I were reduced compared to signals derived from cells that were treated with CPMV-A647E. Nevertheless, both formulations were detectable in HeLa cells at similar levels as reported previously 25-26.
Figure 7.

In vitro evaluations of CPMV-OG488E and eCPMV-OG488I. Representative confocal images depict uptake of CPMV-OG488E (A,C) and eCPMV-OG488I (B,D) by HeLa cells. Side panels in C and D are orthogonal sections from the respective images confirming internalization of VNPs. Both CPMV and eCPMV were tagged with OG488 (green), the cell membrane was stained with wheat germ agglutinin (WGA)-Alexa Fluor 555 (red), and the nucleus was stained with DAPI (blue). Flow cytometry was performed to measure cell uptake (E). Cells to the right of the vertical line were considered positive for A647, and the percent of positive cells for each sample was quantified (F). Unpaired asterisks denote statistical significance as compared to cells only control (* p < 0.05, *** p < 0.001). Difference in fluorescence intensity between eCPMV and CPMV formulations was also statistically significant (p < 0.05).
Finally, the in vivo tumor homing properties of internally labeled eCPMV versus externally labeled CPMV were studied using a nude mouse xenograft model of colon cancer. In a one-pot synthesis reaction, CPMV and eCPMV were conjugated with A647 on their exterior and interior surface, respectively, together with mPEG5000-NHS on their exterior. These reactions yielded formulations with 50 fluorophores and approximately 30% PEGylation, as indicated by UV/vis spectroscopy, denaturing gels, and band analysis (not shown). Integrity of the particles was verified by TEM and SEC (Figures S1 and S2).
NCr-nu/nu mice were used and tumors were induced through subcutaneous injection of HT-29 colon cancer cells. The mice were kept on an alfalfa free diet to reduce tissue auto-fluorescence. Equal amounts of the particles (and thus equal amounts of dye) were injected intravenously (200 μg of eCPMV and 284.3 μg of CPMV) and allowed to circulate for 24 hours for delivery to the tumors. The animals were then sacrificed, and their tissues were collected and imaged ex vivo using a Maestro imaging system (Figure 8). No fluorescence was observed for the PBS control, while there was prominent fluorescence in the liver and some in the spleen for the eCPMV and CPMV particles due to clearing by the reticuloendothelial system (not shown). There was clear tumor homing via the enhanced permeability and retention effect, with fluorescent signal observed for both eCPMV and CPMV. The average signal over the tumor area for each mouse was analyzed using ImageJ. There is some variability between the animals, but the fluorescence intensities from mice injected with the eCPMV formulation appear to be consistently higher, with the difference between the two formulations being statistically significant (p < 0.05). This is an interesting result given the previous fluorescence measurements and flow cytometry studies. Some possible explanations for this difference could be that the display of A647 on the outside of the CPMV somewhat hinders its delivery to the tumor or the exterior dyes may be more easily degraded in vivo.
Figure 8.

Tumor homing of PEG5000E-eCPMV-A647I and PEG5000E-CPMV-A647E. A) Images from Maestro imaging system of tumor tissues under white light (left) and their fluorescent signal (right). B) Quantitative data of average fluorescent signal from the tumor tissues. Asterisks denote statistical significance as compared to PBS control (* p < 0.05, ** p < 0.01). Difference in fluorescence intensity between eCPMV and CPMV formulations was also statistically significant (p < 0.05).
HT-29 tumor cells express surface vimentin and targeting of CPMV to these tumor cells has been previously confirmed in vitro and in vivo 25. PEG was used in our design, as this allows shielding of the particles during circulation and increased tumor homing. Over short time periods (few hours), PEGylation shields CPMV from cell interactions. However, over longer time periods vimentin-specific cell uptake has been observed 25. Based on these previous observations, we propose the eCPMV formulation provides an advantage for in vivo tumor homing applications. The imaging labels can be installed on the interior surface, thus preserving cellular interactions with the native particle surface. Furthermore, eCPMV provides the possibility for additional modifications on the exterior for synthesis of multifunctional nanoparticles for targeted delivery applications.
CONCLUSION
In this study, we successfully established the use of bioconjugation methods for interior cargo loading of CPMV. We illustrated that this method can be used to encapsulate dyes, large PEG polymers, and a variety of negatively and positively charged peptides. From our findings, we hypothesize that factors that may govern the entrance of molecules through the pore and into the cavity of eCPMV include size, charge, and hydrophobicity. The pore size was measured to be 0.75 nm, which excludes larger molecules other than flexible polymers that can adapt their conformation such as PEG. In terms of charge, the greatest extent of labeling was observed using negatively charged dyes. Their diffusion into the cavity may be favored because they mimic the charge of the natural nucleic acid cargo. This trend was not observed for peptide labeling, but the negatively charged FLAG tag also had greater steric hindrance. Finally, hydrophilic molecules such as PEG2000 and MTFB appear to be able to diffuse into the cavity more freely than other molecules, as no difference in labeling between eCPMV and wild-type CPMV was observed.
