

Abstract
Recessively inherited phenotypes are frequent in the Palestinian population, as the result of a historical tradition of marriages within extended kindreds, particularly in isolated villages. In order to characterise the genetics of inherited hearing loss in this population, we worked with West Bank schools for the deaf to identify children with prelingual, bilateral, severe to profound hearing loss not attributable to infection, trauma or other known environmental exposure. Of 156 families enrolled, hearing loss in 17 families (11 per cent) was due to mutations in GJB2 (connexin 26), a smaller fraction of GJB2-associated deafness than in other populations. In order to estimate how many different genes might be responsible for hearing loss in this population, we evaluated ten families for linkage to all 36 known human autosomal deafness-related genes, fully sequencing hearing-related genes at any linked sites in informative relatives. Four families harboured four novel alleles of TMPRSS3 (988ΔA = 352stop), otoancorin (1067A >T = D356V) and pendrin (716T > A = V239D and 1001G > T = 346stop). In each family, all affected individuals were homozygous for the critical mutation. Each allele was specific to one or a few families in the cohort; none were widespread. Since epidemiological tests of association of mutations with deafness were not feasible for such rare alleles, we used functional and bioinformatics approaches to evaluate their consequences. In six other families, hearing loss was not linked to any known gene, suggesting that these families harbour novel genes responsible for this phenotype. We conclude that inherited hearing loss is highly heterogeneous in this population, with most extended families acting as genetic isolates in this context. We also conclude that the same genes are responsible for hearing loss in this population as elsewhere, so that gene discovery in these families informs the genetics of hearing loss worldwide.
Keywords: genetics, genomics, inherited, deafness, hearing loss, mutation, Palestinian, TMPRSS3, pendrin, otoancorin
Introduction
Extended kindreds from highly endogamous communities are ideally suited for identifying genes responsible for clinically important phenotypes. The unique demographic history of the Middle East has led to many such communities. For more than 5,000 years and continuing to the present, the eastern shores of the Mediterranean have seen immigration of people from a wide variety of cultures. Villages were often established by a few extended families and, despite their geographical proximity, remained demographically isolated. For centuries, marriages have been arranged within extended families in these villages, leading to high levels of consanguinity and consequently high frequencies of recessive traits [1,2].
The willingness of these kindreds to participate in research has been of enormous help to geneticists. In order to identify genes responsible for inherited hearing loss, we have worked with consanguineous kindreds from the Palestinian population. As expected, we have found recessive alleles responsible for inherited hearing loss in such families. We have also discovered that an extremely high level of both allelic and locus heterogeneity characterises inherited hearing loss in this population.
Systematic population-wide genomic analysis may be the most effective way to identify all critical genes and alleles for heterogeneous phenotypes in communities with many small isolates. We illustrate such a strategy in this paper. In such populations, most alleles of clinical importance are likely to be rare, found in only one or a few affected families, thus effectively precluding case-control approaches to test associations of alleles with the trait. Functional and bioinformatics approaches can be applied to these situations, and we illustrate such strategies applied to hearing loss phenotypes. The most important consequence of this heterogeneity is that such populations may offer the opportunity to identify and characterise a large number of alleles and genes critical to common phenotypes. They may, therefore, be even more valuable than previously recognised.
Methods
Ascertainment of subjects
Probands are children with prelingual bilateral hearing loss who attend one of the following schools for deaf children: Eftah School for the Deaf in Bethlehem; Al Amal School for the Deaf in Hebron; the Association for the Deaf in Nablus; and the Princess Basma Rehabilitation Center for Deaf Children in Jerusalem. Together, these schools serve nearly all hearing-impaired Palestinian children living on the West Bank. Children were referred to one of us (M.K.) by their teachers. Families were contacted through teachers and social workers. If the family expressed an interest, they were visited at home (by M.K. and A.A.R.) to discuss the project in detail, explaining that the first steps in participation would be physical and audiological examinations to exclude hearing loss due to infection or trauma and to evaluate severity and laterality of the hearing loss. For families who agreed, informed consent was obtained from parents and assent from older children. Controls comprised 100 Palestinian adults with normal hearing, ascertained when blood samples were drawn at West Bank clinics for preventive health services. Controls agreed that their DNA samples could be used anonymously for this project. The project is approved by the Human Subjects Division of the University of Washington, by the Helsinki Committee of Tel Aviv University and by the Human Subjects Committee of Bethlehem University. Single Project Assurances for Tel Aviv University and for Bethlehem University have been obtained from the US Office for Human Research Protection (OHRP). Consenting deaf adults and children, hearing adults and hearing children older than the oldest age of onset of inherited deafness in their family were enrolled.
Genotyping and sequencing
Microsatellite markers within and flanking deafness genes were selected from the Genome Browser or designed using the simple repeat track of the Genome Browser [3]. DNA samples were genotyped and lod scores calculated as previously described [4].
