

Abstract
Similar to other classical science disciplines, immunology has been embracing novel technologies and approaches giving rise to specialised sub-disciplines such as immunogenetics and, more recently, immunogenomics, which, in many ways, is the genome-wide application of immunogenetic approaches. Here, recent progress in the understanding of the immune sub-genome will be reviewed, and the ways in which immunogenomic datasets consisting of genetic and epigenetic variation, linkage disequilibrium and recombination can be harnessed for disease association and evolutionary studies will be discussed. The discussion will focus on data available for the major histocompatibility complex and the leukocyte receptor complex, the two most polymorphic regions of the human immune sub-genome.
Keywords: immunogenomics, immunogenetics, major histocompatibility complex (MHC), recombination, linkage disequilibrium, epigenetics
Introduction
The ongoing 'hide and seek' between pathogens and their host immune systems has led to a molecular arms race which makes immunology one of the most interesting, but also one of the most complex, areas of research. By contrast with the harmless children's game, the outcome of this molecular race determines health versus disease and, in many cases, survival versus death. A prerequisite for immunogenomic research is the availability of comprehensive and fully informative sequence variation and gene maps. A high-quality sequence map of the human genome and successive generations of annotation have been available since 2001 [1,2]. Using this sequence as a reference, global and population-specific variation maps of increasing resolution were subsequently generated by the International SNP Consortium and HapMap Projects [3,4]. Because of the extreme polymorphism encountered in some immune regions, separate efforts were focused on regions such as the major histocompatibility complex (MHC; reviewed by Allcock et al. [5]), the leukocyte receptor complex (LRC; reviewed by Yawata et al. [6]) and others, to complement the global variation map. In addition, several specialised databases, including the Immuno Polymorphism Database [7] and the International Immunogenetics Information System [8], catalogue many of the classical immune genes and their allelic variants. Using immune ontology definitions [9], the first comprehensive gene map and database of the human immune sub-genome was recently reported to consist of 1,562 genes (about 7 per cent of human genes) that are distributed at varying densities across all chromosomes except the Y chromosome [10].
In the following review, some of the benefits and caveats of using a holistic approach to immunogenomic analysis will be discussed with respect to genetic and epigenetic variation and linkage disequilibrium (LD).
Genetic variation
Based on both experimental data [11-15] and inferences from population genetics data [16-18], there is evidence that recombination is not uniformly distributed across the human genome. In addition to the heterogeneity of recombination rates, it was suggested that the human genome could be subdivided into relatively short fragments with little or no evidence of historical recombination [19]. Under this premise, a genome-wide LD map is being constructed by the International HapMap Project, which aims to generate a resource of DNA variation in human populations with different ancestry [3]. In its first phase, which is close to completion, genotyping data at one single nucleotide polymorphism (SNP)/five kilobase (kb) density will provide enough resolution to portray the LD landscape of the genome, including regions of the immune sub-genome.
LD maps of complete chromosomes are already available; [20-22] however, detailed LD patterns at high SNP density covering large chromosomal regions encoding immunerelated gene clusters have long been awaited. LD data have so far been reported for two such clusters: the LRC and the MHC. Within the LRC, the haplotypic nature of the killer immunoglobulin-like receptor (KIR) gene cluster causes a DNA size variation of 120-200 kb as a consequence of the presence/absence of genes in some haplotypes [23-26]. This feature, in addition to intron similarity owing to recent duplications, restricts the availability of informative SNPs for LD studies [27] and poses particular restrictions on large-scale genotyping protocols. Consequently, a detailed LD map for the LRC is not available. For the MHC, a comprehensive LD map is available [16] as a direct consequence of existing SNP resources [28].
Historically, considerable attention has been paid to the MHC region, resulting in a wealth of valuable information in a variety of areas -- that is, origin and maintenance of nucleotide diversity in classical human leukocyte antigen (HLA) genes; gene conversion, average recombination rates, presence and distribution of the recombination hot spots; and extended LD in long-range haplotypes [11,14,15,29-31]. The high-resolution LD map available for the MHC permits the detailed study of the allelic correlation, which can be particularly useful for the selection of tagSNP sets for disease association studies. Since the history of recombination between two SNPs can be estimated using D', DNA segments showing no evidence of historical recombination have been defined as LD-blocks [19]. Consequently, haplotypes within such LD-blocks are likely to share a common ancestral haplotype, and the genealogy of an LD-block might be different from the genealogy of neighbouring LD-blocks. Phylogenetic analysis of haplotypes observed in a ~110 kb LD-block (rs3115569-rs2022533) at the MHC class II-III boundary, the region of lowest recombination rate in the MHC (0.0093 centimorgan [cM]/ megabase [Mb]), surrounded by two recombination hot spots [16], showed that the pattern of nucleotide differences between haplotypes within this LD-block is difficult to account for by mutation only, but could readily be explained by recombination events (Miretti et al., unpublished). Following detailed inspection of the haplotype nucleotide sequence alignment at variable positions (Figure 1B), putative recombination sites can be identified. The subsequent splitting of the LD-block at a recombination site (Figure 1C) resulted in haplotypic networks presenting distinct clustering in both fragments (Figure 1D), which were also dissimilar from that of the complete LD-block. While it needs to be examined in a larger sample of LD-blocks from diverse populations, the presence of recombination within LD-blocks suggests that the occurrence of LD between two SNPs might not be sufficient to unambiguously detect historical recombination beyond the history of the sample population where LD is being measured. Recombination hot spots within LD-blocks -- that is, hot spots that have not left an imprint on LD -- have recently been postulated to be either old hot spots en route to extinction or too young to leave a mark on haplotype diversity in Europeans [18]. Additional experimental data based on sperm cell recombination analysis would help to confirm the signature of recombination identified here and to determine if it is an example of a potentially evolutionarily very young hot spot [18].
