

Abstract
Advances in genome scanning technologies are revealing that copy number variants (CNVs) and polymorphisms, ranging from a few kilobases to several megabases in size, are present in genomes at frequencies much greater than previously known. Discoveries of additional forms of genomic variation, including inversions, insertions, deletions and complex rearrangements, are also occurring at an increased rate. Along with CNVs, these sequence alterations are collectively known as structural variants, and their discovery has had an immediate impact on the interpretation of basic research and clinical diagnostic data. This paper discusses different methods, experimental strategies and technologies that are currently available to study copy number variation and other structural variants in the human genome.
Keywords: copy number and structural variation, genome-scanning techniques
Introduction
The capacity for targeted or en masse detection of variation in the human genome is dictated by the resolution of the available technologies. In the early years of human genetics, variation was detected by studying chromosomes under microscopes, with notable observations of aneuploidy [1-3], heteromorphism [4] and fragile sites [5], to name a few, dominating our knowledge base. The advent of molecular biology, and in particular nucleotide resolution analysis through DNA sequencing and genotyping, led to the discovery, characterisation and mapping of short tandem repeats (STRs; eg di-,triand tetranucleotide microsatellites) [6] and single nucleotide polymorphisms (SNPs) [7,8]. STRs and SNPs, and the technologies used to detect them, are described in numerous comprehensive articles and reviews [9-12].
The latest advances in studying human genome variation have been in the examination of copy number variants (CNVs) (Figures 1 and 2) and other similarly sized structural changes along human chromosomes. In general, this class of variants refers to changes of an intermediate size, between microor minisatellites and microscopically visible changes (usually > 1 kilobase [kb] and < 3 megabases). Several recent investigations have found that these variants are much more frequent in the human genome than previously recognised [13-20] and in-depth descriptions of these variants and their properties can be found in several recent reviews [21-29].
Figure 1.
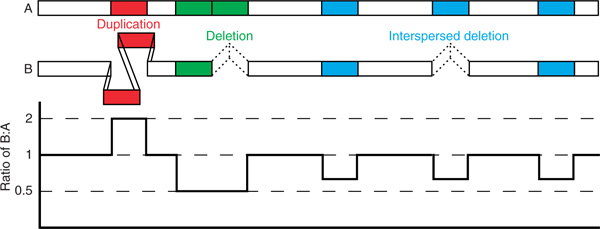
Illustration of copy number differences between two homologous chromosomes (A and B). Coloured boxes indicate copy number changes, including tandem duplication (red) and two types of deletions: deletion of a tandem segment (green) and deletion of an interspersed segment (blue). Below the chromosomes is a line graph showing the results of a comparison of chromosome B with chromosome A. This is an idealised output of the results that could be obtained from a high-resolution comparative genomic hybridisation experiment.
Figure 2.
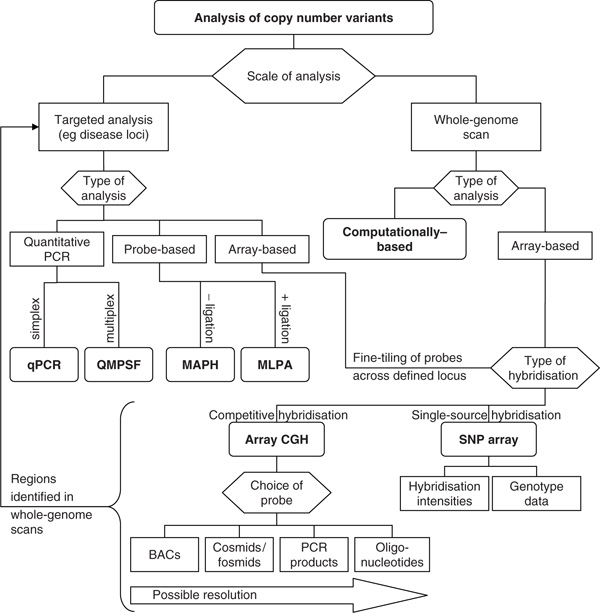
Flowchart illustrating some of the factors that need to be considered when attempting to assess copy number variation. Hexagons are used to designate choices, and rounded rectangles indicate the major techniques that are discussed further in this review. Note the arrow from the bottom of the whole-genome scan that leads back to targeted analysis. This emphasises the fact that copy number variants identified through whole-genome scans can be confirmed or tested directly in large cohorts of individuals using targeted analyses. Abbreviations: BACs, bacterial artificial chromosomes; CGH, comparative genomic hybridisation; MAPH, multiplex amplifiable probe hybridisation; MLPA, multiplex ligation-dependent probe amplification; PCR, polymerase chain reaction; QMPSF, quantitative multiplex PCR of short fluorescent fragments; qPCR, quantitative PCR; SNP, single nucleotide polymorphism.
Investigations of structural variants have been accompanied by a host of newly developed technologies and methodologies, with many of these latest-generation techniques being currently implemented and continually improved upon. As more techniques arise, the most commonly asked question seems to be, 'What is the most appropriate technique or experimental approach to address our specific question?' Several factors can be used to distinguish different techniques and should be considered before embarking on a new study. These include the scope of the technique (does the method assess targeted or genome-wide variation?) and the resolution of the technique (what types and sizes of variants need to be analysed?) (Figure 2). Here, the techniques and methodologies currently available for the analysis of structural variation (in particular, copy number variation) in the human genome are reviewed. Important factors in these analyses are highlighted, while, at the same time, recommendations for these strategies are made -- in some cases based on the authors' own personal experiences. Fluorescence in situ hybridisation (FISH) is not discussed here because it is well established and has been previously reviewed in detail [30]. It should be noted, however, that FISH is often the only way to assess certain forms of structural variation along the chromosomes.
Methods for detecting and scoring copy number variation
The discovery and characterisation of structural variation in the human genome has been driven by advances in methods that allow comprehensive scanning of an entire genome, along with targeted scans of defined loci. Originally, technologies such as Southern blot hybridisation -- using conventional and pulsed-field gel electrophoresis, FISH and microsatellite scanning offered the best methods for revealing changes in DNA copy number. In some cases, particularly for disease diagnostics, these methodologies are still the standard and may continue to offer the only way to correctly resolve a complex rearrangement. Each of these approaches lacks scalability, however, and may also require parental DNA to facilitate data interpretation. Notwithstanding, the prevailing approaches for identifying CNVs have been either array based or quantitative (primarily, polymerase chain reaction [PCR]-based) assays. The former is used primarily for genome scanning, while the latter is used for locus-specific testing (often with multiple loci screened simultaneously) in first-pass and confirmatory screening (for example, to confirm genome scan data). Additional methods for detecting CNVs, including what are referred to as computationally based and genotype-based assays, have come forth recently. A flowchart outlining the available methodologies is given in Figure 2, while some techniques are discussed in greater detail below and illustrated in Figure 3.
Figure 3.