For sequencing candidate genes, primer pairs were designed to amplify and sequence the entire coding regions and splice sites of all isoforms of GJB2 (connexin 26),[4]TMPRSS3,[5] pendrin[6] and otoancorin [7]. Sequencing was carried out using BigDye terminator 3.1 sequencing chemistry (Applied Biosystems) and was then visualised on ABI 3100 sequencers. Sequences were aligned and compared using SeqHelp [8].
Mutations described in the text were genotyped in all probands and controls by sequencing or by polymerase chain reaction (PCR) and restriction enzyme digest. Restriction enzyme digests were made with Mbo II for TMPRSS3.988delA, Hph I for pendrin 1001G >T and Fok I for otoancorin 1067A >T. Pendrin 716T >A was screened by DNA sequencing in probands and controls. All probands and controls were also screened by PCR and restriction enzyme digest for previously published Palestinian and Lebanese deafness alleles of otoferlin,[9]TECTA,[10] otoancorin[7] and whirlin [11]. Restriction enzyme digests were made with Dde I for otoferlin 2416T >A (Y730X), PfiMI for TECTAIVS9(+1)G >A, Cac81 for otoancorin IVS12(+2)T >C and BvbCI for whirlin 2332C >T (R778X). The β-satellite insertion in TMPRSS3 was analysed by PCR, as published [5].
Localisation of GFP chimeras
The pendrin V239D mutant was created by PCR mutagenesis using the QuickChange Site-Directed Mutagenesis Kit (Stratagene) and subcloned into the green fluorescent protein (GFP) expression vector pEYFP-C1 (Clontech). Cell cultures, expression of chimeras and microscopy were carried out as previously described [12].
Results
The cohort presently includes 156 probands and their families. Probands were characterised as having prelingual, bilateral hearing loss that could not be attributed to infection or trauma. No vision problems were present. More subtle syndromic signs in some individuals were detected subsequently, as described below.
Because mutations in the gap junction GJB2 (connexin 26) are responsible for a substantial fraction of recessive inherited hearing loss worldwide,[13] we first sequenced both exons of this gene in genomic DNA from all probands [4]. We also screened for the neighbouring deletion of GJB6 [14]. Of the 156 families in the project, inherited hearing loss in 17 families (11 per cent) was due to mutations in GJB2. Five different mutant alleles of GJB2 were present [4]. The fraction of inherited hearing loss due to GJB2 in this population is somewhat lower than in other populations.
In order to assess genetic heterogeneity of inherited deafness in the population, we chose ten families, which each included at least four relatives with prelingual bilateral hearing loss who were wild-type for GJB2. (An average of six affected individuals per family were sampled.) All living relatives with hearing loss were evaluated by physical examination and audiology to distinguish inherited prelingual hearing loss from age-related hearing loss. Prelingual deafness was consistent with recessive inheritance in each family.
We next carried out a hearing-loss-targeted linkage scan of the ten families, using 68 microsatellite markers within and flanking the 36 autosomal genes known to be responsible for inherited hearing loss (Table 1) [15]. We tested for linkage to genes for either recessive or dominant hearing loss, because different alleles of the same gene may lead to hearing loss by different modes of inheritance. If markers were uninformative, other markers were substituted in that family [3]. We noted whether affected individuals were homozygous for a linked haplotype, but did not exclude families with more than one linked haplotype that might reflect more than one deafness-associated allele.
Table 1.
Genes responsible for inherited deafness (DFN) with neighbouring microsatellite markers used in the subgenome scan.