Our results lead the way for imaging, polymer-mediated drug loading, and peptide-mediated mineralization applications. We have shown that RNA-free eCPMV is necessary to achieve significant dye loading compared to wild-type CPMV and that these interior dye-labeled eCPMV are suitable for fluorescence imaging in vitro and in vivo. eCPMV is able to passively accumulate in tumors through the EPR effect and has a higher signal intensity in vivo than exterior labeled CPMV. In addition to these individual applications, interior modification leaves reactive lysines on the exterior surface free for functionalization with other moieties such as PEG for masking non-specific interactions and prolonging circulation time, targeting ligands to confer tissue-specificity, and contrast agents for magnetic resonance and PET imaging. Interior conjugation is thus the first step in the advancement of the eCPMV platform for further development of multifunctional nanoparticles for in vivo applications. The feasibility to encapsulate biomedically relevant molecules within CPMV has great potential for future therapeutics incorporating tissue-specific targeting, drug delivery, and/or imaging in a single formulation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH/NIBIB grants R00 EB009105 (to NFS), P30 EB011317 (to NFS), Mt. Sinai Foundation (to NSF), a NIH/NIBIB training grant T32 EB007509 (to AMW), a NSF REU training grant EEC-0552804 (to JEM), and Alcoa undergraduate student funding (to ACY). At the John Innes Centre (JIC), the work was supported by BB/J004561/1 from the UK Biotechnology and Biological Sciences Research Council (BBSRC) and the John Innes Foundation (to DJE and GPL), and a BBSRC DTG (to AAAA and PS). Dr. Anouk Dirksen and Prof. Phil Dawson (The Scripps Research Institute) are thanked for providing the R5 peptide.
Footnotes
SUPPORTING INFORMATION AVAILABLE Experimental details of the characterization of eCPMV and CPMV conjugates by TEM and SEC are provided. This information is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES
- 1.Young M, Willits D, Uchida M, Douglas T. Annu. Rev. Phytopathol. 2008;46:361–384. doi: 10.1146/annurev.phyto.032508.131939. [DOI] [PubMed] [Google Scholar]
- 2.Barzon L, Stefani AL, Pacenti M, Palu G. Expert Opin. Biol. Ther. 2005;5:639–662. doi: 10.1517/14712598.5.5.639. [DOI] [PubMed] [Google Scholar]
- 3.Choi VW, McCarty DM, Samulski RJ. Curr. Gene Ther. 2005;5:299–310. doi: 10.2174/1566523054064968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cockrell AS, Kafri T. Mol. Biotechnol. 2007;36:184–204. doi: 10.1007/s12033-007-0010-8. [DOI] [PubMed] [Google Scholar]
- 5.Pokorski JK, Steinmetz NF. Mol. Pharmaceutics. 2011;8:29–43. doi: 10.1021/mp100225y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douglas T, Young M. Science. 2006;312:873–875. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- 7.Yildiz I, Shukla S, Steinmetz NF. Curr. Opin. Biotechnol. 2011;22:901–908. doi: 10.1016/j.copbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aljabali AA, Barclay JE, Butt JN, Lomonossoff GP, Evans DJ. Dalton Trans. 2010;39:7569–7574. doi: 10.1039/c0dt00495b. [DOI] [PubMed] [Google Scholar]
- 9.Aljabali AA, Barclay JE, Lomonossoff GP, Evans DJ. Nanoscale. 2010;2:2596–2600. doi: 10.1039/c0nr00525h. [DOI] [PubMed] [Google Scholar]
- 10.Steinmetz NF, Lomonossoff GP, Evans DJ. Small. 2006;2:530–533. doi: 10.1002/smll.200500453. [DOI] [PubMed] [Google Scholar]
- 11.Sapsford KE, Soto CM, Blum AS, Chatterji A, Lin T, Johnson JE, Ligler FS, Ratna BR. Biosens. Bioelectron. 2006;21:1668–1673. doi: 10.1016/j.bios.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Blum AS, Soto CM, Wilson CD, Cole JD, Kim M, Gnade B, Chatterji A, Ochoa WF, Lin T, Johnson JE, Ratna BR. Nano Lett. 2004;4:867–870. [Google Scholar]
- 13.Steinmetz NF, Ablack AL, Hickey JL, Ablack J, Manocha B, Mymryk JS, Luyt LG, Lewis JD. Small. 2011;7:1664–1672. doi: 10.1002/smll.201000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Nat. Med. 2006;12:354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saunders K, Sainsbury F, Lomonossoff GP. Virology. 2009;393:329–337. doi: 10.1016/j.virol.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 16.Chatterji A, Ochoa W, Paine M, Ratna BR, Johnson JE, Lin T. Chem. Biol. 2004;11:855–863. doi: 10.1016/j.chembiol.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Kaltgrad E, Lin T, Johnson JE, Finn MG. Chem. Biol. 2002;9:805–811. doi: 10.1016/s1074-5521(02)00165-5. [DOI] [PubMed] [Google Scholar]