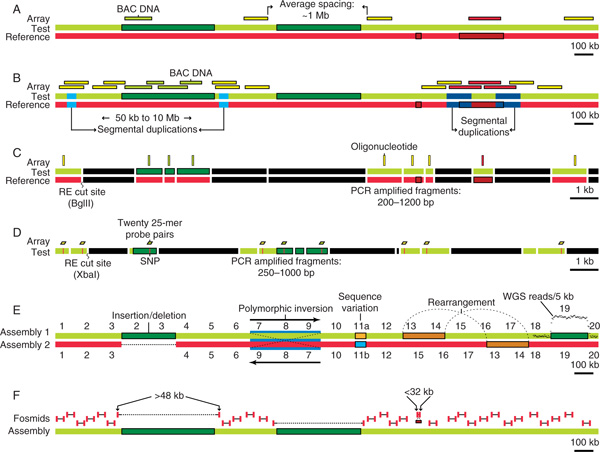
Comparison of methods used to detect novel copy number variations. Three methods employ comparative genomic hybridisation. In these methods, test (green bar) and reference (red bar) DNA are hybridised against: (A) a BAC array; [13] (B) a targeted BAC array; [15] or (C) a representational oligonucleotide array [31]. Probes of the array are drawn above the DNA region to which they hybridise. Darkened boxes within the test or reference sequence bars correspond to duplications only found in one of the two sequences. The colour of the probe indicates the relative hybridisation of the array to the assayed DNA, with yellow representing equal copy number, green representing a duplication of the test region and red representing a duplication of the reference region. (D) The single nucleotide polymorphism (SNP) gene chip can be used as a quantitative array [32] that assays only a test (green) DNA sequence. Quantification of copy number is obtained by comparing the intensity levels of 20 pairs of oligonucleotide probes per SNP (red lines) to data available for a reference set of control individuals. Both (C) and (D) use genomic representations created using restriction enzyme fragmentation followed by adaptor-mediated polymerase chain reaction (PCR). This PCR is optimised to amplify genomic DNA between 200 and 1,200 base pairs (bp). Thus, the regions illustrated are much smaller than in the other methods, demonstrated by their smaller scale. (E) and (F) use in silico genome comparisons. In (E), intra- or inter-assembly comparisons not only detect copy number changes (insertion/deletions; segments 2 and 3), but can also identify inversions (segments 7-9), sequence variations (segments 11a versus 11b) and rearrangements (segments 13 and 14). Segments 18-20 show how whole-genome shotgun (WGS) read depth can be used to detect duplications. A significant increase in the depth of WGS reads per 5 kilobases (kb) is often an indication of duplication (dark green box) in the genome [33]. In (F), the comparison is between the NCBI assembly (green) and a fosmid paired-end sequence (red) derived genome [16]. Fosmid paired-ends are shown above their corresponding sequence in the public assembly. The average size of the fosmid insert is ~40 kb. Significant deviations (< 32 kb or > 48 kb) from this mean could indicate a copy number variant and are highlighted. Abbreviations: BAC, bacterial artificial chromosome; Mb, megabase(s).
Array-based methods for detection of copy number variation
Array-based methods offer the most robust approach to scanning for CNVs in a genome-wide manner. The main differences between available platforms are in the type of DNA molecule found on the array (genomic DNA clones, cDNA, PCR products or oligonucleotides) and the type of hybridisation used (competitive hybridisation versus singlesource hybridisation). These different platforms each have inherent advantages and disadvantages.
Comparative genomic hybridisation
Detection of DNA copy number variation between differentially labelled test and reference genomes has long been feasible using competitive in situ hybridisation to metaphase spreads [34]. This approach, in which fluorescence ratios between the two hybridised DNA sources reveal regions of gain or loss, is referred to as comparative genomic hybridisation (CGH). With the advent of microarrays, sample DNA can now be hybridised to arrays spotted with a multitude of DNA sequences. This not only increases the specificity of CGH, but it also provides a dramatic increase in resolution. Array-based CGH (aCGH) was first described in 1997 [35]. Two years later, the first experiment to employ a genomewide scanning approach utilised an array spotted with cDNA clones [36]. As the technology evolved, bacterial artificial chromosomes (BACs) became the most common type of genomic clone for spotting on the arrays (Figure 3A) [13,15,37-43]. There are several advantages to using BAC clones, including their availability, coverage of the genome, known sequence identifiers (eg completed sequence or clone end-sequence) and FISH-readiness. Moreover, BACs and other large insert clones retain a genomic complexity that often results in a more robust signal-to-noise ratio profile.
There are now several BAC arrays available that provide partial or near-complete genome-wide coverage (genomewide coverage is also referred to as clone 'tiling-path' resolution analysis) [38-42]. There are also commercial BAC arrays available, both for genome-wide screening and for targeting regions involved in microdeletion syndromes (eg Spectral Genomics and Signature Genomics). Additionally, BAC arrays have been designed that target potential rearrangement hotspots that are flanked by highly identical segmental duplications [15] (Figure 3B). BAC arrays do, however, have some inherent disadvantages. First, even with dense coverage, the resolution of BAC arrays is limited by the size of the clones themselves. Since BAC clones are generally 80-200 kb in size, it can be difficult to identify CNVs smaller than ~50 kb. Higher-resolution analysis can be achieved by spotting shorter DNA molecules, such as cosmids, fosmids, cDNA [36], PCR products [44,45] or oligonucleotides on the array [14,31,46-49]. Secondly, to the authors' knowledge, there are currently no available commercial services providing 'tiling-path' resolution arrays. Presumably, this is because of the inherent challenges in manufacturing this type of product. An academic enterprise from the British Columbia Cancer Research Centre http://www.bccrc.ca/arraycgh/arrays.html is the only provider of such arrays, although there may be other vendors that the authors are unaware of. Moreover, some groups offer access to their arrays on a collaborative basis.
Spotting an array with oligonucleotides is one way of achieving an increased resolution. The use of oligonucleotide CGH arrays for copy number detection was first implemented in an assay format called representational oligonucleotide microarray analysis (ROMA) [14,31] (Figure 3C). Similarly to BAC arrays, ROMA utilises differentially labelled test and reference genome DNA competitively hybridised to an array. In order to increase the signal-to-noise ratio, however, the complexity of the input DNA is reduced. This is accomplished by a method called representation or whole-genome sampling [50]. In this method, the genomic DNA is fragmented with a specific restriction enzyme. The resulting fragments are then ligated with adaptors which act as primer binding sites in a subsequent PCR amplification. The amplification conditions are set only to amplify fragments up to ~1.2 kb. The initial version of this method used BglII digestion, which gives rise to an estimated 200,000 fragments in this size range [31]. Oligonucleotides for the array are then designed to match sequences present in the (complexity-reduced) representation sample. The published ROMA arrays have contained 85,000 oligonucleotides that are each 70 base pairs (bp) in length, giving a resolution of ~30 kb throughout the genome. A potential disadvantage of these first-generation ROMA arrays is that, in general, only unique regions in the genome were represented. This means that the ~ 5 per cent of the genome covered by low copy repeats (LCRs -- also called segmental duplications) [33,51] would not generally be assayed, even though a large fraction of the existing copy number variation has been shown to reside in these regions [13,15,52]. The omission of duplicate regions will most likely be remedied in higher resolution arrays, spotted with a larger number of oligonucleotides, which are expected to be forthcoming (ROMA platforms using arrays with > 300,000 probes are now being developed). As reflected in the published literature, the ROMA technology seems to be primarily used by its inventor, Dr Michael Wigler, along with his collaborators, and has not yet been adopted by the majority of users.