Figure 1.
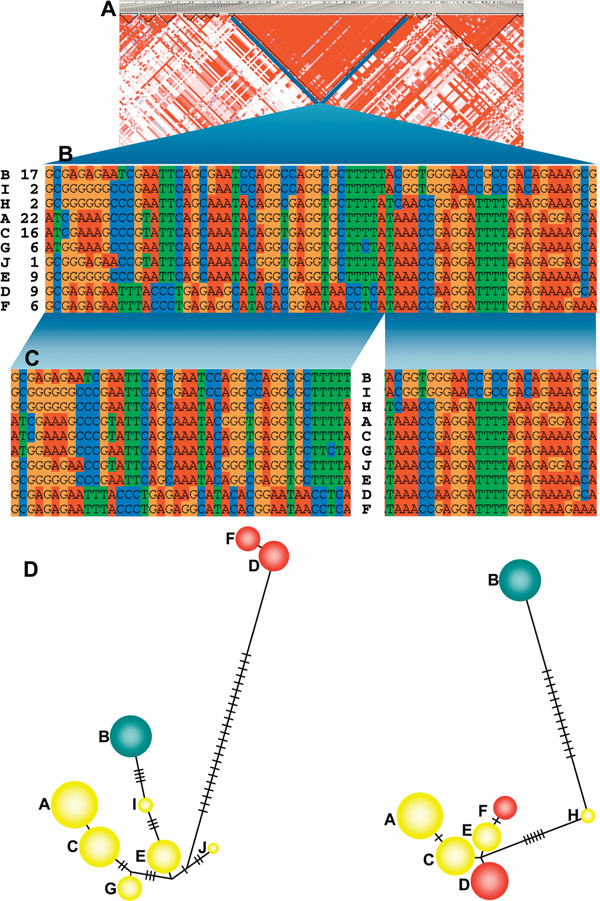
Potential recombination sites within a linkage disequilibrium (LD) block. Patterns of LD for the complete major histocompatibility complex were obtained for a panel of 180 Centre d'Etude du Polymorphisme Humain founder chromosomes [16] and LD-blocks defined according to Gabriel et al. criteria [19] employing Haploview [32]. (A) The region selected for the analysis constitutes an LD-block extending over a ~110 kilobase (kb) DNA segment (National Center for Biotechnology Information built 35 coordinates: 32321766-32431723) containing most of the C6ORF10 gene, with an average space between single nucleotide polymorphisms (SNPs) of 1.6 kb. This region also represents a recombination cold spot -- with recombination rates = 0.00931 centimorgans/megabase, ten times lower relative to that on the neighbouring area -- surrounded by two recombination hot spots identified in NOTCH4 and C6ORF10 genes [16]. (B) Ten haplotypes (rows) observed in this sample, which were recognised based on alleles at 68 variable positions (SNPs, in columns) within the selected LD-block. Haplotype names and frequencies (per cent) are given on the left. (C) A break in the haplotype alignment suggesting the presence of ancestral recombination within the LD-block. Haplotypes were then split at a potential recombination site into two sub-regions (with 41 and 27 SNPs, respectively) aiming to construct a phylogenetic network in order to check for consistency. (D) The distribution of variation among haplotypes as represented by median-joining trees obtained for both sub-regions employing the NETWORK program [33]. Haplotypes F and D are grouped closely related within each network but belong to different clusters when comparing networks from the two sub-regions. The number of substitutions involved in these branching differences could preferably be explained by recombination events rather than owing to mutations occurring between haplotypes. The distribution pattern of these differences adds consistency to this view and strongly suggests the presence of recombination. The circle size is proportional to the haplotype frequency, and the number of substitutions between nodes is represented by the number of lines between them. The site where the haplotypes were split can be changed to few positions on either side of the current site without significantly modifying the topology of the network.
