

Abstract
DNA transposons make up 3% of the human genome, approximately the same percentage as genes. However, because of their inactivity, they are often ignored in favor of the more abundant, active, retroelements. Despite this relative ignominy, there are a number of interesting questions to be asked of these transposon families. One particular question relates to the timing of proliferation and inactivation of elements in a family. Does an ongoing process of turnover occur, or is the process more akin to a life cycle for the family, with elements proliferating rapidly before deactivation at a later date? We answer this question by tracing back to the most recent common ancestor (MRCA) of each modern transposon family, using two different methods. The first method identifies the MRCA of the species in which a family of transposon fossils can still be found, which we assume will have existed soon after the true origin date of the transposon family. The second method uses molecular dating techniques to predict the age of the MRCA element from which all elements found in a modern genome are descended. Independent data from five pairs of species are used in the molecular dating analysis: human–chimpanzee, human–orangutan, dog–panda, dog–cat, and cow–pig. Orthologous pairs of elements from host species pairs are included, and the divergence dates of these species are used to constrain the analysis. We discover that, in general, the times to element common ancestry for a given family are the same for the different species pairs, suggesting that there has been no order-specific process of turnover. Furthermore, for most families, the ages of the common ancestor of the host species and of that of the elements are similar, suggesting a life cycle model for the proliferation of transposons. Where these two ages differ, in families found only in Primates and Rodentia, for example, we find that the host species date is later than that of the common ancestor of the elements, implying that there may be large deletions of elements from host species, examples of which were found in their ancestors.
Keywords: transposons, class II, molecular dating, evolution
Introduction
Three percent of the human genome consists of class II (DNA) transposable elements (Lander et al. 2001), although these sequences are found far less abundantly than class I (RNA) transposable elements, such as Alu (de Koning et al. 2011). However, when we consider that protein-coding gene sequences make up approximately 1.5% of the human genome, and even when including other gene sequences, such as those transcribed into functional RNAs other than mRNAs, only 5–10% of the genome can be accounted for (Pheasant and Mattick 2007), the importance of class II elements’ contribution to the make up of mammalian genomes can be put into perspective. In the human genome, however, all class II elements are currently transpositionally inactive.
In any given genome, the collection of transposable elements of a given family will be connected to a most recent common ancestor element (element MRCA) by a phylogenetic tree, and the changes in elements’ sequences that have happened since the element MRCA can be used to estimate the shape and depth of the tree. That such a tree exists assumes that there has been no recombination between elements at different genomic locations. This may be approximately true, the exceptions being created by the possibility of partial or complete gene conversions creating elements with hybrid origins.
A number of interesting questions can be asked of these transposon families. We are particularly interested in the process of transposons originating in a genome, proliferating and becoming inactive. In particular, we can ask whether the inactivation of elements is somehow linked to their initial spread, such that the nature of their proliferation through genomes and populations has the effect that inactivation follows inevitably (the “life-cycle” model) or whether the elements proliferate to create a population of elements, residing stably in the chromosomes and undergoing a turnover process, with extinction being a subsequent, random event, unlinked to this initial proliferation (the “turnover” model).
The understanding of how this process of spread and inactivation of transposon families occurs has wide-reaching implications for our understanding of noncoding DNA. We wish to know how and why some genomes have more or less noncoding DNA than other closely related species and wish to understand processes occurring in modern organisms, such as the active transposition of class I transposon sequences.
We consider two models for the inactivation of transposable elements. The first model is that of a life cycle for the family of elements. For a given family, there must have, initially, been a single member of that family, in a single host individual. This could have followed a horizontal transfer event. The element will then spread in two dimensions, through the genome by transposition and through the population, as a small subset of elements that have the good fortune to land in effectively neutral locations spread by genetic drift. During these early stages, elements would become scattered across genomic locations, initially all polymorphic in the populations, and would be actively transposing to create new sites. Later, transposition diminishes as a result of changes to the host genome or changes to the elements themselves. Finally, we arrive at a state where there will be no active transposition of the family, as with the human class II elements. Elements can also be physically lost from chromosomes. As an increasing proportion of the elements are fixed in the population, loss must be started by deletion mutations, followed by drift (or possibly weak selection), allowing such chromosomes with deletions to spread to fixation.
This model of a life cycle of elements suggests that the time to the element MRCA of a collection of elements from the human genome may be only slightly more recent than the time of origin of the element family in the genome. Equally, it suggests that the time to the element MRCA for this family’s copies in another descendant of the genome in which the initial proliferation took place, such as a different mammalian order, will be similar to the time to MRCA of the human genome’s element copies.