In addition to the ROMA methodology, companies such as Agilent http://www.agilent.com and NimbleGen http://www.nimblegen.com have generated long oligonucleotide arrays for direct (non-representational) CGH analysis. Agilent now produces an array with ~43,000 60-mer probes with bias toward genic and non-repetitive euchromatic regions [43,53,54]. Experiments using this array can be performed with as little as 100 ng of input DNA per sample and provides a resolution that can discern large chromosomal aberrations but lacks the power to pick up small CNVs. To address the need for greater resolution, Agilent is expecting to release next-generation arrays with ~185,000 and > 300,000 probes early next year. NimbleGen offers both whole-genome and custom-targeted 'fine-tiling' arrays containing 385,000 probes [18,55]. These provide a mean spacing of ~7-8 kb on the whole-genome array and can give a resolution better than 500 bp (with probes as densely spaced as 10 bp apart) in custom fine-tiling arrays. NimbleGen uses probes that vary in size between 45-85 bp, such that their Tms are equalised at 76°C ('isothermal'), thus providing more uniform probe performance. In the Nimble-Gen business model, however, the NimbleGen staff performs all of the hybridisation experiments, requiring customers to supply 1-3 μg of purified sample and reference genomic DNA. Data analysis is also heavily based on company-designed algorithms. Although this may be preferable for many laboratories, some may find it difficult to provide such a relatively large quantity of DNA. Other laboratories will not be able to provide DNA because of research ethics restrictions governing how their DNA samples can be handled and disseminated.
SNP chips
A slightly different approach to using oligonucleotide arrays for copy number detection is to use the hybridisation intensities from SNP genotyping platforms, such as those made by Affymetrix or Illumina [32,56-62]. Although both of these approaches use oligonucleotides spotted on an array, the SNP arrays differ from CGH in a number of fundamental ways. Most notably, the SNP arrays were originally designed for genotyping, so they do not directly compare two DNA sources through competitive hybridisation, as is the case with CGH. Instead, hybridisation intensities from a single DNA source are adapted to provide information about copy number through subsequent comparisons with a set of reference values from control individuals.
With the Affymetrix SNP arrays, input DNA preparation is similar to ROMA in that the DNA to be hybridised is reduced in complexity by restriction digestion followed by PCR-based amplification of fragments in a specific size range. These DNA fragments are then hybridised to an array where each SNP is represented by 20 probe pairs representing the match and mismatch alleles [57] (Figure 3D). Comparison of the intensity values from these match and mismatch probes to reference values from control individuals is used to acquire copy number data. The highest resolution array currently available assays roughly 500,000 SNPs, with an average spacing of ~ 6 kb. This high density of probes allows consecutive SNPs to be used to estimate copy number and effectively increases the signal-to-noise ratio. Although the distribution of probes along the euchromatic regions is relatively uniform, there is under-representation within segmentally duplicated regions. As reflected in numerous publications demonstrating applicability, the most common publicly available algorithms used to extract the copy number intensity data include the Dchip [63] and CNAT [57] packages.
Instead of spotting oligonucleotides on glass slides, Illumina attaches probes to beads. The coated beads are pooled and then positioned randomly on an array, giving approximately 30-fold feature redundancy. Because the beads are positioned randomly, a series of hybridisations are needed to identify the probe locations along the array. Illumina has two genotyping platforms that can be used for the detection of CNVs: GoldenGate and Infinium. Whereas GoldenGate can be used to multiplex genotyping experiments (up to 1,536-plex), the newer Infinium platform -- which utilises 100 K or 317 K 'beadchips' -- is the most viable option for wholegenome analyses. In this platform, input DNA is hybridised to the probes, this is followed by a single base extension at the SNP with differentially labelled nucleotides. Like the Affymetrix SNP arrays, the intensity values from this hybridisation are compared with values from control individuals to gain information about copy number. At the time of writing, there were only a few studies that used the Illumina platforms to identify CNVs. It is expected that this will soon change, however, as more initiatives incorporate copy number identification into the experimental design of their genetic studies [64].
Genotype-based methods for identifying deletions
Along with hybridisation intensities, SNP platforms also generate genotype data. These genotypes, when generated from multiple related individuals, afford additional opportunities to assess genomic deletions. With a single individual, genotyping will not detect deletions due to the fact that hemizygosity will be miscalled as homozygosity for the present allele (or, in the case of homozygous deletion, a null or failed genotype will be observed). By contrast, if parent-offspring trios are analysed, losses of heterozygosity [65,66] can be discovered when Mendelian inheritance is violated. These can lend support to regions identified as deletions when assessing hybridisation intensities. Naturally, non-Mendelian inheritance can also signify other genomic alterations, including segmental or complete uniparental isodisomy or heterodisomy [67,68].
Additionally, large collections of genotype data, such as those generated by the International HapMap Consortium, can be used to identify deletion polymorphisms [18-20]. Two recent studies have utilised this resource to identify deletions in the human genome by looking for violations of Mendelian inheritance, Hardy-Weinberg disequilibrium or null genotypes [19,20]. An earlier study of patient samples offers a prototypic example of how this approach can be used in a disease study [69]. Certain regions of the genome have extensive marker coverage, giving this approach a high resolution with the power to detect relatively small (as small as 1 kb) deletions.
Interestingly, many of the deletions appear to be in high linkage disequilibrium (LD) with neighbouring SNPs [18,20,70]. This could indicate that they have arisen on a single chromosome from an ancestral population. This is important, as it allows deletion carrier status to be inferred from SNP genotypes and further allows the investigation of associations between deletions and disease. It is also expected that other CNVs, such as insertions/deletions of regions with copy number greater than two, will show significant LD with neighbouring SNPs. To date, however, no reports have been published to support this assumption. Additionally, LD associations are expected to be more complex in regions that have many copies (such as segmental duplications), making complex disease association analyses more difficult.
Quantitative methods for locus-specific testing
Quantitative methods offer an effective approach to assessing variation at targeted loci. These can be used to assess copy number changes at known or proposed disease regions, often with high-throughput screening of large cohorts of samples. Alternatively, these methods can be used to confirm regions that have been identified in whole-genome scans using arraybased approaches. In general, the major difference between quantitative methods is in how they analyse the input DNA; do they directly assay the genomic DNA with PCR or do they first hybridise with a targeted probe? These differences are discussed in more detail below.
Quantitative PCR
Real-time quantitative PCR (qPCR) has been used for many years in the quantification of gene expression [71]. The basis of qPCR is that the rate of amplification is proportionate to the number of template copies. By monitoring the amplification in 'real-time', it is possible to determine when the PCR reaction is in the exponential phase of amplification. It is in this phase that quantification of the starting template occurs; however, it is also necessary to use a control region with known copy number to adjust for variable starting amounts between samples. There are several types of qPCR assay, but they are all based on the same basic principle: an increase in PCR product is manifested as an increase in fluorescence, which can be monitored throughout the PCR reaction. Although most available protocols for real-time qPCR work well for scoring deletions and duplications [72,73], they are, generally, not suitable for multiplexing. To facilitate scoring of multiple target regions in a single experiment, some novel approaches have been developed. These assays are similar in that the final product is separated by size within a gel, which is inherently more amenable to multiplexing than spectral separation of products.