Overall recombination rates within the MHC are known to be lower than across chromosome 6 and the genome average [34,35], and regions of relatively high recombination rates (hot spots) and low recombination rates (cold spots) have been described in males based on microsatellite typing [15]. The fine-scale experimental recombination analysis in a 200 kb segment within the MHC class II region revealed that a few hot spots shape the distribution of LD throughout the segment, that hot spots tend to be delimited to short DNA segments (usually < 5 kb) and that a portion of the recombination is contributed by gene conversion [11,31,36,37]. The question therefore arises as to whether the recombination pattern observed in this segment can be extended to the complete MHC and, ultimately, if it represents a general characteristic of the genome. The heterogeneous model of recombination seems to be a general feature of the genome [17], where the landscape is moulded by the local distribution and intensity of recombination. According to the recombination rates inferred from population genetic data [16], the experimentally verified recombination hot spots in the 200 kb segment of the MHC class II region [11] might not represent a paradigm for the entire MHC, as their intensities are ~ 2-100 times higher than hot spots in the rest of the MHC. Figure 2 shows the uneven distribution of hot spots across the complete MHC -- integrated with gene annotation, LD-blocks and tagSNP distribution, as represented by the GLOVAR genome browser http://www.glovar.org -- and also that the hot spot intensity is far from being uniform. Additional large-scale experimental evidence of hot spot intensity and distribution is required to more accurately assess the concordance with predictions based on population genetic data. Importantly, hot spot features will significantly influence the selection of tagSNPs for disease association studies involving any particular region of the immune sub-genome.
Figure 2.
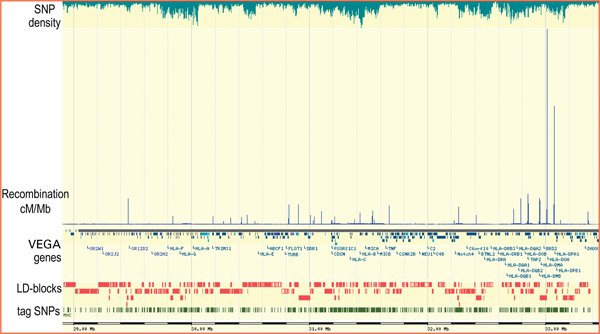
Data integration in the major histocompatibility complex (MHC) region as represented by the GLOVAR genome browser www.glovar.org showing the distribution and intensity of recombination hot spots relative to gene annotation, linkage disequilibrium blocks and tag single nucleotide polymorphism distribution. Note that recombination hot spots within the MHC class II region are comparatively more intense than in the rest of the MHC. Most gene names were excluded owing to space constraints.
From the detailed distribution of recombination and LD across the MHC, it would be possible to exploit the common variation observed in approximately 80 per cent of the MHC sequence, which is in high LD, to conduct association studies. Even with the current SNP density (1 SNP/1.9 kb), however, between 10-27 per cent of the DNA sequence in the MHC sub-regions is contained in regions of low LD, where tagSNP efficiency -- the number of genotyped markers divided by the number of tagging SNPs [38] -- would not entail any benefit, meaning that almost every SNP would need to be typed. Figure 2 illustrates the dependence of the tagSNP distribution on the SNP density and LD pattern, which is particularly variable across the MHC. Long regions of strong LD are common in the extended class I sub-region, where 75 tagSNPs are sufficient to recover haplotypic information provided by 408 SNPs. Conversely, 204 tagSNPs are necessary to recover the equivalent information from 466 SNPs in the class II sub-region, where the LD pattern is more interrupted and the decay of LD is more pronounced [16]. Also, the tagSNP sets are derived from studies which mostly dismiss loci of minor allele frequency (MAF; < 5 per cent), which seem to be present at increased frequency in low LD regions [39], thus excluding rare variants from the analyses. This raises the question of whether failure to detect disease variants simply results from the exclusion of SNPs with rare alleles from the analysis (discussed below). Furthermore, tagging effectiveness -- the proportion of 'hidden' SNPs being detected in LD with SNPs from the tagging set -- can be substantially increased by incorporating SNPs with rare alleles (MAF < 5 per cent); this effect was even more prominent in low LD regions [39].
Epigenetic variation
Much less is known about epigenetic variation, both in terms of causality and function. The most frequent and stable form of epigenetic variation is differential DNA methylation. DNA methylation, which was first discovered in 1948, occurs naturally at the carbon-5 position of cytosine (5-methyl cytosine) at CpG dinucleotides [40]. In subsequent years, it was proposed that DNA methylation played an important role in the regulation of gene expression [41,42] and disease aetiology, particularly cancer [43]. The discovery of CpG islands (sequences enriched in cytosine-guanosine dinucleotides) suggested candidate regions in the genome for epigenetic modulation [44]. DNA methylation occurs predominantly at CpG sites [45] and, in addition to gene regulation, is also involved in phenomena such as X-chromosome inactivation in female mammals, parent-of-origin-specific, mono-allelic gene expression (imprinting) and epigenetic reprogramming in mammalian development [46]. The key technology for detecting 5-methyl cytosine by DNA sequencing was developed in 1992, and is known as bisulphite sequencing [47].