| Chromosome | Distance from pter (MB) | Gene | DFN locus | Size of gene (kb) | Flanking markers and kb spanned | ||
|---|---|---|---|---|---|---|---|
| 1 p36.31 | 6.4 | ESPN | B36 | 36 | D1S253 | D1S2731 | 342 |
| 1 p34.3 | 34.9 | GJB3 | A2 | 5 | D 1S1570 | D1S496 | 190 |
| 1 p34.2 | 40.9 | KCNQ4 | A2 | 55 | D1S2706 | D1S1188 | 273 |
| 2 p23.3 | 26.6 | OTOF | B9 | 102 | D2S2350 | D2S174 | 112 |
| 3 p21.31 | 46.7 | TMIE | B6 | 9 | AC(46.635) | TG(46.779) | 144 |
| 4 p16.1 | 6.4 | WFS1 | A6/A14 | 33 | TCCC(6.457) | D4S431 | 77 |
| 5 q31.3 | 140.9 | DIAPH1 | A1 | 104 | D5S2119 | D5S2010 | 254 |
| 5 q32 | 145.7 | POU4F3 | A15 | 1 | D 5S2099 | D5S2033 | 628 |
| 6 p21.32 | 33.2 | COL11A2 | A13 | 30 | D6S2414 | D6S1701 | 449 |
| 6 q14.1 | 76.6 | MY O6 | B37/A22 | 167 | AC (76.528) | GT(76.674) | 146 |
| 6 q23.2 | 133.7 | EYA4 | A10 | 288 | D6S975 | D6S1722 | 517 |
| 7 p15.3 | 24.5 | DFN A5 | A5 | 51 | D7S2444 | D7S2493 | 477 |
| 7 q22.1 | 102.6 | SLC26A5 (Prestin) | B61 | 93 | D7S2509 | D7S2504 | 478 |
| 7 q31.1 | 106.9 | SLC26A4 (Pendrin) | B4 | 57 | D7S496 | D7S2459 | 177 |
| 8 q22.3 | 102.6 | TFCP2L3 | A28 | 177 | D8S521 | D8S1046 | 761 |
| 9 q21.13 | 72.4 | TMC1 | B7/B11/A36 | 315 | D9S1837 | D9S1876 | 48 |
| 9 q32 | 114.3 | WHRN | B31 | 103 | D9S1824 | D9S1855 | 619 |
| 10 p12.1 | 26.4 | MY03A | B30 | 278 | D10S2481 | D10S1775 | 60 |
| 10 q21.1 | 56.0 | PCDH15 | B23 | 980 | D10S546 | D10S1642 | 321 |
| 10 q22.1 | 73.0 | CDH23 | B12 | 420 | D10S584 | D10S1694 | 152 |
| 11 p15.1 | 17.5 | USH1C (Harmonin) | B18 | 51 | D11S902 | D11S4138 | 268 |
| 11 q13.5 | 76.6 | MY07A | B2/A11 | 87 | D11S4179 | D11S4186 | 573 |
| 11 q23.3 | 120.5 | TECTA | B21/A8/A12 | 88 | D11S4089 | D11S4107 | 60 |
| 12 q13.3 | 55.7 | MY O1A | A48 | 22 | D12S1644 | D12S1691 | 1 |
| 13 q12.11 | 19.7 | GJB2 (Connexin26) | B1/A3 | 5 | D 13S1316 | D13S175 | 167 |
| 13 q12.11 | 19.7 | GJB6 (Connexin31) | B1/A3 | 9 | D 13S1316 | D13S175 | 167 |
| 14 q12 | 30.4 | COCH | A9 | 16 | D14S1021 | D14S54 | 256 |
| 15 q15.3 | 41.7 | STRC | B16 | 19 | TG(41.690) | TG(41.707) | 17 |
| 16 p12.2 | 21.2 | CRYM | A40 | 20 | D16S3045 | TTA(21.603) | 661 |
| 16 p12.2 | 21.6 | OTOA | B22 | 82 | D16S3045 | TTA(21.603) | 661 |
| 17 p11.2 | 18.0 | MY015A | B3 | 71 | AC (17.958) | AA T(18.013) | 55 |
| 17 q25.3 | 77.1 | ACTG1 | A20/A26 | 3 | G T(77.007) | CA (77.107) | 100 |
| 19 q13.33 | 55.4 | MYH14 | A4 | 107 | D19S866 | D19S904 | 22 |
| 21 q22.13 | 36.8 | CLDN14 | B29 | 20 | D21S1252 | D21S167 | 369 |
| 21 q22.3 | 42.7 | TMPRSS3 | B8/B10 | 25 | D21S1225 | GT(43.277) | 740 |
| 22 q12.3 | 35.1 | MYH9 | A1720 | 107 | D22S1173 | D22S283 | 115 |
| 22 q13.1 | 36.4 | TRIOBP | B28 | 89 | TG(36.464) | CA (36.494) | 30 |
DFNA loci are DFN loci associated with dominant (A) hearing loss; DFNB loci are DFN loci associated with recessive (B) hearing loss.
In six families, hearing loss was not linked to any known hearing-related gene. The model for linkage was recessive fully penetrant hearing loss with no sporadics; marker allele frequencies were estimated from the families in the sample. By genotyping all flanking markers shown in Table 1, linkage could be excluded for all known genes by negative lod scores; however, no one marker was consistently sufficiently informative to exclude linkage. Genes for hearing loss in these families will be identified by genome-wide linkage analysis and positional cloning. In four other families, hearing loss was linked to locales of known deafness genes: TMPRSS3 on chromosome 21q23, pendrin on 7q31 and otoancorin on 16p12 (Figure 1).
Figure 1.