The simplest of these assays is called quantitative multiplex PCR of short fluorescent fragments (QMPSF) [74,75]. PCR assays are designed to amplify up to ten target regions in parallel, with each product varying in length. One primer for each target is labelled with a 6-FAM (6-carboxyfluorescein) moiety, while the other primer carries a short stabilising tail sequence. PCR amplification is stopped within the exponential phase and products of different size are separated by electrophoresis. Each product is represented by a peak, and the peak height -- relative to a reference -- correlates with the amount of product. Because each reaction requires a labelled primer, experiments can become expensive if many regions are tested. To lower the cost, an alternative protocol has been developed [76], in which all primers are designed with a specific hexa-decamer tail sequence. These tails then serve as templates in a subsequent amplification reaction, where universal labelled primers are used. Although this adds an extra step to the protocol, only a single FAM-labelled primer is needed, irrespective of the number of targets.
Probe-based multiplex assays
Multiplex amplifiable probe hybridisation (MAPH) [75,77-79] and multiplex ligation-dependent probe amplification (MLPA) [80-82] allow differences in copy number to be detected based on quantification of probes specific for a target region.
In MAPH, the test DNA is denatured, bound to a nylon filter and then hybridised with probes specific for the target region. Each probe has a different length but they all carry identical tail sequences, allowing subsequent amplification with fluorescently-labelled universal primers. After amplification, products are separated by size and quantified based on the fluorescence intensity ratio of target compared with control regions. Up to 40 loci can be interrogated simultaneously [83]. The main disadvantage of MAPH is the amount of work and optimisation needed to obtain a robust probe set. Each probe has to be cloned in order to add the universal tail sequences. Once a probe set has been developed, however, it can be used for high-throughput screening of all exons of a specific gene (eg DMD [84]) for deletions or duplications in large patient cohorts.
MLPA is different from MAPH in that it is performed in solution, it is, however, still dependent on probe amplification. In MLPA, pairs of probes are made for each target. The two probes in each pair are designed to hybridise adjacent to each other at the target region. Through a DNA ligation step, a contiguous probe molecule is created. The probes carry a tail sequence that serves as a template for universal fluorescentlylabelled primers in a subsequent amplification step. The resulting products can then be separated and quantified in the same manner as in MAPH. In the initial protocol for MLPA, each probe was cloned in a vector. A more recent advance demonstrates that synthesised probes work equally well, but there is a limit to the size of the probes that can be produced. This, however, can be overcome by introducing two different universal primer pairs, each labelled with a different fluorescent marker, thereby allowing for separation of the final products based on both size and wavelength of fluorescence [85,86]. MLPA has been successful using up to 40 probes in a single experiment [80]. As with MAPH, once a probe set has been developed, it works reproducibly in screening large cohorts of samples [82]. There are more than 50 commercially available pre-tested MLPA probe kits designed for many of the known microdeletion syndromes, as well as for genes where intragenic deletions and duplications are common causes of disease http://www.mrc-holland.com.
Computationally-based methods for detecting structural variants
While the above techniques physically assay DNA molecules to assess copy number variation, it is also possible to evaluate genomes in silico by comparing DNA sequences. As more sequence data become available, this option will become more viable and popular. Three main strategies have emerged, utilising different types of sequence data: (a) sequence assemblies (Figure 3E); (b) clone end sequences (Figure 3F); and (c) sequence read depths (Figure 3E), although these methods are largely limited to the analysis of data that are already publicly available from large-scale sequencing initiatives. This is due to the current, hugely prohibitive cost of generating full sequence coverage or redundant clone library end-sequence data from an individual's DNA.
Whole-genome or chromosome assemblies have the benefit of being able to detect practically any type of variation, even down to the single nucleotide. This provides an advantage over array-based methods, where the resolution is dependent on the density of probes spotted on the array. In this strategy, sequence assemblies from two sources are compared computationally, allowing differences in sequence, copy number or orientation to be annotated. Although the majority of these strategies compare two human assemblies, such as the two distinct and near-complete assemblies of chromosome 7 [87-89] and the HLA region [90], it is also possible to detect large variations by comparing the human genome with its closest living relative, the chimpanzee. Although these interspecies comparisons primarily look for sequence variation between species, they can also identify polymorphic differences, such as inversions [17] and copy number differences [91].
Using clone end-sequences (eg fosmids) from a genomic library constructed from a single genome is another sensitive approach that allows for the detection of variants as small as 8 kb in one study [16]. In this technique, end-sequences are anchored to the public genome assembly. The distance between the two ends can then be calculated, giving an observed or computed size of the clone. Because the approximate physical size of the clone is known (in the case of fosmids, the size is typically ~40 kb), large deviations between the computed and physical sizes can represent variations between the two genomes. Although some large deletions can be identified, this approach generally does not readily allow the detection of copy number increases larger than 40 kb. On top of copy number variation, this method is capable of detecting some inversions by looking for end-sequences that have an incorrect orientation with respect to the public assembly. Along with genome assembly sequence comparisons, this represents the only published method (to the authors' knowledge) for genome-wide investigation of inversions. There is currently a National Institutes of Health-led largescale genome initiative to end-sequence fosmid clones from numerous libraries prepared from different HapMap samples aimed at discovering structural variation in the human genome.
Following the example of scanning for segmental duplications [33], the third approach to assessing sequence-read depth will probably become more relevant when whole-genome shotgun sequencing of multiple genomes becomes standard practice [92]. The rationale behind this technique is that regions of the assembly with greater read depth may be present in multiple copies and, in certain instances, the copy number of the region may vary between individuals.
In all three of these sequence analysis strategies, the accuracy of the data will only be as good as the quality of the assemblies available and confirmation using a secondary method (eg quantitative methods listed above) will probably be required. It is also noteworthy that the study of SNPs located in LCRs [93], some being paralogous sequence variants [52], can also be used to detect simple and complex copy number differences arising from duplication, deletion or gene conversion.
Clinical diagnostic implications
The ultimate aim of genetic diagnostics is to evaluate the genomic content of a cell or group of cells as completely and accurately as possible. The objective is to provide either diagnostic insight into the phenotype of a patient or, alternatively, to provide predictive or prognostic insight into the patient's disease or developmental outcome. The upshot is that there have always been some implicit assumptions as to what constitutes a normal genome and a concomitant normal phenotype and hence, by extension, what would constitute an abnormal genome and a deleterious phenotype. For example, the location of the abl gene and BCR locus (either by molecular genetic or cytogenetic means) on chromosomes 9q34 and 22q11.2, respectively, is considered normal, whereas their co-localisation on a Philadelphia chromosome is considered abnormal and suggestive of chronic myelogenous leukaemia. Similarly, the presence of two copies of chromosome 21 in a metaphase preparation is considered consistent with a normal karyotype, whereas the presence of a third copy would be consistent with Down syndrome.
Whereas the majority of approved diagnostic tests query specific genomic loci of confirmed pathogenicity, such as in the previously mentioned examples, others assess loci only suspected to be associated with disease (eg some sub-telomeric deletions in mental retardation) [94-100]. In the latter scenario, if a genomic variant is found at the locus in question in a proband, current convention assumes that such a variation is pathogenic if: (i) there is an obvious accompanying phenotypic abnormality in the proband and (ii) the variant is absent in both parents. If one of these parameters is untrue, however, the diagnostician faces a dilemma as to the potential pathogenicity of the genomic aberration [101]. The discovery of the phenomena of CNVs, particularly in the context of wholegenome array-based analyses, will challenge conventional understanding of the genome and necessitate a cautionary note regarding assumptions about phenotype/genotype associations. Many questions will emerge, such as: is the CNV detected in a proband truly the causative genomic aberration, or is it a benign CNV which deflected our attention from the more subtle malignant genomic aberration?' 'Is that genomic variant serendipitously detected in a normal healthy individual during the course of an unrelated genetic test cause for concern? Is it a prognosticator of a late-onset disease or, again, simply a benign structural variant?' 'Is that copy number polymorphism present in greater than 1 per cent of the population truly benign, or does it, in fact, have a clinical utility in identifying disease susceptibility?'