The need to consider epigenetic variation alongside genetic variation in the context of disease has been highlighted by the finding of high discordance rates in monozygotic twin studies and numerous other studies, confirming that epigenetic factors play a decisive role in the aetiology of virtually all human pathologies (reviewed by Robertson and Wolffe) [48]. Therefore, when the SNP Consortium announced in 1999 that it would generate a first-generation genetic variation map of the human genome [49], the time and opportunity seemed right to generate a first-generation epigenetic (methylation) variation map alongside -- which is what the Human Epigenome Consortium announced that it would do [50]. One aspect of the Human Epigenome Project (HEP) that is of particular interest in the context of this review, is the aim and ability to identify methylation variable positions (MVPs), which, together with SNPs, promise to significantly advance our ability to understand and diagnose human disease. MVPs are defined as differentially methylated CpG sites that have the statistical power to discriminate, for example, between biological states such as active versus inactive or healthy versus diseased.
As a result of the HEP and other studies, a detailed genomic methylation map is already available for the MHC [51] and models for the epigenetic control of the KIR expression repertoire have been proposed (reviewed by Uhrberg [52]). For the MHC, methylation profiles have been generated essentially for all expressed genes, demonstrating tissue-specificity (eg for the C2 locus) and inter-individual heterogeneity (eg for the TNF locus). Previously, it has been shown that transcription profiles can be associated with specific haplotypes [53], with epigenetic states [54] or even with specific epi-alleles in the absence of DNA variation [55]. Overall, the methylation profile of the MHC appears to be strongly bimodal, with good correlation between hypo- and hypermethylation within the upstream regions of genes and their transcriptional activity (Figure 3). For the LRC, fewer data are available and are mostly restricted to the KIR genes. KIR genes exhibit a high degree of polymorphism and are expressed in a clonally restricted fashion [56]. Each cell expresses a mono-allelic repertoire which is highly variable with respect to number and combination of receptors. KIR genes are regulated by three types of promoters, corresponding to the three different modes of expression: one for KIR2DL4, which is constitutively expressed; one for KIR3DL3, which is expressed at a very low level; and one which is common to all clonally distributed KIR genes [57]. Given the absence of promoter variability among the clonally distributed KIR genes, it is, in fact, likely that the variegated expression of these genes is mainly regulated epigenetically, and a model has been proposed to this effect [52]. The model proposes four stages of sequential DNA and histone modifications leading to the observed mosaicism of clonally restricted KIR expression but requires some still unproven processes, including locus- and allele-specific DNA demethylation.
Figure 3.
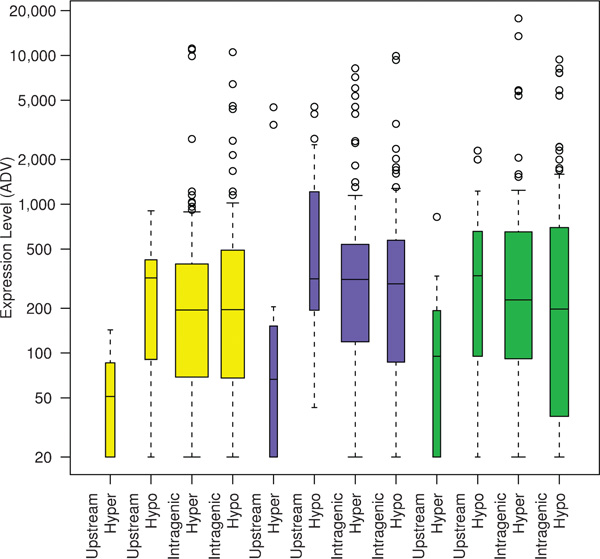
Correlation between DNA methylation and gene expression. DNA methylation data derived from the Human Epigenome Pilot Project were grouped according to 5'-UTR being methylated or unmethylated and the body of the gene being methylated or unmethylated. Each group was then plotted against the corresponding Genomics Institute of the Novartis Research Foundation (GNF) http://symatlas.gnf.org/SymAtlas expression level, which is set to anything below 200 as not being expressed and anything above 200 as being expressed. This cluster analysis shows that genes with methylated 5'-UTRs are silent and genes with unmethylated 5'-UTRs are expressed. In this study, no correlation was found between expression and the methylation level within the body of the gene. Reproduced from Rakyan et al. [51]
Compared with genetic data, the ability to analyse and interpret epigenetic data is still fairly simplistic, although enormous progress has been made, particularly in the past few years. What is becoming increasingly clear, however, is that the two are inextricably intertwined, and efforts towards an integrated (epi)genetic approach to common disease are well underway [58,59].