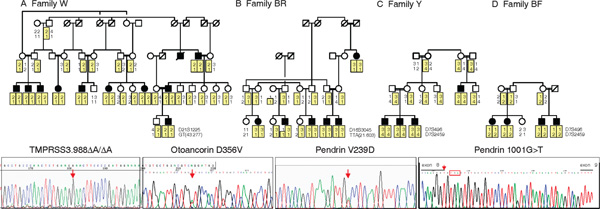
Novel mutations responsible for inherited deafness in four kindreds. In each pedigree, filled symbols represent individuals with severe, bilateral, prelingual hearing loss. The mutation found in each family is illustrated by a sequence below the appropriate pedigree, with mutations indicated on the sequences by red arrows. (a) In Family W, hearing loss is linked to markers flanking the serine protease TMPRSS3 on chromosome 21q22.3. Sequence of TMPRSS3 in affected members of the family revealed frameshift 988ΔA (357stop), which abrogates serine protease activity. As predicted by linkage data, all affected relatives in Family W are homozygous for TMPRSS3.988ΔA and all unaffected relatives are heterozygous or wild-type. (b) In Family BR, hearing loss is linked to markers flanking otoancorin on chromosome 16p12.2. The sequence of otoancorin in affected members of the family revealed missense mutation 1067A > T (D356V), a highly non-conservative change that is likely to disrupt the otoancorin transmembrane structure. All affected relatives in Family BR are homozygous for otoancorin 1067A > T (D356V) and all unaffected relatives are heterozygous or wild-type. D356V is the first deafness-associated missense mutation reported in otoancorin. (c) In Family Y, hearing loss is linked to markers flanking pendrin on chromosome 7q31.1. The sequence of pendrin in affected members of the family revealed missense mutation 716T > A (V239D), which leads to cellular mislocalisation of pendrin protein (see Figure 2). All affected relatives in Family Y are homozygous for pendrin 716T > A (V239D) and all unaffected relatives are heterozygous or wild-type. (d) In Family BF, hearing loss is also linked to markers flanking pendrin, but to different alleles than were linked to the phenotype in Family Y. The sequence of pendrin in affected members of Family BF revealed 1001G > T. RT-PCR of pendrin from lymphoblasts of affected relatives of Family BF indicates that 1001G > T alters splicing, leading to insertion of 41 intronic base pairs into the pendrin message and a premature stop at codon 346 (red box). By homology with other sulphate transporters, truncation at this site would abrogate sulphate transporter activity of the protein. All affected relatives in Family BF are homozygous for pendrin 1001G > T and all unaffected relatives are heterozygous or wild-type.
TMPRSS3
Family W included 11 individuals with prelingual, bilateral, severe to profound hearing loss, with thresholds poorer than 85 decibels (dB) at all frequencies. All ten living affected individuals were homozygous at markers D21S1225 and GT(42.677), which flank TMPRSS3 (Figure 1A). The lod score for linkage of hearing loss to the D21S1225-GT(42.67) haplotype was 9.26. We sequenced TMPRSS3 in an affected child from Family W and identified 988delA, a frameshift mutation in exon 10 leading to a stop at codon 357. All relatives of Family W were genotyped for 988delA: all affected individuals were homozygous for the mutation and all hearing individuals were heterozygous carriers or wild-type.
TMPRSS3 is a transmembrane serine protease, with a trypsin-like protease domain extending from residues 217 - 444 and conserved active sites at histidine 257, aspartic acid 304 and serine 401 [5]. Truncation at residue 357 would eliminate the active site at serine 401 and much of the domain, abrogating TMPRSS3 serine protease activity. A signalling pathway in the inner ear controlled by proteolytic cleavage includes activation by TMPRSS3 of the epithelial sodium channel (ENaC) [16]. Truncated TMPRSS3 of Family W could not activate ENaC.
Disruptions of proteolysis are associated with a wide variety of genetic disorders,[17] but TMPRSS3 was the first protease associated with hearing loss, demonstrated by mutations that defined DFNB8 and DFNB10 [5]. The DFNB10 mutation, found in a Palestinian family, was remarkable, consisting of the insertion of 18 complete β-satellite repeat monomers, which are normally present in mobile tandem arrays of up to several hundred kilobases (kb) on the short arms of acrocentric chromosomes. The β-satellite mutation was not present in any probands in our series, nor in any hearing controls in our series. In addition to all affected members of Family W, three other probands in our series were homozygous for 988delA = 357stop, one hearing control was also heterozygous for this mutation.
Otoancorin
Family BR included six individuals with prelingual, bilateral, moderate to severe hearing loss. Five affected individuals were homozygous for markers D16S3045 and TTA(21.603), which flank otoancorin. The sixth affected individual was homozygous at TTA(21.603) and heterozygous at D16S3045, possibly reflecting an ancestral recombination event (Figure 1B). The lod score for linkage of hearing loss to the D16S3045 -TTA(21.603) haplotype was 3.49. We sequenced otoancorin in a deaf individual and identified mis-sense mutation 1067A > T, which leads to substitution of valine for aspartic acid at residue 356 (D356V). All six affected individuals were homozygous for D356V and all hearing individuals were heterozygous for the mutation or wild-type.
Otoancorin was shown to be associated with hearing loss as the result of a single mutation, a presumptive splice variant IVS12(+2)T > C, identified in a Palestinian family [7]. Otoancorin IVS12(+2)T > C did not appear among any probands in our series, nor among any Palestinian hearing controls. Similarly, the Family BR mutation D356V did not appear among any other probands in our series, but one control was a heterozygous carrier of the allele.