In the authors' estimation, the major factors influencing which of the current methods will be used broadly for the detection of CNVs in a clinical diagnostic (or basic research) setting will depend on many factors including accuracy, specificity, set-up time, assay cost, the extent of a genome required to be assayed and requirements for sample input. Broad-based implementation, including those at the regulatory level, will also be influenced by patent restrictions. The flowchart in Figure 2 summarises some factors that need to be considered regarding the technologies and approaches described in this review. To obtain the most comprehensive analysis of both the microscopic and sub-microscopic copy number variation of a genome, it is the authors' belief that the likely paradigm that will prevail will consist of: (i) a karyotype and array-based scan for global assessment of balanced and copy number-type unbalanced variants, respectively, followed by (ii) locus-specific confirmation using targeted FISH or quantitative PCR-based approaches. Obviously, where simple alterations are being tested for in defined phenotypes, such as the dosage-related microdeletion and duplication genomic disorders [102,103], only those techniques yielding locus-specific data would be required.
Unresolved issues and recommendations
Currently, there is no single approach that will allow all types of copy number or other structural variants to be identified. This is underscored by the surprisingly small degree of overlap between the published datasets, which in some cases assess identical samples [28]. Short of comparing 'finished' sequence assemblies generated from unique donor (and preferably haploid) sources [26], a multitude of approaches will be required to generate and validate variants in a comprehensive manner. There is also the possibility that new technologies, such as those that utilise direct counting and/or sequencing of single DNA molecules, will provide superior resolution of CNVs [92,104]. Not only would these analyses be high resolution and high throughput but they could be highly informative, in that they would provide more accurate copy numbers, as opposed to the current techniques, which only annotate CNVs as gains or losses. Notwithstanding, the current repertoire of technologies has facilitated numerous significant studies of chromosome structure [105], disease [64,100] and clinical diagnostics [98,106,107].
For a newcomer to this field, the authors recommend reading papers relevant to the biological question at hand and then assessing which approach(es) is/are most conducive to success for that project. Even the best-funded laboratories will not be able to establish and maintain the complete range of technologies, so selection of one or a few platforms will need to be made. In a way, the copy number and structural variation field is currently in the same state of flux as the SNP genotyping field was in the late 1990s to early 2000s. A general trend in technologies seems to be moving towards the development of long (> 60-mers) oligonucleotide arrays containing very high probe coverage uniformly distributed across the genome (eg Agilent, Nimblegen platforms and others). It will be very interesting to see how the market dictates the success and evolution of this type of platform versus SNP-based approaches (eg Affymetrix and Illumina), which also allow extraction of copy number information but have the added information of genotypes. There will also be progress in identifying surrogate SNPs [19,70] or other markers for the more complex structural variants, allowing typing in large sample collections. In some cases, the technology used will be dictated by what equipment already exists in local (core) laboratories. The authors consider this first review as providing a snapshot of what is happening in the field and foresee that a second edition summary in a year or so would look entirely different, due to rapid advances in the field.
When investing in the technologies described in this paper, there are considerations that extend outside those of most typical laboratory management decisions. For example, beyond the rather straightforward choice of targeted or genome-wide experiments (Figure 2) and then selection of the corresponding reagent (eg types of primers or microarrays), one must consider the training level of technicians, specialised equipment (eg hybridisation ovens, scanners), and extended warranty and service support for some expensive equipment that may require constant upgrading. Moreover, in the authors' experience, the weakest links in the current state of the technologies are the algorithms available for mining accurate structural variation data. Investments in this area are growing, such that interpretation of any data generated now will only improve.
Many other practical issues are worth discussing, including:
(1) DNA must be of very high-quality preparation and, for genetic studies, preferably from peripheral blood (lymphoblastoid lines can introduce some transformationderived alterations). Low-quality DNA will lead to major problems, whereas whole-genome amplified DNA seems to be suitable; [108-110] however, this assertion will require more analysis to confirm that copy number ratios are maintained.
(2) At present, for comparative array CGH approaches, there is no standard hybridisation control DNA sample that has been adopted (although some experiments compare an 'average' genome by using pooled DNA samples). Differences between control DNA can cause various problems in correlating results between projects. In a positive step, at a recent Wellcome Trust-sponsored meeting, a recommendation was made to contact the Genome Standards Department of the British Standards Institute regarding appropriate reference standards for CGH.
(3) There are issues surrounding the content and quality of data in control databases. Currently, most structural variation data are available from two databases, namely the Database of Structural Variants http://projects.tcag.ca/variation/[13] and the Human Structural Variation Database http://humanparalogy.gs.washington.edu/structuralvariation/; [15] however, neither of these datasets is currently displayed by the major genome browsers (NCBI, UCSC). The number of samples and entries in these databases are still rudimentary but are expected to grow substantially in 2006. The quality of the data in the databases is fairly heterogeneous, with only a small proportion of variants being validated using a second technology. This will become even more of an issue as large genotyping datasets become a significant source for discovery of new structural variants [64].
(4) There are still limitations to all of the technologies. Most approaches readily allow resolution of a single gain or loss in copy number away from diploidy. With more complex deviations, however, where the normal copy number may be five or six, the interpretation becomes increasingly imprecise.
(5) The accuracy of mapping the breakpoints of a structural variant can be fairly wide-ranging, due to limitations inherent in the technique or the density and distribution of probes on an array. Moreover, because many of the breakpoint regions can overlap segmental duplications or gaps in the human sequence assembly, more detailed targeted analysis is often required.
This review focuses on methodologies for the identification of CNVs in the human genome. If the goal is explicitly to fine-map the precise content and boundaries of the variable regions, however, additional experiments are required. These include the elucidation of the DNA sequence content in each copy of a region. Due to the nature of their design, certain approaches are more amenable to this type of analysis. For example, in the fosmid end-sequencing strategy, fosmid clones overlapping the variable interval are automatically available for sequencing experiments. Also, some of these duplicated sequences may already be present in the human genome raw sequences (as seen in the human sequence read-depth analysis [33]), but are collapsed into a single copy in the current genome build. For some variations, however, such as duplications > 100 kb, this process could be more tedious and possibly require extensive library preparation and/or screening, mapping and re-sequencing.
The field of structural variation has now taken centre stage in human genetics and genomics research. The strengths and weaknesses of the different experimental and technical approaches discussed in this review, and also new ones, will be borne out through thorough scientific investigation. As more studies are undertaken, in particular through the investigation of the association between structural variants and disease [111-113], a greater understanding of the nature of the human genome and its variability in the population will be achieved. As in many other genomic studies, advances in the analysis of CNVs and structural variants in the human genome will set the precedent for the study of genomes in other species.