Application of immunogenomic data for disease association studies
Major progress in genotyping technology has improved our ability to harvest genotypic information from a comprehensive number of loci and has laid down the foundation for the genome-wide common variation survey [3], enhancing our understanding of human genetic variation. Current genotyping costs, however, make genome-wide association studies still challenging for the large sample sizes that will be required for adequately powered studies. The interdependencies of marker numbers, allele frequency, LD, cohort size and power in disease association studies have recently been reviewed [60-62] and will therefore not be discussed in depth here. Essentially, genome-wide studies are still restricted by the allelic spectra underlying complex diseases. The frequency distribution of the disease variants and the proportion of the trait variance for which they are responsible determine the potential power of genetic association studies and, therefore, the feasibility of the study. Disease susceptibility variants associated with Mendelian disorders tend to be modelled by purifying selection and present low population frequencies ( < 1 per cent) [63]. Thus, rare alleles -- also including lowfrequency, mildly deleterious variants together with those alleles in high LD -- might be under-represented in genomic LD surveys and tagSNP sets excluding loci with MAF < 5 per cent. It has been proposed, however, that genetic risk for common diseases and most of the clinically important traits are determined by the joint contribution of diseasepredisposing high-frequency alleles that are shared between unrelated affected individuals in the population: the common-disease/common-variant (CD/CV) model [60,64,65]. A less extreme model based on the divergence of the allelic spectra of disease susceptibility variants relative to that of all variants has been proposed for evaluating the impact of allele architecture on common diseases [61]. Ultimately, the applicability of the CD/CV model in disease association studies will eventually rely on the selected markers and on the relationship between the four parameters that affect the apparent size at the marker locus, namely the odds ratio of the disease allele, the disease allele frequency, the marker allele frequency and the LD between the marker and the disease locus (reviewed by Zondervan and Cardon [62]).
Diseases related to defective immune response, such as autoimmune disorders, might comply with the CD/CV model of allelic frequency distribution for susceptibility loci, showing increased allele frequency after being under positive selection for infectious disease resistance or heterozygote advantage [61,66]. Direct extrapolations to the immune sub-genome and autoimmune diseases could be elusive, however; that is, it would be difficult to prove that alleles having an effect on the function of an immune gene -- and also contribute to the risk of autoimmune diseases -- are beneficial on a population level because they increase the diversity in the immune response [67]. In fact, the overall level of polymorphism in immune genes is, in general, similar to that of non-immune genes [10], and only a few loci among the genes constituting the immune sub-genome largely contribute to the diversity of the immune response -- disregarding loci with somatic recombination. In addition, modes of selection maintaining extreme allelic diversity levels -- heterozygote advantage, balanced selection -- have been proposed for these few loci, including the classical HLA class I and II and KIR genes [29,30,66,68]. Consequently, it is probably premature to generalise any disease model for immune genes. A recent study shows, for instance, that multiple disease loci with predominant disease variants are not necessarily required to develop a complex immune disorder [69]. It has been shown that sarcoidosis -- a multi-systemic immune disorder initially associated with several classical HLA markers -- is associated with a single disease locus independently responsible for most of the predisposing influence [69]. The authors of this study demonstrated that a single transition in exon 5 of the BTNL2 gene, which alters a splice site and causes a premature stop codon, has a profound effect on the mature protein structure and function.
A number of autoimmune disease-susceptibility loci have been mapped to the MHC and LRC regions [70-72]. Autoimmunity and cancer are associated with the progressive decline in immune functions, such as decline in T cell function, dysregulation of T cell apoptosis and immune senescence during ageing [73,74]. Tolerance induction of T cells is mediated by cytotoxic T lymphocyte antigen 4. Polymorphism in this inhibitory receptor has also been associated with autoimmune diseases [75,76].
Outlook
Since the term haplotype was coined in 1967 [77], a vast amount of literature has been published employing haplotypes to construct genetic and LD maps, which have enabled the emerging field of immunogenetics to make major contributions to immunology, population genetics and medicine [78]. For the MHC, a haplotype hierarchy has been defined based on pedigree and population genetic data, where physically linked classical HLA genes constitute 'blocks' of relatively short DNA segments containing alleles in LD at different loci, namely HLA-B/C, TNF, complotype and HLA-DQB/DRB. Combinations of these four basic blocks are frequently found in LD, constituting 'conserved extended haplotypes' (CEH) [79,80] or 'ancestral haplotypes' [81]. More recently, local fine-scale LD blocks based on high-density SNP typing have been described and meiotic recombination hot spots inferred from population genetic data. The integration of long-range LD in CEH and local fine-scale LD data will provide new and exciting opportunities to explore the evolution and disease associations of the immune sub-genome. While the jury is still out on the validity of the CD/CV model, the recently announced Wellcome Trust Case Control Consortium (WTCCC) is likely to provide the answer to this ongoing debate. The WTCCC proposes to analyse 19,000 samples for common genetic variants in eight common diseases using data generated by the HapMap Consortium. Integration of the genetic and epigenetic datasets discussed here is a necessary next step towards a more holistic approach to immunological research, which is one of the aims of the recently formed International Immunomics Society, IMMIS http://research.i2r.a-star.edu.sg/IIMMS/. The IMMIS was formed with the main objective of promoting the science of immunomics, which is the interdisciplinary field spanning immunology, immunoinformatics, genomics, proteomics, bioinformatics and related scientific fields.