- 18.Steinmetz NF, Lomonossoff GP, Evans DJ. Langmuir. 2006;22:3488–3490. doi: 10.1021/la060078e. [DOI] [PubMed] [Google Scholar]
- 19.Steinmetz NF, Evans DJ, Lomonossoff GP. ChemBioChem. 2007;8:1131–1136. doi: 10.1002/cbic.200700126. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q, Lin T, Johnson JE, Finn MG. Chem. Biol. 2002;9:813–819. doi: 10.1016/s1074-5521(02)00166-7. [DOI] [PubMed] [Google Scholar]
- 21.Hong V, Presolski SI, Ma C, Finn MG. Angew. Chem., Int. Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunel FM, Lewis JD, Destito G, Steinmetz NF, Manchester M, Stuhlmann H, Dawson PE. Nano Lett. 2010;10:1093–1097. doi: 10.1021/nl1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Lin T, Tang L, Johnson JE, Finn MG. Angew. Chem., Int. Ed. 2002;41:459–462. doi: 10.1002/1521-3773(20020201)41:3<459::aid-anie459>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Raja KS, Janda KD, Lin T, Finn MG. Bioconjugate Chem. 2003;14:38–43. doi: 10.1021/bc025587g. [DOI] [PubMed] [Google Scholar]
- 25.Steinmetz NF, Cho CF, Ablack A, Lewis JD, Manchester M. Nanomedicine. 2011;6:351–364. doi: 10.2217/nnm.10.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G, Manchester M. PLoS Pathog. 2009;5:e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinmetz NF, Maurer J, Sheng H, Bensussan A, Maricic I, Kumar V, Braciak TA. Cancers. 2011;3:2870–2885. doi: 10.3390/cancers3032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aljabali AAA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. Mol. Pharmaceutics. 2012 doi: 10.1021/mp3002057. in press, DOI: 10.1021/mp3002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Destito G, Yeh R, Rae CS, Finn MG, Manchester M. Chem. Biol. 2007;14:1152–1162. doi: 10.1016/j.chembiol.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Z, Chen K, Yildiz I, Dirksen A, Fischer R, Dawson PE, Steinmetz NF. Nanoscale. 2012;4:3567–3576. doi: 10.1039/c2nr30366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wellink J. Methods Mol. Biol. 1998;81:205–209. doi: 10.1385/0-89603-385-6:205. [DOI] [PubMed] [Google Scholar]
- 32.Montague NP, Thuenemann EC, Saxena P, Saunders K, Lenzi P, Lomonossoff GP. Hum. Vaccines. 2011;7:383–390. doi: 10.4161/hv.7.3.14989. [DOI] [PubMed] [Google Scholar]
- 33.Lin T, Chen Z, Usha R, Stauffacher CV, Dai JB, Schmidt T, Johnson JE. Virology. 1999;265:20–34. doi: 10.1006/viro.1999.0038. [DOI] [PubMed] [Google Scholar]
- 34.Brewer CF, Riehm JP. Anal. Biochem. 1967;18:248–255. [Google Scholar]
- 35.Ren Y, Wong SM, Lim LY. Bioconjugate Chem. 2007;18:836–843. doi: 10.1021/bc060361p. [DOI] [PubMed] [Google Scholar]
- 36.Pokorski JK, Breitenkamp K, Liepold LO, Qazi S, Finn MG. J. Am. Chem. Soc. 2011;133:9242–9245. doi: 10.1021/ja203286n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Svergun DI, Ekstrom F, Vandegriff KD, Malavalli A, Baker DA, Nilsson C, Winslow RM. Biophys. J. 2008;94:173–181. doi: 10.1529/biophysj.107.114314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Gennes PG. Adv. Colloid Interface Sci. 1987;27:189–209. [Google Scholar]
- 39.Aljabali AA, Shah SN, Evans-Gowing R, Lomonossoff GP, Evans DJ. Integr. Biol. 2011;3:119–125. doi: 10.1039/c0ib00056f. [DOI] [PubMed] [Google Scholar]
- 40.Dirksen A, Dawson PE. Bioconjugate Chem. 2008;19:2543–2548. doi: 10.1021/bc800310p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aljabali AA, Sainsbury F, Lomonossoff GP, Evans DJ. Small. 2010;6:818–821. doi: 10.1002/smll.200902135. [DOI] [PubMed] [Google Scholar]
- 42.Sainsbury F, Saunders K, Aljabali AA, Evans DJ, Lomonossoff GP. ChemBioChem. 2011;12:2435–2440. doi: 10.1002/cbic.201100482. [DOI] [PubMed] [Google Scholar]
- 43.Nieba L, Nieba-Axmann SE, Persson A, Hämäläinen M, Edebratt F, Hansson A, Lidholm J, Magnusson K, Karlsson AF, Plückthun A. Anal. Biochem. 1997;252:217–228. doi: 10.1006/abio.1997.2326. [DOI] [PubMed] [Google Scholar]
- 44.Green NM. Biochem. J. 1963;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leong HS, Steinmetz NF, Ablack A, Destito G, Zijlstra A, Stuhlmann H, Manchester M, Lewis JD. Nat. Protoc. 2010;5:1406–1417. doi: 10.1038/nprot.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh P, Thomas DM, Estrada MN, Powell E, Finn MG, Manchester M. Virology. 2005;343:224–235. doi: 10.1016/j.virol.2005.08.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.