Acknowledgements
Research is supported by funds from The Centre for Applied Genomics, Hospital for Sick Children, Genome Canada/Ontario Genomics Institute, the Canadian Institutes of Health Research (CIHR) and the McLaughlin Centre for Molecular Medicine. L.F. is supported by the Swedish Medical Research Council, A.R.C is supported through a scholarship from the Natural Science and Engineering Research Council and S.W.S is an Investigator at CIHR and International Scholar of the Howard Hughes Medical Institute.
References
- Jacobs PA, Baikie AG, Court Brown WM, Strong JA. The somatic chromosomes in mongolism. Lancet. 1959;1:710. doi: 10.1016/s0140-6736(59)91892-6. [DOI] [PubMed] [Google Scholar]
- Edwards JH, Harndean DG, Cameron AH. et al. A new trisomic syndrome. Lancet. 1960;1:787–790. doi: 10.1016/s0140-6736(60)90675-9. [DOI] [PubMed] [Google Scholar]
- Patau K, Smith DW, Therman E. et al. Multiple congenital anomaly caused by an extra autosome. Lancet. 1960;1:790–793. doi: 10.1016/s0140-6736(60)90676-0. [DOI] [PubMed] [Google Scholar]
- Maegenis RE, Donlon TA, Wyandt HE. Giemsa-11 staining of chromosome 1: A newly described heteromorphism. Science. 1978;202:64–65. doi: 10.1126/science.694520. [DOI] [PubMed] [Google Scholar]
- Lubs HA. A marker X chromosome. Am J Hum Genet. 1969;21:231–244. [PMC free article] [PubMed] [Google Scholar]
- Edwards A, Civitello A, Hammond HA, Caskey CT. DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am J Hum Genet. 1991;49:746–756. [PMC free article] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J. et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Mir KU, Southern EM. Sequence variation in genes and genomic DNA: Methods for large-scale analysis. Annu Rev Genomics Hum Genet. 2000;1:329–360. doi: 10.1146/annurev.genom.1.1.329. [DOI] [PubMed] [Google Scholar]
- Syvanen AC. Accessing genetic variation: Genotyping single nucleotide polymorphisms. Nat Rev Genet. 2001;2:930–942. doi: 10.1038/35103535. [DOI] [PubMed] [Google Scholar]
- Kwok PY, Gu Z. Single nucleotide polymorphism libraries: Why and how are we building them? Mol Med Today. 1999;5:538–543. doi: 10.1016/S1357-4310(99)01601-9. [DOI] [PubMed] [Google Scholar]
- Kwok PY, Chen X. Detection of single nucleotide polymorphisms. Curr Issues Mol Biol. 2003;5:43–60. [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN. et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J. et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Locke DP, McGrath SD. et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun E, Sharp AJ, Bailey JA. et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- Feuk L, MacDonald J, Tang T. et al. Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies. PLoS Genet. 2005;1:e56. doi: 10.1371/journal.pgen.0010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Andrews TD, Carter NP. et al. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- Hinds DA, Kloek AP, Jen M. et al. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Hadnott TN, Perry GH. et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- Carter NP. As normal as normal can be? Nat Genet. 2004;36:931–932. doi: 10.1038/ng0904-931. [DOI] [PubMed] [Google Scholar]
- Buckley PG, Mantripragada KK, Piotrowski A. et al. Copy-number polymorphisms: Mining the tip of an iceberg. Trends Genet. 2005;21:315–317. doi: 10.1016/j.tig.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Check E. Human genome: Patchwork people. Nature. 2005;437:1084–1086. doi: 10.1038/4371084a. [DOI] [PubMed] [Google Scholar]
- Lee C. Vive la difference! Nat Genet. 2005;37:660–661. doi: 10.1038/ng0705-660. [DOI] [PubMed] [Google Scholar]
- van Ommen GJ. Frequency of new copy number variation in humans. Nat Genet. 2005;37:333–334. doi: 10.1038/ng0405-333. [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Nadeau JH, Lee C. Genetics: Copies count. Nature. 2006;439:798–799. doi: 10.1038/439798a. [DOI] [PubMed] [Google Scholar]
- Eichler EE. Widening the spectrum of human genetic variation. Nat Genet. 2006;38:9–11. doi: 10.1038/ng0106-9. [DOI] [PubMed] [Google Scholar]
- Freeman JL, Perry GH, Feuk L, Copy number variation: New insights in genome diversity. Genome Res. 2005. in press . [DOI] [PubMed]
- Speicher MR, Carter NP. The new cytogenetics: Blurring the boundaries with molecular biology. Nat Rev Genet. 2005;6:782–792. doi: 10.1038/nrg1692. [DOI] [PubMed] [Google Scholar]
- Lucito R, Healy J, Alexander J. et al. Representational oligonucleotide microarray analysis: A high-resolution method to detect genome copy number variation. Genome Res. 2003;13:2291–2305. doi: 10.1101/gr.1349003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignell GR, Huang J, Greshock J. et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004;14:287–295. doi: 10.1101/gr.2012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA. et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Kallioniemi A, Kallioniemi OP, Sudar D. et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–821. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- Solinas-Toldo S, Lampel S, Stilgenbauer S. et al. Matrix-based comparative genomic hybridization: Biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997;20:399–407. doi: 10.1002/(SICI)1098-2264(199712)20:4<399::AID-GCC12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Perou CM, Alizadeh AA. et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- Locke DP, Segraves R, Nicholls RD. et al. BAC microarray analysis of 15q11-q13 rearrangements and the impact of segmental duplications. J Med Genet. 2004;41:175–182. doi: 10.1136/jmg.2003.013813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishkanian AS, Malloff CA, Watson SK. et al. A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet. 2004;36:299–303. doi: 10.1038/ng1307. [DOI] [PubMed] [Google Scholar]
- de Vries BB, Pfundt R, Leisink M. et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson K, Heidenblad M, Strombeck B. et al. High-resolution genome-wide array-based comparative genome hybridization reveals cryptic chromosome changes in AML and MDS cases with trisomy 8 as the sole cytogenetic aberration. Leukemia. 2006;20:840–846. doi: 10.1038/sj.leu.2404145. [DOI] [PubMed] [Google Scholar]
- Garnis C, Lockwood WW, Vucic E. et al. High resolution analysis of non-small cell lung cancer cell lines by whole genome tiling path array CGH. Int J Cancer. 2006;118:1556–1564. doi: 10.1002/ijc.21491. [DOI] [PubMed] [Google Scholar]
- Shadeo A, Lam WL. Comprehensive copy number profiles of breast cancer cell model genomes. Breast Cancer Res. 2006;8:R9. doi: 10.1186/bcr1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake N, Shimokawa O, Harada N. et al. BAC array CGH reveals genomic aberrations in idiopathic mental retardation. Am J Med Genet A. 2006;140:205–211. doi: 10.1002/ajmg.a.31098. [DOI] [PubMed] [Google Scholar]
- Dhami P, Coffey AJ, Abbs S. et al. Exon array CGH: Detection of copy-number changes at the resolution of individual exons in the human genome. Am J Hum Genet. 2005;76:750–762. doi: 10.1086/429588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantripragada KK, Tapia-Paez I, Blennow E. et al. DNA copy-number analysis of the 22q11 deletion-syndrome region using array-CGH with genomic and PCR-based targets. Int J Mol Med. 2004;13:273–279. [PubMed] [Google Scholar]
