

Abstract
Inference of population structure from genetic markers is helpful in diverse situations, such as association and evolutionary studies. In this paper, we describe a two-stage strategy in inferring population structure using multilocus genotype data. In the first stage, we use dimension reduction methods such as singular value decomposition to reduce the dimension of the data, and in the second stage, we use clustering methods on the reduced data to identify population structure. The strategy has the ability to identify population structure and assign each individual to its corresponding subpopulation. The strategy does not depend on any population genetics assumptions (such as Hardy-Weinberg equilibrium and linkage equilibrium between loci within populations) and can be used with any genotype data. When applied to real and simulated data, the strategy is found to have similar or better performance compared with STRUCTURE, the most popular method in current use. Therefore, the proposed strategy provides a useful alternative to analyse population data.
Keywords: population structure, subpopulation, singular value decomposition, dimension reduction, clustering
Introduction
Information about the population structure of species is useful in a variety of situations, such as admixture mapping, subspecies classification, genetic barrier detection and evolutionary study [1-5]. For example, anthropologists may have the debris of ancient people, supplied by archaeologists, and want to learn about the relationship between the ancient people and modern populations to infer the evolutionary history of human beings. Population structure can be identified based on visible characteristics such as language, culture, physical appearance and geographical region. But this can be subjective and may bear no relevance to genetics [3]. In other situations, the presence of population structure may constitute a practical nuisance. In association studies, case-control design is often used to identify genetic variants underlying complex traits by comparing allele frequencies between unrelated individuals who are affected and those who are unaffected. The presence of population structure can lead to spurious associations between a candidate marker and a phenotype, however, as a result of population structure in the sample [6,7]. In forensic studies, the identification of reference groups is very important, but this can be difficult when population structure exists [4,8]. In all of these situations, the first step is to identify population substructure.
Pritchard et al. introduced a model-based clustering method to infer population structure and assign individuals to populations using multilocus genotype data [3]. They used a Bayesian formulation to generate the posterior distribution using a Markov chain Monte Carlo (MCMC) method based on Gibbs sampling. Their main modelling assumptions were Hardy-Weinberg equilibrium within populations and linkage equilibrium between loci within each population [3]. This is the predominant method currently used in genetic studies. Some other methods have been proposed,[9-12] but all are model-based (parametric) methods. These methods have their own advantages and disadvantages. They all have model assumptions because of their parametric nature.
Here, we describe a two-stage strategy for inferring population structure, which is an alternative and complementary approach to STRUCTURE[3,10] for exploring data. In the first stage, we use methods such as singular value decomposition (SVD) to reduce the dimension of data and then perform clustering on the reduced data. This two-stage strategy is widely used in knowledge induction and representation to determine similarities between the meaning of words and passages by analysis of large text corpora [13,14]. Our method does not use assumptions such as Hardy-Weinberg equilibrium for populations or linkage equilibrium for loci. Here we show that our method is faster and has comparable (when model assumptions hold) or better performance (when model assumptions fail) than STRUCTURE when applied to real and simulated data [3,10].
In the next section, we describe the strategy and methods we use and some of the advantages of the approach we take. We illustrate our method with examples and make comparisons with STRUCTURE in the Results section. In the Discussion section, we highlight issues in the methods, the potential use of the methods, and future work.
Methods
It is well known that cluster analysis is difficult in high-dimensional space because standard clustering algorithms such as the Expectation-Maximization (EM) algorithm[15] and the K-means method are probably trapped in local minima [13,16]. Although many initialisation methods have been proposed to deal with this problem, they have had only limited success [13]. Therefore, a two-stage procedure seems valuable: first reduce the dimension of the original space and then cluster in the reduced (low-dimensional) space. In general, any dimension reduction methods and clustering methods can be plugged into this two-stage framework. In this paper, we use SVD as the dimension-reduction method, and the mixture model and K-means as the clustering methods. We also propose a non-parametric clustering method, which can be viewed as a variant of the K-means method, for small sample sizes.