The second model describes a process of turnover, where the transposable elements within a family inactivate randomly sometime after their insertion into the genome, whereas other copies, at least initially, continue to transpose. The process of turnover is continuous from the origin to the family until the entire family has become inactive. The process of turnover is likely to create a phylogenetic tree of elements with a time to element MRCA that is considerably more recent than the time to the origin of the family. Figure 1 shows the expected differences in the phylogenetic trees of elements sampled from different mammalian orders (red and blue). Under the turnover model, the elements will have continued to transpose and be lost within individual orders, such that MRCA elements for a given genome are order specific, and much more recent than the MRCA of all the elements of the family from all the orders that now possess it. For the life cycle model, although some transpositions and losses have continued within the orders, the MRCA of elements sampled within the orders (blue and red) is also the MRCA of all the elements from all the orders. Although we have described these processes as distinct, there is really a spectrum of possibilities, at the ends of which lie our two models.
Fig. 1.

Diagram showing phylogenies resulting from a life cycle or turnover process. Example phylogenetic trees expected from the lifecycle or turnover processes. Red and blue lines represent elements from different mammalian orders. The MRCA of elements from each mammalian order fall at different times from each other in the turnover process but at the same time in the lifecycle process. The life cycle process also has an MRCA much closer to the origin of the family than in the turnover process.
The origin of class II transposable elements has received little attention. A previous study (Pace and Feschotte 2007) looked into the origin and the extinction dates of class II transposons, using genomes and divergence dates available at the time. Here, we build upon this study, using the greater number of genomes now available, to suggest likely times of origin for the class II transposon families, through the identification of species divergence events, which can be inferred to have occurred after the origin of an element family shared by two species. Thus, we see which extant species share the element family and thus must descend from the species MRCA in which the element first proliferated. In this, we assume an absence of horizontal transfer save for an event that could have introduce the family into a single ancestral mammalian genome and thus do not account for multiple horizontal transmission events (Pace et al. 2008). This method of transposon family dating gives us a time span within which we can assume the elements originated. In addition, we look at the time to the element MRCA from which all extant sequences in the human genome, or other genomes, descended.
These methods may not be predicting the date of the same event. The analysis using occurrence in modern genomes to extrapolate back to the species MRCA is likely to predict a date close to the actual origin of the family. However, wholesale deletions of elements from a genome may have had the result that the element family can no longer be detected in some descendants of the ancestral species that first possessed it. Similar predictions of the time to MRCA from each method would lend weight to the life cycle hypothesis, whereas if the molecular dating prediction of the element MRCA is more recent than the species MRCA predicted by the analysis of presence in host species, this would lend weight to the turnover hypothesis.
The prediction of the element MRCA is carried out using BEAST (Drummond and Rambaut 2007), a Monte Carlo Markov Chain (MCMC) molecular dating technique from population genetics. The analysis predicts the date of the earliest element from which all the modern elements are descended. Using the life cycle model, we would assume that this date was close to that of the origin of the family; however, the turnover model would allow this date to be much later.
Although transposable elements are nonstandard data for use in BEAST, molecular dating techniques were used successfully in a previous study (Hellen and Brookfield 2012) to discover the time to the element MRCA of the Golem transposable element and its deletion products, Golem_A and Golem_B. BEAST and other molecular dating techniques have been shown to be highly reliant on the sequences used and the constraints placed upon them (Hug and Roger 2007; Hugall et al. 2007; Rutschmann et al. 2007). The use of multiple independent lineages should reduce any effects of changing rates of evolution, or other events, such as large-scale deletions, which may alter the predicted origin date. Here, we analyze the same mammalian transposable element families in three different orders: primates, carnivora, and artiodactyla. All the families examined are present in the human genome. Other lineages, such as rodentia, were dismissed due to the fragmented nature of the transposable elements still present in the genomes of modern organisms. Further lineages, such as perissodactyla, were dismissed due to the lack of genomes from pairs of recently diverged organisms, making the discovery of orthologous sequences difficult. As more genomes are sequenced and made publically available, it will be possible to widen this type of analysis to include other lineages.
Materials and Methods
Predicted Time to Species MRCA by Presence in Modern Organisms
Consensus sequences for human class II transposable elements were retrieved from the Repbase database (Jurka et al. 2005). All transposons that were found in the human genome using repeat masker through the UCSC genome browser (Dreszer et al. 2012) were initially included in the analysis.