- Barrett MT, Scheffer A, Ben-Dor A. et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc Natl Acad Sci USA. 2004;101:17765–17770. doi: 10.1073/pnas.0407979101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan C, Zhang Y, Leo C. et al. High-resolution global profiling of genomic alterations with long oligonucleotide microarray. Cancer Res. 2004;64:4744–4748. doi: 10.1158/0008-5472.CAN-04-1241. [DOI] [PubMed] [Google Scholar]
- Urban AE, Korbel JO, Selzer R. et al. High-resolution mapping of DNA copy alterations in human chromosome 22 using high density tiling oligonucleotide arrays. Proc Natl Acad Sci USA. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Ijssel P, Tijssen M, Chin SF. et al. Human and mouse oligonucleotide-based array CGH. Nucleic Acids Res. 2005;33:e192. doi: 10.1093/nar/gni191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy GC, Matsuzaki H, Dong S. et al. Large-scale genotyping of complex DNA. Nat Biotechnol. 2003;21:1233–1237. doi: 10.1038/nbt869. [DOI] [PubMed] [Google Scholar]
- Cheung J, Estivill X, Khaja R. et al. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 2003;4:R25. doi: 10.1186/gb-2003-4-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman D, White SJ, Potter S. et al. Complex SNP-related sequence variation in segmental genome duplications. Nat Genet. 2004;36:861–866. doi: 10.1038/ng1401. [DOI] [PubMed] [Google Scholar]
- Wang Y, Barbacioru C, Hyland F. et al. Large scale real-time PCR validation on gene expression measurements from two commercial long-oligonucleotide microarrays. BMC Genomics. 2006;7:59. doi: 10.1186/1471-2164-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Reynies A, Geromin D, Cayuela JM. et al. Comparison of the latest commercial short and long oligonucleotide microarray technologies. BMC Genomics. 2006;7:51. doi: 10.1186/1471-2164-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer RR, Richmond TA, Pofahl NJ. et al. Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH. Genes Chromosomes Cancer. 2005;44:305–319. doi: 10.1002/gcc.20243. [DOI] [PubMed] [Google Scholar]
- Zhao X, Richmond TA, Pofahl NJ. et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.CAN-03-3308. [DOI] [PubMed] [Google Scholar]
- Huang J, Wei W, Zhang J. et al. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genomics. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KK, Tsang YT, Shen J. et al. Allelic imbalance analysis by high-density single-nucleotide polymorphic allele (SNP) array with whole genome amplified DNA. Nucleic Acids Res. 2004;32:e69. doi: 10.1093/nar/gnh072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Fan JB, Campbell D. et al. High-throughput SNP genotyping on universal bead arrays. Mutat Res. 2005;573:70–82. doi: 10.1016/j.mrfmmm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Arinami T, Ohtsuki T, Ishiguro H. et al. Genomewide high density SNP linkage analysis of 236 Japanese families supports the existence of schizophrenia susceptibility loci on chromosomes 1p, 14q, and 20p. Am J Hum Genet. 2005;77:937–944. doi: 10.1086/498122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Rayner W, Morris AP. et al. An evaluation of HapMap sample size and tagging SNP performance in large-scale empirical and simulated data sets. Nat Genet. 2005;37:1320–1322. doi: 10.1038/ng1670. [DOI] [PubMed] [Google Scholar]
- Steemers FJ, Change W, Lee G. et al. Whole-genome genotyping with the single-base extension assay. Nat Methods. 2006;3:31–33. doi: 10.1038/nmeth842. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. In: The Analysis of Gene Expression Data: Methods and Software. Parmigiani G, et al, editor. Springer, New York, NY; 2003. DNA-Chip Analyzer (dChip) [Google Scholar]
- Feuk L, Marshall CR, Wintle RF, Scherer SW. Structural variants: Changing the landscape of chromosomes and design of disease studies. Hum Mol Genet. 2006;15(Suppl 1):R57–R66. doi: 10.1093/hmg/ddl057. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Tanenbaum DM, Daly MJ. et al. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol. 2000;18:1001–1005. doi: 10.1038/79269. [DOI] [PubMed] [Google Scholar]
- Mei R, Galipeau PC, Prass C. et al. Genome-wide detection of allelic imbalance using human SNPs and high-density DNA arrays. Genome Res. 2000;10:1126–1137. doi: 10.1101/gr.10.8.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce SK, Leinonen R, Lindgren CM. et al. Global analysis of uniparental disomy using high-density genotyping arrays. J Med Genet. 2005;42:847–851. doi: 10.1136/jmg.2005.032367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altug-Teber O, Dufke A, Poths S. et al. A rapid microarray based whole genome analysis for detection of uniparental disomy. Hum Mutat. 2005;26:153–159. doi: 10.1002/humu.20198. [DOI] [PubMed] [Google Scholar]
- Yu CE, Dawson G, Munson J. et al. Presence of large deletions in kindreds with autism. Am J Hum Genet. 2002;71:100–115. doi: 10.1086/341291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman TL, Rieder MJ, Morrison VA. et al. High-throughput genotyping of intermediate-size structural variation. Hum Mol Genet. 2006;15:1159–1167. doi: 10.1093/hmg/ddl031. [DOI] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Bieche I, Olivi M, Champeme MH. et al. Novel approach to quantitative polymerase chain reaction using real-time detection: Application to the detection of gene amplification in breast cancer. Int J Cancer. 1998;78:661–666. doi: 10.1002/(SICI)1097-0215(19981123)78:5<661::AID-IJC22>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Ponchel F, Toomes C, Bransfield K. et al. Real-time PCR based on SYBR-green I fluorescence: An alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003;3:18. doi: 10.1186/1472-6750-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonnier F, Raux G, Wang Q. et al. Detection of exon deletions and duplications of the mismatch repair genes in hereditary nonpolyposis colorectal cancer families using multiplex polymerase chain reaction of short fluorescent fragments. Cancer Res. 2000;60:2760–2763. [PubMed] [Google Scholar]
- Vaurs-Barriere C, Bonnet-Dupeyron MN, Combes P. et al. Golli-MBP copy number analysis by FISH, QMPSF and MAPH in 195 patients with hypomyelinating leukodystrophies. Ann Hum Genet. 2006;70:66–77. doi: 10.1111/j.1529-8817.2005.00208.x. [DOI] [PubMed] [Google Scholar]
- Casilli F, Di Rocco ZC, Gad S. et al. Rapid detection of novel BRCA1 rearrangements in high-risk breast-ovarian cancer families using multiplex PCR of short fluorescent fragments. Hum Mutat. 2002;20:218–226. doi: 10.1002/humu.10108. [DOI] [PubMed] [Google Scholar]