Otoancorin is located at the interface between the apical surface of the sensory epithelia and its overlying acellular gels, and is expressed only in the inner ear [7]. It remains attached to the apical plasma membrane of epithelial cells via seven transmembrane helices. Residue 356 lies between the first and second transmembrane helices and is perfectly conserved in all otoancorin homologues that can be predicted from current genomic sequences (human, chimpanzee, dog, mouse, rat, chicken and Xenopus) [18]. D356V is a highly non-conservative substitution at this site. D356V is predicted by Tmpred[19] to disrupt the transmembrane domain structure and by the program 'Sorting Intolerant From Tolerant'[20] not to be a tolerated substitution. Otoancorin D356V is likely to be the second deafness-associated mutation and the first deafness-associated mis-sense mutation in this gene.
Pendrin (SLC26A4)
Family Y included four individuals with prelingual, bilateral, severe to profound hearing loss. All were homozygous at markers D7S496 and D7S2459, which flank pendrin (Figure 1C). In Family Y, the lod score for linkage of hearing loss to the D7S496-D7S2459 haplotype was 2.11, close to the maximum lod score possible in this family. We sequenced pendrin in an affected child from family Y and identified 716T >A in exon 6, which causes a substitution of aspartic acid for valine at amino acid 239 (V239D). All relatives were genotyped for this mutation: all four affected individuals were homozygous for V239D and all hearing individuals were either heterozygous or wild-type.
Mutations in pendrin can cause either non-syndromic hearing loss or Pendred syndrome,[6] which, in addition to sensorineural hearing loss, includes thyroid enlargement (goitre) and temporal bone abnormalities of the inner ear, ranging from isolated enlargement of the vestibular aqueduct (EVA) to Mondini dysplasia--a complex malformation of the cochlear spiral [21]. Therefore, we evaluated the temporal bones of the affected children of Family Y by computed tomography (CT) scan. As Figure 2A indicates, the vestibular aqueduct is enlarged. There was no clinical evidence of thyroid abnorm-alities and serum thyroid-stimulating hormone and thyroxine concentrations in the four affected individuals were normal.
Figure 2.
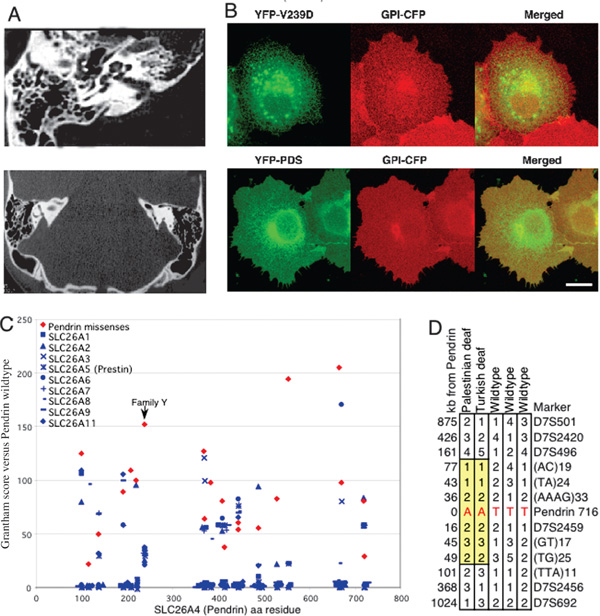
Clinical, biological and evolutionary analyses of pendrin 716T > A (V239D) from Family Y. (a) CT scan of the temporal bones of a child from Family Y who is homozygous for 716T > A (V239D) reveals enlargement of the vestibular aqueduct, or EVA (red arrows). EVA is characteristic of Pendred syndrome. All four homozygous relatives in Family Y have prelingual, severe hearing loss, with thresholds > 85 decibels at all frequencies. There was no clinical indication of goitre and serum thyroid-stimulating hormone and thyroxine levels were normal. (b) Intracellular localisation and trafficking of YFP-tagged mutant (V239D) and wild-type (PDS) pendrin, prepared as previously described [12,23]. Living COS7 cells were transiently transfected with YFP-V239D mutant pendrin (green) and with CFP-GPI (glycosylphosphatidylinositol [GPI] tagged with crimson fluorescent protein [CFP], red), which localises to the Golgi apparatus and the plasma membrane [12]. After incubation, cells were visualised by confocal microscopy [12]. Merged confocal images (yellow) indicate that pendrin V239D is retained in the endoplasmic reticulum, whereas wild-type pendrin (PDS) colocalises with GPI to the Golgi apparatus and plasma membrane. (c) The predicted severity of V239D in Family Y was compared with other disease-associated mis-sense mutations in pendrin (red diamonds) and to wild-type sequences of other human SLC26A anion transporters at homologous sites (blue symbols) using Grantham's amino acid difference formula [29]. Some, but not all, pendrin mis-sense mutations are more diverged from wild-type pendrin than are other SLC26A transporters. By this measure, V239D is among the most divergent mis-sense mutations of pendrin, particularly given the conservation of other SLC26A proteins at this site. (d) Palestinian and Turkish individuals with the 716A (239D) mutation share an extended haplotype of 126 -260 kilobases flanking pendrin (yellow boxes), based on markers polymorphic among wild-type (716T) haplotypes. Pendrin 716T > A has not been observed in any other Palestinian families with inherited hearing loss or Turkish individuals with Pendred syndrome,[30] suggesting that these two families may share a recent common ancestor.