The consensus sequences were used to carry out Ensembl BLAT (Flicek et al. 2012) searches in well-annotated, publically available, genomes. “Near Exact” matches were deemed to be the most suitable for transposable element searches; however, a brief exploration of the results when using tightened or relaxed criteria showed no difference in the number of elements retrieved. We are, therefore, happy that we have retrieved as many elements as possible using a homology search-based method. Transposable elements where no more than 25 hits could be found at <1.0 e−70 were excluded from the analysis as these were unlikely to provide enough examples for accurate molecular dating. This resulted in the retention of 29 consensus sequences. The presence or absence of matches to the consensus sequences allowed a rough origin date to be assigned to each sequence. Transposons were clustered into groups with similar predicted origin times using hierarchical clustering, implemented through the statistical language R (version 2.11.1) (http://www.R-project.org.). A binary assignment of presence/absence, in each of the genomes available through Ensembl, was used to remove the influence of the relative abundance of the transposable element in the genomes.
Predicted Time to Element MRCA by Molecular Dating
Pairs of orthologous transposon sequences were discovered using a reciprocal BLAT search (Kent 2002) through the UCSC genome browser. A rough estimate of synteny was also used to confirm orthology of pairs by checking that matches were on syntenous chromosomes according the Ensembl (Flicek et al. 2012). For each transposon consensus sequence, all human BLAT hits were paired with chimpanzee orthologs and with orangutan orthologs. If elements belonging to the transposon family were found in the cow genome, these hits were paired with pig orthologs, and if they were observed in the dog genome, the hits were paired with both cat orthologs and with panda orthologs. Only human, cow, and dog sequences with a BLAT e value of <1.0 e−70 when queried with the Repbase consensus were included.
Separate alignments were created for each transposon family, for each of the sets of orthologous pairs. Alignments were used in BEAST (Drummond and Rambaut 2007) analyses with a Yule tree structure and a strict clock. Previous analysis with the Golem transposon family (Hellen and Brookfield 2012) had determined that the transposable element data showed no significant difference in predictions when using strict or relaxed clocks but that the strict clock gave predictions with a smaller associated error. Intermediate dates were assigned to the phylogeny, using a normal distribution, at the divergence point between orthologs for each of the transposon elements. Dates chosen were the mean divergence times found in Timetree.org (Hedges et al. 2006; Kumar and Hedges 2011) (table 1). Standard deviations were chosen to include the majority of dates found in previous analyses and reported in Timetree.org.
Table 1.
Dates and Standard Deviations Assigned to the Divergence Point of Paired Orthologs to Constrain the BEAST Analysis.
| Organism 1 | Organism 2 | Mean Age | Standard Deviation |
|---|---|---|---|
| Human | Chimp | 6.1 | 0.5 |
| Human | Orangutan | 15.4 | 1 |
| Cow | Pig | 63.5 | 2 |
| Dog | Panda | 44 | 2 |
| Dog | Cat | 57.5 | 2 |
Evolution of Transposon Families
Each of the consensus sequences used was BLASTed (Altschul et al. 1990) against a local database consisting of all the consensus sequences in this analysis. Sequences found to have similarity with each other were grouped together to allow further analysis. Groups of similar sequences were aligned using a global alignment algorithm, and the NCBI OrfFinder (http://www.ncbi.nlm.nih.gov/projects/gorf/) was used to detect ORFs in autonomous transposon sequences.
Variation in Rates of Evolution
An analysis was carried out to test for systematic differences in the evolutionary rates predicted by data consisting of different pairs of species, for example, do the analyses carried out using primate species show a lower rate of evolution than those carried out using carnivora species? Paired t-tests were used to compare the distribution of evolutionary rates resulting from analyses using each of the pairs of species. The evolutionary rate results of every pair of species were compared against the results from every other pair. Only results from transposon families where the analysis was carried out using both pairs of species were used.
Further t-tests were used to compare the evolutionary rates between families. All the evolutionary rates for each transposon family, predicted using different species pairs, were compared with the evolutionary rates for the other families. Only analyses where all five sets of species pairs could be used in the BEAST analyses were included.
To determine whether there was a relationship between the predicted evolutionary rates and the predicted origin of the ancestral element, a Pearson correlation coefficient was calculated between the predicted rates of evolution and the dates given for the origin of the ancestral element. This was to determine whether the BEAST predictions were more reliable when using larger data sets. For each of these analyses, all BEAST results were pooled. All statistical analyses were carried out using R (version 2.11.1).
Results
Predicted Time to Species MRCA by Presence in Modern Organisms
BLAT searches were carried out in each of the publically available, well-annotated genomes in Ensembl (Flicek et al. 2012). The results of this allow estimates to be made of the origin of each family of transposable elements, using the assumption that the time to the MRCA of the species whose genomes contain the family (species MRCA) corresponds to the time of the origin of the element family and that the transposon that originated in the species MRCA is the ancestor element of all element copies in all modern genomes that contain the family. The families were clustered using hierarchical clustering to determine groups that appear to have originated at a similar time point. The transposable element families have been divided into six main groups on this basis (fig. 2).