Acknowledgements
We wish to thank V. Rakyan for reading the manuscript and C. Tyler-Smith for suggestions. M.M. was supported by a Wellcome Trust Sanger postdoctoral fellowship and S.B. was supported by The Wellcome Trust.
References
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature. 2001;409:928–933. doi: 10.1038/35057149. [DOI] [PubMed] [Google Scholar]
- Allcock RJN, Atrazhev AM, Beck S. et al. The MHC haplotype project: A resource for HLA-linked association studies. Tissue Antigens. 2002;59:520–521. doi: 10.1034/j.1399-0039.2002.590609.x. [DOI] [PubMed] [Google Scholar]
- Yawata M, Yawata N, McQueen K. et al. Predominance of group A KIR haplotypes in Japanese associated with diverse NK cell repertoires of KIR expression. Immunogenetics. 2002;54:543–550. doi: 10.1007/s00251-002-0497-x. [DOI] [PubMed] [Google Scholar]
- Robinson J, Waller MJ, Stoehr P, Marsh SGE. IPD -- The Immuno Polymorphism Database. Nucl Acids Res. 2005;33(Suppl 1):D523–D526. doi: 10.1093/nar/gki032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP, Giudicelli V, Kaas Q. et al. IMGT, the international ImMunoGeneTics information system(R) Nucl Acids Res. 2005;33(Suppl 1):D593–D597. doi: 10.1093/nar/gki065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowsdale J, Parham P. Defense strategies and immunityrelated genes. Eur J Immunol. 2004;34:7–17. doi: 10.1002/eji.200324693. [DOI] [PubMed] [Google Scholar]
- Kelley J, de Bono B, Trowsdale J. IRIS: A database surveying known human immune system genes. Genomics. 2005;85:503–511. doi: 10.1016/j.ygeno.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Ritchie A, Neumann R. High resolution analysis of haplotype diversity and meiotic crossover in the human TAP2 recombination hotspot. Hum Mol Genet. 2000;9:725–733. doi: 10.1093/hmg/9.5.725. [DOI] [PubMed] [Google Scholar]
- Cullen M, Erlich H, Klitz W, Carrington M. Molecular mapping of a recombination hotspot located in the 2nd intron of the human Tap2 locus. Am J Hum Genet. 1995;56:1350–1358. [PMC free article] [PubMed] [Google Scholar]
- Cullen M, Noble J, Erlich H. et al. Characterization of recombination in the HLA class II region. Am J Hum Genet. 1997;60:397–407. [PMC free article] [PubMed] [Google Scholar]
- Cullen M, Perfetto SP, Klitz W. et al. High-resolution patterns of meiotic recombination across the human major histocompatibility complex. Am J Hum Genet. 2002;71:759–776. doi: 10.1086/342973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miretti MM, Walsh EC, Ke XY. et al. A high-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am J Hum Genet. 2005;76:634–646. doi: 10.1086/429393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GAT, Myers SR, Hunt S. et al. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R, Panayi M. et al. Human recombination hot spots hidden in regions of strong marker association. Nature Genetics. 2005;37:601–606. doi: 10.1038/ng1565. [DOI] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H. et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Dawson E, Abecasis GR, Bumpstead S. et al. A first-generation linkage disequilibrium map of human chromosome 22. Nature. 2002;418:544–548. doi: 10.1038/nature00864. [DOI] [PubMed] [Google Scholar]
- Phillips MS, Lawrence R, Sachidanandam R. et al. Chromosome-wide distribution of haplotype blocks and the role of recombination hot spots. Nat Genet. 2003;33:382–387. doi: 10.1038/ng1100. [DOI] [PubMed] [Google Scholar]
- De la Vega FM, Isaac H, Collins A. et al. The linkage disequilibrium maps of three human chromosomes across four populations reflect their demographic history and a common underlying recombination pattern. Genome Res. 2005;15:454–462. doi: 10.1101/gr.3241705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrberg M, Valiante NM, Shum BP. et al. Human diversity in killer cell inhibitory receptor genes. Immunity. 1997;7:753–763. doi: 10.1016/S1074-7613(00)80394-5. [DOI] [PubMed] [Google Scholar]
- Uhrberg M, Parham P, Wernet P. Definition of gene content for nine common group B haplotypes of the Caucasoid population: KIR haplotypes contain between seven and eleven KIR genes. Immunogenetics. 2002;54:221–229. doi: 10.1007/s00251-002-0463-7. [DOI] [PubMed] [Google Scholar]
- Hsu KC, Chida S, Dupont B, Geraghty DE. The killer cell immunoglobulin-like receptor (KIR) genomic region: Gene-order, haplotypes and allelic polymorphism. Immunol Rev. 2002;190:40–52. doi: 10.1034/j.1600-065X.2002.19004.x. [DOI] [PubMed] [Google Scholar]