Pendrin is a chloride -iodide transporter; mutations associated with Pendred syndrome can cause its retention in the endoplasmic reticulum [22]. In order to test the consequences of the V239D mis-sense mutation on intracellular localisation of pendrin, we evaluated trafficking of wild-type pendrin and of the V239D mutant protein in living cells using GFP chimeras [22,23]. As shown in Figure 2B, the V239D mutant pendrin is retained in the endoplasmic reticulum, whereas the wild-type protein targets the plasma membrane. Retention of pendrin in the endoplasmic reticulum is a major mechanism for Pendred syndrome,[12] suggesting that V239D is probably the pathogenic mutation in Family Y.
We also developed a method for evaluating the severity of mis-sense mutations in pendrin, based on conservation across the large SLC26A family of solute carrier proteins and the growing number of known disease-associated pendrin missense mutations [6,24-28]. We compared the severity of mis-sense changes in pendrin with naturally occurring variation across all proteins in the family. Figure 2C illustrates these comparisons as a 'diversity plot', calibrated against wild-type pendrin. For each amino acid site at which a pendrin mis-sense mutation has been reported, we plotted the amino acid difference based on Grantham's formula[29] for all naturally occurring amino acid differences in any SLC26A transporter. Then we compared scores of pendrin mis-senses to scores of naturally occurring variants. The mis-sense mutation V239D is a less conservative substitution than any naturally occurring variant at the homologous site, and is among the least conservative changes observed at any site in the SLC26A gene family.
Pendrin V239D did not occur in other Palestinian probands with hearing loss, nor among Palestinian controls; however, pendrin 716T >A (V239D) appeared in a Turkish brother and sister with prelingual hearing loss, hypothyroid and EVA [30]. The Palestinian and Turkish haplotypes harbouring 716A >T were identical for microsatellite markers spanning 262 kb, whereas other haplotypes in this region in these families were diverse (Figure 2D). The mutation may have a common ancestor, although the families are not aware that they are related. The difference between normal thyroid function in the affected individuals in Family Y versus hypothyroidism in the affected individuals in the Turkish family is not surprising, given the intra-familial variability of Pendred syndrome described for other mis-sense mutations [26,31].
In Family BF, the lod score for linkage of hearing loss to the D7S496-D7S2459 haplotype was 1.66, which was not statistically significant but was close to the maximum possible for this family. Sequencing genomic DNA from Family BF revealed a different mutation in pendrin. Four children with prelingual, bilateral, severe to profound hearing loss were homozygous at markers D7S496 and D7S2459 but for different alleles than in Family Y (Figure 1D). We sequenced pendrin in an affected child from Family BF and identified 1001G >T, a splice mutation in the last base pair of exon 8. All affected individuals in Family BF were homozygous for 1001G >T and all hearing individuals were either heterozygous for the mutation or wild-type.
Reverse transcriptase-PCR (RT-PCR) of the pendrin message from lymphoblast cDNA of a homozygous child revealed aberrant splicing, leading to insertion of 41 bp of intron 8 into the pendrin message and a premature stop at codon 346. The region of pendrin homologous to other sulphate transporters includes the site of the truncation, suggesting that this mutation would abrogate sulphate transporter activity. The Family BF mutation 1001G >T has not been previously reported. Mutation of the adjacent base pair, 1001(+1)G >A, however, which also leads to 346stop, is a founder mutation among English families with Pendred syndrome, probably including those families in whom the syndrome was originally described [24,32].
Discussion
Inherited hearing loss in Middle Eastern populations is highly heterogeneous, both in the number of genes involved and in the number of alleles at each gene. Of the ten families screened for linkage of deafness to all genes known to influence hearing loss, none carried any of the more than 100 previously identified alleles. All families with hearing loss who were wild-type for GJB2 harboured novel alleles, either in known genes (four families) or in genes yet to be identified (six families). Alleles responsible for hearing loss in this population are individually very rare.