Fig. 2.

Hierarchical clustering of transposons by presence/absence in Ensembl genomes. Transposon families have been divided into six groups using a hierarchical clustering algorithm implemented in R (version 2.11.1), based on a binary assessment of whether the family can be found through BLAT searching of each Ensembl genome: P + R (present in Primates and Rodentia), P + (present in Primate and some other species but no other whole order), Primate (present in Primates), Therian (found in all eutherian and marsupial species), Eutherian (found in all eutherian species), and Eutherian subset (found in a large number of, but not all, eutherian species). The horizontal line shows the point at which we have cut the graph to provide the groupings of elements.
Therian Transposons
These transposons are found in all genomes analyzed, with the exception of the platypus (Ornithorhynchus anatinus) genome, in which none of our transposon sequences were identified. We can assume that the origin of the transposon was at some point before the marsupial–eutherian divergence (van Rheede et al. 2006) (Timetree.org mean: 163.9 Ma). but after the therian–monotreme divergence (Timetree.org mean: 167.4 Ma). We assume that the lack of sequences in the platypus genome is because of an origin date after the therian–monotreme divergence; however, it is possible that, due to the ancient nature of this event, any sequences which originated before this point have since been deleted or evolved so far as to not be identified in BLAT searches of the platypus genome. This uncertainty is compounded by the lack of availability of other monotreme genomes. Suggested origin based on Timetree.org: 163.9–167.4 Ma.
Eutherian Transposons
These transposons can be found in most of the genomes analyzed but not in the marsupial genomes. As with the Therian transposons, we cannot tell by looking at the genomes of modern organisms whether the transposon originated later than the marsupial–eutherian divergence date or whether these early sequences can no longer be detected. Suggested origin: 94.4–163.9.
Eutherian Subset
The transposons belonging to the eutherian subset are found in a large number of the mammalian species analyzed, but not all, placing their likely origin just after the eutherian species start to diverge. There are several orders in which elements can be found in some organisms, but not others, such as afrotheria, insectivora, and rodentia. This suggests that the transposon sequences may have been lost from certain lineages at a later date. The small number, or, in some cases, lack, of sequences found in the rodentia genomes supports this hypothesis, as any transposon originating in the MRCA of primate and carnivore organisms would also be expected to be present in rodents (Bininda-Emonds et al. 2007). One possible reason for this patchy distribution is the variation in evolutionary rates found within orders (Gissi et al. 2000; Douzery et al. 2003). For example, mouse and rat are known to have much higher rates than squirrel, and it is, therefore, possible that the transposon families found in the squirrel genome were also present in a mouse ancestor but that the sequences have undergone large changes and are no longer recognizable using this method. However, such a patchy distribution could also, in principle, have resulted from horizontal transfers. Suggested origin: 90–163.9.
Primate +
This group contains sequences from three transposon families, which are found in all primate species analyzed, in guinea pig (Cavia porcellus), cow (Bos taurus), megabat (Pteropus vampyrus), and dolphin (Tursiops truncates). Assuming this is not an effect of badly annotated genomes, it would seem likely that the transposon sequences have either been deleted in the other species or have been horizontally inserted into these genomes at a later date. Only primate orthologs can be used to date the sequences due to the lack of a suitable comparison species for the cow elements. However, the molecular dating of the transposon family should allow a hypothesis to be constructed about how this unusual pattern of transposon presence came to occur. Further discussion of the Primate + family can be found in the Evolution of Transposon Families section. Suggested origin: 90.0–163.9.
P + R
The P + R group of transposons is found in all species analyzed from both the primate and rodent orders but not any other eutherian orders. As the current literature mostly agrees that the Primate and Rodentia orders diverged more recently than either diverged from the carnivora, artiodactyla, and other eutherian orders (Bininda-Emonds et al. 2007), it is likely that these transposons originated between the time of the divergence of primates plus rodentia (and lagomorphs) from the other eutherian species and the divergence between the primates and rodents themselves. Suggested origin: 92.4–94.4.
Primate Transposons
Some of the transposons studied occur only in primate genomes, either all primates, or a subset of those analyzed. All these transposons can be found in at least human, chimpanzee, and orangutan and can, therefore, be used in the analysis.
Although these transposable elements have been designated as primate specific, it is interesting to note that they appear to also be found, in small numbers, in the guinea pig genome; however, they are not found in any of the other rodentia genomes and so are not classified as P + R transposons. Whether this is the result of an origin earlier than the primate divergence or of horizontal transfer is unknown, but it may be possible to decide between these two hypotheses through the molecular dating analysis. Suggested origin: 42.6–94.4.