- Parham P. MHC class I molecules and kirs in human history, health and survival. Nat Rev Immunol. 2005;5:201–214. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- Norman PJ, Cook MA, Carey BS. et al. SNP haplotypes and allele frequencies show evidence for disruptive and balancing selection in the human leukocyte receptor complex. Immunogenetics. 2004;56:225–237. doi: 10.1007/s00251-004-0674-1. [DOI] [PubMed] [Google Scholar]
- Stewart CA, Horton R, Allcock RJN. et al. Complete MHC haplotype sequencing for common disease gene mapping. Genome Res. 2004;14:1176–1187. doi: 10.1101/gr.2188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;336:167–170. doi: 10.1038/336167a0. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M. Nucleotide substitution at major histocompatibility complex class II loci: Evidence for overdominant selection. Proc Natl Acad Sci USA. 1989;86:958–962. doi: 10.1073/pnas.86.3.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, May CA. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nat Genet. 2004;36:151–156. doi: 10.1038/ng1287. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bandelt H, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J. et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Mungall AJ, Palmer SA, Sims SK. et al. The DNA sequence and analysis of human chromosome 6. Nature. 2003;425:805–811. doi: 10.1038/nature02055. [DOI] [PubMed] [Google Scholar]
- Kauppi L, Stumpf MPH, Jeffreys AJ. Localized breakdown in linkage disequilibrium does not always predict sperm crossover hot spots in the human MHC class II region. Genomics. 2006. in press . [DOI] [PubMed]
- Kauppi L, Sajantila A, Jeffreys AJ. Recombination hotspots rather than population history dominate linkage disequilibrium in the MHC class II region. Hum Mol Genet. 2003;12:33–40. doi: 10.1093/hmg/ddg008. [DOI] [PubMed] [Google Scholar]
- Ke X, Durrant C, Morris AP. et al. Efficiency and consistency of haplotype tagging of dense SNP maps in multiple samples. Hum Mol Genet. 2004;13:2557–2565. doi: 10.1093/hmg/ddh294. [DOI] [PubMed] [Google Scholar]
- Ke X, Miretti MM, Broxholme J. et al. A comparison of tagging methods and their tagging space. Hum Mol Genet. 2005;14:2757–2767. doi: 10.1093/hmg/ddi309. [DOI] [PubMed] [Google Scholar]
- Hotchkiss R. The quantitative separation of purines, pyrimidines and nucleosides by paper chromatography. J Biol Chem. 1948;168:315–332. [PubMed] [Google Scholar]
- Riggs A. X inactivation, differentiation, and DNA methylation. Cytogenetics and cell genetics. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- Holliday R, Pugh J. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–232. doi: 10.1126/science.1111098. [DOI] [PubMed] [Google Scholar]
- Feinberg A, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- Bird A. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. 1995. pp. 20–27. [DOI] [PubMed]
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Frommer M, McDonald L, Millar D. et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson K, Wolffe A. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- Masood E. As consortium plans free SNP map of human genome. Nature. 1999;398:545–546. doi: 10.1038/19126. [DOI] [PubMed] [Google Scholar]
- Beck S, Olek A, Walter J. From genomics to epigenomics: A loftier view of life. Nat Biotechnol. 1999;17:1144. doi: 10.1038/70651. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Hildmann T, Novik KL. et al. DNA methylation profiling of the human major histocompatibility complex: A pilot study for the Human Epigenome Project. PLoS Biol. 2004;2:e405. doi: 10.1371/journal.pbio.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrberg M. Shaping the human NK cell repertoire: An epigenetic glance at KIR gene regulation. Mol Immunol. 2005;42:471–475. doi: 10.1016/j.molimm.2004.07.029. [DOI] [PubMed] [Google Scholar]
- Knight JC, Keating BJ, Rockett KA, Kwiatkowski DP. In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nature Genetics. 2003;33:469–475. doi: 10.1038/ng1124. [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Cooper WN. et al. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: Interaction between genotype and epigenotype. Hum Mol Genet. 2004;13:247–255. doi: 10.1093/hmg/ddh013. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Blewitt ME, Druker R. et al. Metastable epialleles in mammals. Trends Genet. 2002;18:348–351. doi: 10.1016/S0168-9525(02)02709-9. [DOI] [PubMed] [Google Scholar]
- Santourlidis S, Trompeter HI, Weinhold S. et al. Crucial role of DNA methylation in determination of clonally distributed killer cell Ig-like receptor expression patterns in NK cells. J Immunol. 2002;169:4253–4261. doi: 10.4049/jimmunol.169.8.4253. [DOI] [PubMed] [Google Scholar]