Allelic heterogeneity of recessive diseases in the Palestinian population is well documented [2]. As pointed out by Zlotogora,[2] the many unique mutations among patients with autosomal recessive diseases in this population reflect the high rate of de novo deleterious mutations occurring in all human genomes [33]. De novo recessive alleles are generally not detectable because the associated phenotypes are not expressed in outbred populations. The appearance of multiple novel alleles of known deafness-related genes also suggests that hearing loss in this population is genetically similar to hearing loss worldwide, and that the phenotype is simply more common in this population because of consanguinity.
More than 50 years before the studies of Mendel, Darwin or Galton, Joseph Adams recognised the importance of isolated communities to the study of human disease, noting that 'endemic peculiarities may be found in certain sequestered districts' [34]. A very large number of the discoveries of modern human genetics have been as the result of studies of such 'sequestered' populations [35]. Historical demography of isolated populations may lead to the occurrence of population-specific alleles of relatively high frequency, such as the ancient mutations responsible for diseases found in the Ashkenazi Jewish population,[36,37] and also many population-specific alleles that are individually rare, each present in only one a few extended families, as described here.
Given the availability of genomics tools, it is now possible to develop gene discovery strategies best suited to the demography of each community. For phenotypes with a high level of allele and locus heterogeneity, identified in extended consanguineous families, we developed the following strategy. (1) For any gene with relatively widespread mutations (eg GJB2), we sequenced all affected individuals in all families. It was important to fully sequence such genes, rather than only screening known mutations, because both old and new alleles are likely to appear [4]. It was also important to sequence all affected individuals, not only probands, because more than one gene for hearing loss might segregate in a family. (2) For families which are wild-type at the commonly mutant genes, we tested for linkage of the phenotype to all known disease genes by a 'subgenome scan' consisting of markers intragenic or immediately flanking all genes known to be associated with the phenotype. Because there are a large number of known genes for hearing loss, we first genotyped two such markers at each site, then added additional markers as necessary. (3) In each potentially linked family, we fully sequenced the known gene linked to the phenotype in an affected relative and an obligate carrier. Clearly, it was not sufficient to genotype only known disease alleles because it is likely that new families will harbour new alleles. Also, although in this project each novel allele was homozygous in affected individuals, it is certainly possible that deaf individuals in some families will be compound heterozygotes. (4) We screened each mutation in all probands and in a small series (100) of Palestinian controls to determine allele frequencies, but mutations were individually too rare for casecontrol comparisons to have adequate statistical power for epidemiological analyses. Therefore, to evaluate the consequence of mis-sense mutations, we developed and used both functional and bioinformatics approaches.
In conclusion, extended consanguineous kindreds prove extremely valuable for the identification of novel genes for otherwise intractable, highly heterogeneous phenotypes. Often extended kindreds in our series evaluated thus far, four families carried novel mutations in known genes and six families are likely to harbour novel mutations in novel genes for hearing loss. The rate of discovery of new genes related to any phenotype will eventually reach an asymptote. The ratio of mutations in known versus unknown genes in the present study (4:6), however, suggests that in this cohort of 156 families, a large number of novel genes for hearing loss still await discovery.
Acknowledgements
We thank the families and the staff of the Palestinian schools for children with hearing loss for their enthusiastic participation. This project was supported by NIH R01 DC005641.
References
- Jaber L, Halpern GJ, Shohat T. Trends in the frequencies of consanguineous marriages in the Israeli Arab community. Clin Genet. 2000;58:106–110. doi: 10.1034/j.1399-0004.2000.580203.x. [DOI] [PubMed] [Google Scholar]
- Zlotogora J. Molecular basis of autosomal recessive diseases among the Palestinian Arabs. Am J Med Genet. pp. 176–182. [DOI] [PubMed]
- Kent WJ, Sugnet CW, Furey TS. et al. The Human Genome Browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Walsh V, Vreugde S. et al. From flies' eyes to our ears: Mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci USA. 2002;99:7518–7523. doi: 10.1073/pnas.102091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott HS, Kudoh J, Wattenhofer M. et al. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- Everett LA, Glaser B, Beck JC. et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- Zwaenepoel I, Mustapha M, Leibovici M. et al. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc Natl Acad Sci USA. 2002;99:6240–6245. doi: 10.1073/pnas.082515999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Lynch ED, King MC. SeqHelp: A program to analyze molecular sequences utilizing common computational resources. Genome Res. 1998;8:306–312. doi: 10.1101/gr.8.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen-Salmon M. et al. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- Mustapha M, Weil D, Chardenoux S. et al. An alpha-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum Mol Genet. 1999;8:409–412. doi: 10.1093/hmg/8.3.409. [DOI] [PubMed] [Google Scholar]
- Mburu P, Mustapha M, Varela A. et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet. 2003;34:421–428. doi: 10.1038/ng1208. [DOI] [PubMed] [Google Scholar]
- Rotman-Pikielny P, Hirschberg K, Maruvada P. et al. Retention of pendrin in the endoplasmic reticulum is a major mechanism for Pendred syndrome. Hum Mol Genet. 2002;11:2625–2633. doi: 10.1093/hmg/11.21.2625. [DOI] [PubMed] [Google Scholar]
- Nance WE, Kearsey MJ. Relevance of connexin deafness (DFNB1) to human evolution. Am J Hum Genet. 2004;74:1081–1087. doi: 10.1086/420979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo I, Villamar M, Moreno-Pelayo MA. et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346:243–249. doi: 10.1056/NEJMoa012052. [DOI] [PubMed] [Google Scholar]
- Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage. http://webhost.ua.ac.be/hhh/ Accessed 15th July, 2005.