Predicted Time to Element MRCA by Molecular Dating
The BEAST analyses, using human–chimp orthologs, predict ancestral element dates, which mostly fall within the bounds of the species MRCA predictions made using the presence of the family in modern genomes and timetree.org mean divergence estimates (fig. 3). The Primate + group is predicted to have occurred at a similar time to the Therian and Eutherian subset groups. This would imply that the occurrence of the elements in certain organisms, but not others which are closely related, is due to the loss of elements from certain lineages rather than horizontal transfer of the element into these species.
Fig. 3.

Molecular dating of the MRCA of transposable elements using human–chimp orthologs. Predicted dates for the time to the element MRCA for each transposon family using human–chimp divergence dates as a constraint. Error bars show the period of time between the lower and upper bounds of the highest posterior density interval (HPD) for each BEAST analysis, a range that contains 95% of the sampled values. Points represent the mean value. Dark-gray-shaded regions represent the predicted range of values predicted for the species MRCA analysis using timetree.org mean values. Light-gray-shaded regions show the range of values predicted for the analysis of the species MRCA using highest and lowest published values from timetree.org, and transposon families in each category have been divided using dashed lines.
Of those which do not fall within the mean estimates calculated using the species MRCA, all but those in the “Primate + Rodent” group are within the upper and lower published estimates for these divergence dates. The earlier than expected dates for element MRCA found for the “Primate + Rodent” group are confirmed when using the human–orangutan orthologs but cannot be assessed using any other lineages, because of the lack of examples in modern genomes. These early origin dates, coupled with a lack of examples in most modern eutherian species, may imply that the latter is the result of a large-scale deletion of these elements in species not on the primate–rodentia lineage. An alternative explanation for this is that multiple horizontal transmission events took place. Horizontal transfers could have had the effect that the element MRCA being dated existed outside the mammals, and its descendants were subsequently introduced into primates and rodents only. A final possibility is suggested by the large errors shown for the element MRCA predictions—it is possible that the true values actually lie at the younger end of these error bars, which would make the problem much less acute.
For the majority of transposon families, the human–chimpanzee and human–orangutan analyses show overlapping prediction ranges and often very similar mean values. However, when all analyses for a family are taken in to account, we often see a large range of possible values for the predicted time to origin (fig. 4). In some families, we find that predictions given by analysis of one species pair do not overlap with predictions given by another analysis of the same family based on a different species pair. This raises the possibility that the element MRCAs occasionally differ between orders. Thus, a later carnivora or artiodactyla prediction can be explained by the smaller data sets that are usually available for species pairs within these orders and an assumption that part of the tree was lost, leading to the dating of an ancestral element, which existed later than the ancestral element being dated when using the larger primate data sets. However, the explanation for an earlier time inferred using nonprimate data than is expected from the primate data is less clear. It may be that as a result of the longer time to the divergence point between the two species, the extrapolated evolutionary rate is less accurate. Alternatively, it may be the effect of the smaller numbers of orthologs found in the pairs of organisms with an earlier divergence time, leading to less robust data sets that are prone to error.
Fig. 4.

Molecular dating of the time to element MRCA using primate, carnivora, or artiodactyla orthologous pairs. Predicted dates for the time to element MRCA of each transposon family, using human–chimp, human–orangutan, dog–panda, dog–cat, and cow–pig divergence dates as constraints. Error bars show the period of time between the lower and upper bounds of the highest posterior density interval (HPD) for each analysis, a range that contains 95% of the sampled values. Points represent the mean value. Dark-gray-shaded regions represent the predicted range of values predicted for the species MRCA analysis using timetree.org mean values. Light-gray-shaded regions show the range of values predicted for the analysis of the species MRCA using highest and lowest published values from timetree.org, and transposon families in each category have been divided using dashed lines. Successive transposon families’ estimates are colored in red or in black for clarity.
Evolution of Transposon Families
An all-against-all Basic Local Alignment Search Tool (BLAST) search of the consensus transposon sequences from the different transposon families showed little evidence of their having evolved from a common ancestor family, except for a few cases (fig. 5). The majority of these transposons are related through deletion events, often leading to the removal of the internal ORF region and the establishing of a nonautonomous element. However, some transposons, such as Charlie2A and Charlie2B, have highly similar terminal regions with differing, but not deleted, internal regions. These changes may be due to the effect of partial gene conversion.
Fig. 5.