- Armour JA, Sismani C, Patsalis PC, Cross G. Measurement of locus copy number by hybridisation with amplifiable probes. Nucleic Acids Res. 2000;28:605–609. doi: 10.1093/nar/28.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox EJ, Atia T, Cross G. et al. High throughput screening of human subtelomeric DNA for copy number changes using multiplex amplifiable probe hybridisation (MAPH) J Med Genet. 2002;39:790–795. doi: 10.1136/jmg.39.11.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox EJ, Akrami SM, Armour JA. DNA copy number analysis by MAPH: Molecular diagnostic applications. Expert Rev Mol Diagn. 2002;2:370–378. doi: 10.1586/14737159.2.4.370. [DOI] [PubMed] [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijen R. et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langerak P, Nygren AO, Schouten JP, Jacobs H. Rapid and quantitative detection of homologous and non-homologous recombination events using three oligonucleotide MLPA. Nucleic Acids Res. 2005;33:e188. doi: 10.1093/nar/gni187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalic T, Vossen RH, Coffa J. et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet. 2005;13:1231–1234. doi: 10.1038/sj.ejhg.5201465. [DOI] [PubMed] [Google Scholar]
- Armour JA, Rad IA, Hollox EJ. et al. Gene dosage analysis by multiplex amplifiable probe hybridization. Methods Mol Med. 2004;92:125–139. doi: 10.1385/1-59259-432-8:125. [DOI] [PubMed] [Google Scholar]
- White S, Kalf M, Liu Q. et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. 2002;71:365–374. doi: 10.1086/341942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SJ, Vink GR, Kriek M. et al. Two-color multiplex ligation-dependent probe amplification: Detecting genomic rearrangements in hereditary multiple exostoses. Hum Mutat. 2004;24:86–92. doi: 10.1002/humu.20054. [DOI] [PubMed] [Google Scholar]
- Stern RF, Roberts RG, Mann K. et al. Multiplex ligation-dependent probe amplification using a completely synthetic probe set. Biotechniques. 2004;37:399–405. doi: 10.2144/04373ST04. [DOI] [PubMed] [Google Scholar]
- Scherer SW, Cheung J, MacDonald JR. et al. Human chromosome 7: DNA sequence and biology. Science. 2003;300:767–772. doi: 10.1126/science.1083423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SW, Green ED. Human chromosome 7 circa 2004: A model for structural and functional studies of the human genome. Hum Mol Genet. 2004;13(Spec No 2):R303–R313. doi: 10.1093/hmg/ddh231. [DOI] [PubMed] [Google Scholar]
- Hillier LW, Fulton RS, Fulton LAA. et al. The DNA sequence of human chromosome 7. Nature. 2003;424:157–164. doi: 10.1038/nature01782. [DOI] [PubMed] [Google Scholar]
- Beck S, Trowsdale J. The human major histocompatability complex: Lessons from the DNA sequence. Annu Rev Genomics Hum Genet. 2000;1:117–137. doi: 10.1146/annurev.genom.1.1.117. [DOI] [PubMed] [Google Scholar]
- Newman TL, Tuzun E, Morrison VA. et al. A genome-wide survey of structural variation between human and chimpanzee. Genome Res. 2005;15:1344–1356. doi: 10.1101/gr.4338005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Mitra RD, Varma C, Church GM. Advanced sequencing technologies: Methods and goals. Nat Rev Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- Estivill X, Cheung J, Pujana MA. et al. Chromosomal regions containing high-density and ambiguously mapped putative single nucleotide polymorphisms (SNPs) correlate with segmental duplications in the human genome. Hum Mol Genet. 2002;11:1987–1995. doi: 10.1093/hmg/11.17.1987. [DOI] [PubMed] [Google Scholar]
- Kok K, Dijkhuizen T, Swart YE. et al. Application of a comprehensive subtelomere array in clinical diagnosis of mental retardation. Eur J Med Genet. 2005;48:250–262. doi: 10.1016/j.ejmg.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Rooms L, Reyniers E, Kooy RF. Subtelomeric rearrangements in the mentally retarded: A comparison of detection methods. Hum Mutat. 2005;25:513–524. doi: 10.1002/humu.20185. [DOI] [PubMed] [Google Scholar]
- Buckley PG, Mantripragada KK, Benetkiewicz M. et al. A full-coverage, high-resolution human chromosome 22 genomic microarray for clinical and research applications. Hum Mol Genet. 2002;11:3221–3229. doi: 10.1093/hmg/11.25.3221. [DOI] [PubMed] [Google Scholar]
- Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet. 2003;12(Spec No 2):R145–R152. doi: 10.1093/hmg/ddg261. [DOI] [PubMed] [Google Scholar]
- Rickman L, Fiegler H, Shaw-Smith C. et al. Prenatal detection of unbalanced chromosomal rearrangements by array-CGH. J Med Genet. 2006;43:353–361. doi: 10.1136/jmg.2005.037648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitman TJ, Dong R, Vyse TJ. et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- Vissers LE, Veltman JA, van Kessel AG. et al. Identification of disease genes by whole genome CGH arrays. Hum Mol Genet. 2005;14(Spec No 2):R215–R223. doi: 10.1093/hmg/ddi268. [DOI] [PubMed] [Google Scholar]
- Rosenberg C, Knijnenburg J, Bakker E. et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: Clinical significance of imbalances present both in affected children and normal parents. J Med Genet. 2006;43:180–186. doi: 10.1136/jmg.2005.032268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC. et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Chen KS, Manian P, Koeuth T. et al. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- Service RF. Gene sequencing The race for the $1000 genome. Science. 2006;311:1544–1546. doi: 10.1126/science.311.5767.1544. [DOI] [PubMed] [Google Scholar]
- Woodfine K, Carter NP, Dunham I, Fiegler H. Investigating chromosome organization with genomic microarrays. Chromosome Res. 2005;13:249–257. doi: 10.1007/s10577-005-1504-5. [DOI] [PubMed] [Google Scholar]
- Wong A, Lese Martin C, Heretis K. et al. Detection and calibration of microdeletions and microduplications by array-based comparative genomic hybridization and its applicability to clinical genetic testing. Genet Med. 2005;7:264–271. doi: 10.1097/01.GIM.0000160076.14102.EC. [DOI] [PubMed] [Google Scholar]
- Ylstra B, van den Ijssel P, Carvalho B. et al. BAC to the future! or oligonucleotides: A perspective for micro array comparative genomic hybridization (array CGH) Nucleic Acids Res. 2006;34:445–450. doi: 10.1093/nar/gkj456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazani AA, Arneson NC, Warren K. et al. Limited tissue fixation times and whole genomic amplification do not impact array CGH profiles. J Clin Pathol. 2006;59:311–315. doi: 10.1136/jcp.2005.029777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little SE, Vuononvirta R, Reis-Filho JS. et al. Array CGH using whole genome amplification of fresh-frozen and formalin-fixed, paraffin-embedded tumor DNA. Genomics. 2006;87:298–306. doi: 10.1016/j.ygeno.2005.09.019. [DOI] [PubMed] [Google Scholar]
- Wang G, Maher E, Brennan C. et al. DNA amplification method tolerant to sample degradation. Genome Res. 2004;14:2357–2366. doi: 10.1101/gr.2813404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Caignec C, Boceno M, Saugier-Veber P. et al. Detection of genomic imbalances by array based comparative genomic hybridisation in fetuses with multiple malformations. J Med Genet. 2005;42:121–128. doi: 10.1136/jmg.2004.025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobanputra V, Sebat J, Troge J. et al. Application of ROMA (representational oligonucleotide microarray analysis) to patients with cytogenetic rearrangements. Genet Med. 2005;7:111–118. doi: 10.1097/01.GIM.0000153661.11110.FB. [DOI] [PubMed] [Google Scholar]
- Bejjani BA, Saleki R, Ballif BC. et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: Is less more? Am J Med Genet A. 2005;134:259–267. doi: 10.1002/ajmg.a.30621. [DOI] [PubMed] [Google Scholar]