- Guipponi M, Vuagniaux G, Wattenhofer M. et al. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet. 2002;11:2829–2836. doi: 10.1093/hmg/11.23.2829. [DOI] [PubMed] [Google Scholar]
- Richard I. The genetic and molecular bases of monogenic disorders affecting proteolytic systems. J Med Genet. 2005;42:529–539. doi: 10.1136/jmg.2004.028118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovine L, Park J, Wassarman PM. Sequence similarity between stereocilin and otoancorin points to a unified mechanism for mechanotransduction in the mammalian inner ear. BMC Cell Biol. 2002;3:28–31. doi: 10.1186/1471-2121-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W. TMbase: A database of membrane spanning protein segments. Biol Chem. 1993;374:166. [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser GR, Morgans ME, Trotter W. The syndrome of sporadic goitre and congenital deafness. Quart J Med. 1960;29:279–295. [PubMed] [Google Scholar]
- Shepshelovich J, Goldstein-Magal L, Globerson A. et al. Protein synthesis inhibitors and the chemical chaperone TMAO reverse endoplasmic reticulum perturbation induced by overexpression of the iodide transporter pendrin. J Cell Sci. 2005;118:1577–1586. doi: 10.1242/jcs.02294. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Metcalfe RA, Watson PF. et al. Mutations of the PDS gene. encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: Implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab. 2002;87:1778–1784. doi: 10.1210/jc.87.4.1778. [DOI] [PubMed] [Google Scholar]
- Coyle B, Reardon W, Herbrick JA. et al. Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol Genet. 1998;7:1105–1112. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- Li XC, Everett LA, Lalwani AK. et al. A mutation in PDS causes non-syndromic recessive deafness. Nat Genet. 1998;18:215–217. doi: 10.1038/ng0398-215. [DOI] [PubMed] [Google Scholar]
- Masmoudi S, Charfedine I, Hmani M. et al. Pendred syndrome: Phenotypic variability in two families carrying the same PDS missense mutation. Am J Med Genet. 2000;90:38–44. doi: 10.1002/(SICI)1096-8628(20000103)90:1<38::AID-AJMG8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Usami S, Abe S, Weston MD. et al. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188–192. doi: 10.1007/s004390050933. [DOI] [PubMed] [Google Scholar]
- Van Hauwe P, Everett LA, Coucke P. et al. Two frequent missense mutations in Pendred syndrome. Hum Mol Genet. 1998;7:1099–1104. doi: 10.1093/hmg/7.7.1099. [DOI] [PubMed] [Google Scholar]
- Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- Tekin M, Akcayoz D, Comak E. et al. Screening the SLC26A4 gene in probands with deafness and goiter (Pendred syndrome) ascertained from a large group of students of the schools for the deaf in Turkey. Clin Genet. 2003;64:371–374. doi: 10.1034/j.1399-0004.2003.00144.x. [DOI] [PubMed] [Google Scholar]
- Napiontek U, Borck G, Muller-Forell W. et al. Intrafamilial variability of the deafness and goiter phenotype in Pendred syndrome caused by a T416P mutation in the SLC26A4 gene. J Clin Endocrinol Metab. 2004;89:5347–5351. doi: 10.1210/jc.2004-1013. [DOI] [PubMed] [Google Scholar]
- Pendred V. Deaf-mutism and goitre. Lancet. 1896;II:532. [Google Scholar]
- Eyre-Walker A, Keightley PD. High genomic deleterious mutation rates in hominids. Nature. 1999;397:344–347. doi: 10.1038/16915. [DOI] [PubMed] [Google Scholar]
- Adams J. J Callow. London, UK; 1815. A Philosophical Treatise on the Hereditary Peculiarities of the Human Race. [Google Scholar]
- Peltonen L, Palotie A, Lange K. Use of population isolates for mapping complex traits. Nat Rev Genet. 2000;1:182–190. doi: 10.1038/35042049. [DOI] [PubMed] [Google Scholar]
- Ostrer H. A genetic profile of contemporary Jewish populations. Nat Rev Genet. 2001;2:891–898. doi: 10.1038/35098506. [DOI] [PubMed] [Google Scholar]
- Risch N, Tang H, Katzenstein H. et al. Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am J Hum Genet. 2003;72:812–822. doi: 10.1086/373882. [DOI] [PMC free article] [PubMed] [Google Scholar]