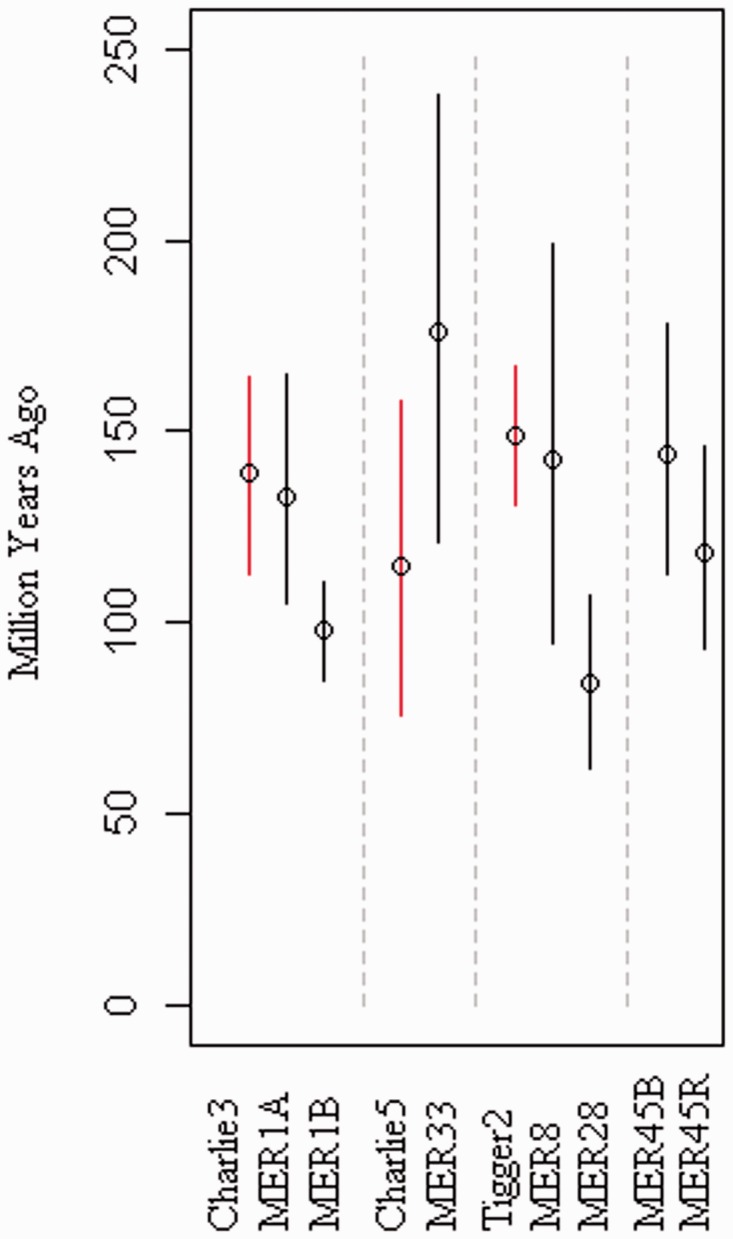
Comparison of the time to element MRCA for related transposon families. Time to element MRCA, for each transposon family, predicted by BEAST using human–chimpanzee orthologs. Transposon families are grouped by likely homology and divided using a dashed line. Transposons marked in red contain a recognizable ORF.
An analysis of the, BEAST-predicted, times to element MRCA for related transposon families provides a timescale for the creation of the related deletion products. Charlie3 and Tigger2 are both predicted to have arisen >100 Ma with one deletion product each, MER1A and MER8, respectively, originating soon afterward and a further deletion product occurring much later. The mean predicted origins for Charlie5 and its assumed deletion product, MER33, do not show the expected pattern of a later time to ancestral element for the MER33 family than for the Charlie 5 family. However, once the upper and lower bounds of the highest posterior density interval have been considered, it is still possible to suppose that Charlie5 originated first and that MER33 is a later deletion product. This is still the most likely scenario as MER33 contains a fragment of the ORF found in Charlie5, reducing the likelihood that the nonautonomous version occurred first, becoming autonomous upon the insertion of an ORF from another transposable element.
It is also possible that the autonomous elements are more highly constrained, evolving more slowly and appearing to be younger. This would explain some of the unusual predictions of origin date. However, we do not have clear evidence for differential rates, and the large errors and thus overlap in the time estimates are consistent with an early Charlie5 followed by a later MER33 deletion product.
The creation of MER1A and MER1B through deletion events occurring in Charlie3 is particularly interesting as these are the three transposon families that make up the Primate + group, which exhibits an unexpected pattern of presence/absence in mammalian genomes. A closer look at the blast hits retrieved and the alignments created using the consensus sequences for each of these families show that all the cow, bat, and dolphin hits in MER1A and MER1B align to the terminal sections, those sections which are in common with Charlie3. It therefore seems like a much more likely solution that the strange pattern of presence/absence occurred only in Charlie3, and the similarity between sequences has had the effect of making MER1A and MER1B appear to be present in these extra species.