- Trompeter HI, Gomez-Lozano N, Santourlidis S. et al. Three structurally and functionally divergent kinds of promoters regulate expression of clonally distributed killer cell Ig-like receptors (KIR), of KIR2DL4, and of KIR3DL3. J Immunol. 2005;174:4135–4143. doi: 10.4049/jimmunol.174.7.4135. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–358. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Murrell A, Rakyan VK, Beck S. From genome to epigenome. Hum Mol Genet. 2005;14(Suppl 1):R3–R10. doi: 10.1093/hmg/ddi110. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- Wang WYS, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: Theoretical and practical concerns. Nat Rev Genet. 2005;6:109–118. doi: 10.1038/nrg1522. [DOI] [PubMed] [Google Scholar]
- Zondervan KT, Cardon LR. The complex interplay among factors that influence allelic association. Nat Rev Genet. 2004;5:89–100. doi: 10.1038/nrg1270. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Cox NJ. The allelic architecture of human disease genes: Common disease-common variant... or not? Hum Mol Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. doi: 10.1016/S0168-9525(01)02410-6. [DOI] [PubMed] [Google Scholar]
- Blangero J. Localization and identification of human quantitative trait loci: King Harvest has surely come. Curr Opin Genet Dev. 2004;14:233–240. doi: 10.1016/j.gde.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Nelson GW, Martin MP, Gladman D. et al. Cutting edge: Heterozygote advantage in autoimmune disease: Hierarchy of protection/susceptibility conferred by HLA and killer Ig-like receptor combinations in psoriatic arthritis. J Immunol. 2004;173:4273–4276. doi: 10.4049/jimmunol.173.7.4273. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Jager PLD. Applying a new generation of genetic maps to understand human inflammatory disease. Nat Rev Immunol. 2005;5:83–91. doi: 10.1038/nri1532. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Evolution of the human killer cell inhibitory receptor family. Mol Phylogenet Evol. 2002;25:330–340. doi: 10.1016/S1055-7903(02)00255-5. [DOI] [PubMed] [Google Scholar]
- Valentonyte R, Hampe J, Huse K. et al. Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat Genet. 2005;37:357–364. doi: 10.1038/ng1519. [DOI] [PubMed] [Google Scholar]
- Barcellos LF, Oksenberg JR, Begovich AB. et al. HLA-DR2 dose effect on susceptibility to multiple sclerosis and influence on disease course. Am J Hum Genet. 2003;72:710–716. doi: 10.1086/367781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orru S, Giuressi E, Carcassi C. et al. Mapping of the major psoriasis-susceptibility locus (PSORS1) in a 70-kb interval around the corneodesmosin gene (CDSN) Am J Hum Genet. 2005;76:164–171. doi: 10.1086/426948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton R, Wilming L, Rand V. et al. Gene map of the extended human MHC. Nat Rev Genet. 2004;5:889–899. doi: 10.1038/nrg1489. [DOI] [PubMed] [Google Scholar]
- Gupta S. Molecular mechanisms of apoptosis in the cells of the immune system in human aging. Immunol Rev. 2005;205:114–129. doi: 10.1111/j.0105-2896.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- Hsu HC, Scott DK, Mountz JD. Impaired apoptosis and immune senescence -- Cause or effect? Immunol Rev. 2005;205:130–146. doi: 10.1111/j.0105-2896.2005.00270.x. [DOI] [PubMed] [Google Scholar]
- Gough SCL, Walker LSK, Sansom DM. CTLA4 gene polymorphism and autoimmunity. Immunol Rev. 2005;204:102–115. doi: 10.1111/j.0105-2896.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- Ueda H, Howson JMM, Esposito L. et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- Cepellini R, Curtoni E, Mattiuz P, Genetics of Leukocyte Antigens: A Family study of Segregation and Linkage, Munksgaard, Copenhagen, Denmark. 1967.
- Klein J. Seeds of time: Fifty years ago Peter A. Gorer discovered the H-2 complex. Immunogenetics. 1986;24:331–338. doi: 10.1007/BF00377947. [DOI] [PubMed] [Google Scholar]
- Awdeh ZL, Raum D, Yunis EJ, Alper CA. Extended HLA/complement allele haplotypes: Evidence for T/t-like complex in man. Proc Natl Acad Sci USA. 1983;80:259–263. doi: 10.1073/pnas.80.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunis EJ, Larsen CE, Fernandez-Vina M. et al. Inheritable variable sizes of DNA stretches in the human MHC: Conserved extended haplotypes and their fragments or blocks. Tissue Antigens. 2003;62:1–20. doi: 10.1034/j.1399-0039.2003.00098.x. [DOI] [PubMed] [Google Scholar]
- Dawkins R, Leelayuwat C, Gaudieri S. et al. Genomics of the major histocompatibility complex: Haplotypes, duplication, retroviruses and disease. Immunol Rev. 1999;167:275–304. doi: 10.1111/j.1600-065X.1999.tb01399.x. [DOI] [PubMed] [Google Scholar]