Variation in Rates of Evolution
The evolutionary rates predicted by the BEAST analyses are found to follow a normal distribution (Shapiro–Wilk; P > 0.05) with a mean value of 1.261 e−3 per base per My (fig. 6).
Fig. 6.

The effect of the number of orthologous pairs used in the prediction of evolutionary rate. Each point represents one BEAST analysis, comparing the predicted evolutionary rate against the number of orthologous pairs used in the analysis; data from different families and using different orthologous pairs have been pooled. The black dotted line shows the mean rate, and the red dotted lines show mean ± 2 standard deviations.
The idea of a molecular clock that is constant across eutherian species is a contentious one, and our analysis suggests variation across both transposon families. The difference in evolutionary rates between analyses of the same transposon family, using different orthologous pairs from the primate, carnivora or artiodactlya orders, was not found to be significant (t-test; P > 0.05), whereas the differences among rates in different transposon families were found to be significant. This implies that the evolutionary rate of each transposon family is fairly constant in all organisms but that different families may have slightly different evolutionary rates. The difference in rates between families is possibly due to the environments surrounding the elements, such as their presence in or near a coding or regulatory region, affecting the base evolutionary rate, due to selective constraints, if indeed the elements have in some cases evolved cis-acting functions.
More variation can be seen across the analyses carried out using a smaller number of orthologs than those with a higher number, where the evolutionary rates predicted are close to the mean rate. A threshold of 40 pairs of orthologs can be used to reduce the variability in evolutionary rates, however, for many families, it was not possible to identify this many orthologous pairs of elements. The difference in evolutionary rates, shown by analyses with a small number of orthologs, appears to be due to a small number of pairs of orthologs with a much higher or lower rate predicted for that branch. This is likely to be due to the selection pressures that the environment in which the element has been inserted has been subjected to. In larger analyses, where the clock is averaged over a greater number of branches, this effect is muted. An exception to this is found in the Tigger 7 analysis using 163 pairs of human–chimp orthologs, where the rate of evolution is estimated to be higher than expected. The human–orangutan analysis of the same transposon family gives a lower rate of 1.17 e−3, which implies that there may an external factor increasing the rate of evolution in the human or, more probably, chimpanzee Tigger 7 sequences.
The majority of the evolutionary rates predicted in the analysis fall between the global molecular clock (Kumar and Subramanian 2001) and noncoding (Soojin et al. 2002) rates suggested in the literature (fig. 7).
Fig. 7.

The effect of differing evolutionary rates on the prediction of the element MRCA. Plot showing the mean rate of evolution (clock.rate) and the mean time to element MRCA (treemodel.rootHeight) for each of the BEAST analyses; data from different families and using different orthologous pairs have been pooled. The colors show the pairs of orthologs used in the order: human–chimp (red), human–orangutan (orange), dog–panda (dark green), dog–cat (light green), and cow–pig (blue). Dotted lines show mammalian evolutionary rates suggested in the literature.
A strong negative correlation can be seen between the evolution rate and the predicted time to element MRCA, particularly for the earliest predicted dates (Pearson; r = −0.64, P < 0.05). The majority of element MRCA dates, later than 200 Ma, a date which all our transposable elements are likely to originated after, are associated with an evolutionary rate similar to global evolutionary rates, which have been reported in the literature. The correlation between evolutionary rate and time to element MRCA is not expected. Instead, we assume this to be an artifact caused by the small amount of data in many of the analyses, allowing sequence pairs with higher, or lower, than expected, evolutionary rates to push the estimate of the origin date from its true position. If, by chance, the orthologs from a particular species pair were unexpectedly similar, this would lower the estimate of the evolutionary rate and raise the estimate of the time to the element MRCA for the family.
Discussion
The recent expansion in the number of annotated and publically available genomes has allowed a more detailed analysis of the dates at which class II transposon families have originated than has previously been conducted. By analysis of the presence or absence of a particular family in the genomes of modern organisms, an assumption can be made about the most recent common ancestor species (species MRCA) in which the family originated, and a date can be attached to the timescale in which this ancestor is likely to have lived. However, the wide range of predicted dates for the divergences of some organisms, particularly those such as the marsupial–eutherian divergence, attaches a large error to many of these predictions.
Alongside this method, molecular dating techniques were also used, to date the occurrence of the ancestral element from which all modern family elements descended (element MRCA). Although in some cases this technique suggested a wide range of origin dates, the likelihood of any date being correct can be calculated from the number of occasions it is predicted by the MCMC analysis. Our predictions have shown that, for the majority of cases, the prediction of the time to element MRCA is similar to the range of dates predicted for the species MRCA of extant elements.
This observation is consistent with the concept of a life cycle of the proliferation of the elements followed by inactivation, rather than an ongoing process of turnover, many tens of millions of years after the elements’ origin. If the latter were to have happened, the time to element MRCA in a given genome would be expected to be much more recent than the time to the species MRCA of the host organisms that now contain the elements (fig. 1).
Where the two predicted origin dates differ, we find that, counter intuitively, the prediction of the element MRCA is earlier than that for the species MRCA. In a number of cases, this can be traced to a lowered or elevated rate of evolution predicted due to a small data set. However, for certain families of elements, this appears to be an accurate timing, consistent with the collection of species for which the MRCA is estimated being a subset of those whose ancestors initially possessed the element, as a result of the deletion of transposable elements from certain lineages. Hence, the species MRCA now identified is more recent than the MRCA of all the species whose ancestors had the element. Alternatively, in these cases, there could have been horizontal transfers into the two orders. This appears to be particularly true for the group of transposons thought, from the genome analysis, to only occur in primate and rodentia species but predicted by the molecular dating analysis to have occurred at an earlier time, consistent with the date of ancestry for the transposons that can be found in all mammalian species.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council (research grant reference: BB/H009884/1).
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bininda-Emonds ORP, Cardillo M, Jones KE, MacPhee RDE, Beck RMD, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. The delayed rise of present-day mammals. Nature. 2007;446:507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- de Koning APJ, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7:e1002384. doi: 10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douzery EJ, Delsuc F, Stanhope MJ, Huchon D. Local molecular clocks in three nuclear genes: divergence times for rodents and other mammals and incompatibility among fossil calibrations. J Mol Evol. 2003;57:S201–S213. doi: 10.1007/s00239-003-0028-x. [DOI] [PubMed] [Google Scholar]
- Dreszer TR, Karolchik D, Zweig AS, et al. (29 co-authors) The UCSC genome browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, et al. (57 co-authors) Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gissi C, Reyes A, Pesole G, Saccone C. Lineage-specific evolutionary rate in mammalian mtDNA. Mol Biol Evol. 2000;17:1022–1031. doi: 10.1093/oxfordjournals.molbev.a026383. [DOI] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- Hellen EHB, Brookfield JFY. Investigation of the origin and spread of a mammalian transposable element based on current sequence diversity. J Mol Evol. 2012;73:287–296. doi: 10.1007/s00239-011-9475-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug LA, Roger AJ. The impact of fossils and taxon sampling on ancient molecular dating analyses. Mol Biol Evol. 2007;24:1889–1897. doi: 10.1093/molbev/msm115. [DOI] [PubMed] [Google Scholar]
- Hugall AF, Foster R, Lee MS. Calibration choice, rate smoothing, and the pattern of tetrapod diversification according to the long nuclear gene RAG-1. Syst Biol. 2007;56:543–563. doi: 10.1080/10635150701477825. [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. TimeTree2: species divergence times on the iPhone. Bioinformatics. 2011;27:2023–2024. doi: 10.1093/bioinformatics/btr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Subramanian S. Mutation rates in mammalian genomes. Proc Natl Acad Sci U S A. 2001;99:803–808. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, et al. (256 co-authors) Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Pace JK, Feschotte C. The evolutionary history of human DNA transposons: evidence for intense activity in the primate lineage. Genome Res. 2007;17:422–432. doi: 10.1101/gr.5826307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace JK, Gilbert C, Clark MS, Feschotte C. Repeated horizontal transfer of a DNA transposon in mammals and other tetrapods. Proc Natl Acad Sci U S A. 2008;105:17023–17928. doi: 10.1073/pnas.0806548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pheasant M, Mattick JS. Raising the estimate of functional human sequences. Genome Res. 2007;17:1245–1253. doi: 10.1101/gr.6406307. [DOI] [PubMed] [Google Scholar]
- Rutschmann F, Eriksson T, Salim KA, Conti E. Assessing calibration uncertainty in molecular dating: the assignment of fossils to alternative calibration points. Syst Biol. 2007;56:591–608. doi: 10.1080/10635150701491156. [DOI] [PubMed] [Google Scholar]
- Soojin Y, Elsworth D, Wen-Hsiung L. Slow molecular clocks in Old World monkeys, apes, and humans. Mol Biol Evol. 2002;19:2191–2198. doi: 10.1093/oxfordjournals.molbev.a004043. [DOI] [PubMed] [Google Scholar]
- van Rheede T, Bastiaans T, Boone DN, Hedges SB, de Jong WW, Madsen O. The platypus is in its place: nuclear genes and indels confirm the sister group relation of monotremes and therians. Mol Biol Evol. 2006;23:587–597. doi: 10.1093/molbev/msj064. [DOI] [PubMed] [Google Scholar]