Dimension reduction
SVD is widely used in knowledge induction and representation and information retrieval. For example, SVD plays a key role in latent semantic analysis (LSA) or latent semantic indexing (LSI). The semantic dimensions are thought to contain redundant and noisy information, which can be separated out and should be ignored. Bartell et al. showed that the document representations given by LSI are equivalent to the optimal representations found when solving a particular multidimensional scaling problem in which the given inter-object similarity information is provided by the inner product similarities between the documents themselves [17]. LSI automatically computes a much smaller semantic subspace from the original text collection. This improves recall and precision in information retrieval; information filtering or text classification; word sense disambiguation; word sorting and relatedness judgments; the prediction of learning from text; and summarising skills [14,18]. The effectiveness of LSI in empirical studies is often attributed to the reduction of noise, redundancy and ambiguity [14,18,19]. By introducing a dual probabilistic model based on similarity concepts, Ding showed that semantic correlations could be characterised quantitatively by their statistical significance -- that is, the likelihood [18]. He further showed that LSI is the optimal solution of the model and proved the existence of the optimal semantic subspace. This model explains theoretically the performance improvements observed for LSI.
Mathematically speaking, SVD is a matrix decomposition technique. A real-valued m-by-n matrix (say X) can be represented uniquely (up to certain trivial rearrangements of columns and subspace rotations, in the case of duplicated singular values) as the product of three matrices:
| (1) |
where both U and V are column orthonormal and S is a diagonal matrix of singular values [20]. There is a direct relationship between SVD and principal component analysis (PCA) when PCA is performed from the covariance matrix using the following equations:
| (2) |
| (3) |
If each row of X is normalised (centred and unitary), the covariance matrix ∑ of data X is XXT. We know that:
where A is an orthonormal matrix and the λ values are the eigenvalues of ∑. The decomposition is unique up to some trivial column rearrangements. Matrix A contains the principal components of columns of X. From equations (2) and (4), we can see that the left singular vectors U are the same as the principal components of columns of X. Similarly, the right singular vectors V are the same as the principal components of rows of X.
Clustering
We choose to use two clustering methods: one is mixture 4, proposed by Figueiredo et al., based on the mixture model,[21] the other is K-means. The advantages of mixture 4 are that it is capable of selecting the number of components (ie the number of clusters) and that it is relatively robust to the initialisation of the parameters. Figueiredo et al. [21] used the following finite mixture models:
where Y = [Y1,..., Yd]T, a d-dimensional random variable with y = [y1,...,yd]T being a realisation of Y; α1,...,αk are the mixing probabilities; each θm is the set of parameters defining the mth component; and θ = {θ1,..., θk, α1,...,αk} is the complete set of parameters needed to specify the mixture. They used the minimum message length criterion and the component-wise EM algorithm to integrate estimation and model selection in a single algorithm [22]. This method can avoid another well-known drawback of the EM algorithm for mixture fitting -- namely, the possibility of convergence towards a singular estimate at the boundary of the parameter space [21].
K-means is a commonly used non-parametric clustering method, but it has some drawbacks. We propose a clustering method (density-based mean clustering [DBMC]), which is a variant of K-means but can avoid some of its drawbacks. The details of DBMC are provided in Appendix 1.
Number of subpopulations
There are many methods for estimating the number of clusters which can also be used for estimating the number of subpopulations. Zhu et al. showed that Bayesian information criterion (BIC) from their mixture model performed better than STRUCTURE in inferring the number of subpopulations [23]. All of these methods can be integrated into the clustering procedure.
Missing data imputation
It is not uncommon to have missing values in genetic studies. Such data can be manually flagged and excluded from subsequent analyses [24]. Many analytical methods, such as PCA or SVD, require complete matrices [25]. Although some studies reported dealing with SVD/PCA with missing data,[26-29] they often rely on specific probabilistic models and have limited generalisability. Although one solution to the missing data problem is to repeat the experiment, and this method has been used in the validation of microarray analysis algorithms,[30] this strategy may be too expensive and impractical for most studies. Therefore, we need to estimate the missing values from non-experimental methods.
There is little published literature concerning missing value estimations for genotype data from human populations. The uniqueness of this issue is that the genotype data are categorical by nature. Note that Sen and Churchill[31] and Broman et al. [32] discussed using the EM algorithm and the hidden Markov model to deal with missing genotypes, but they were mostly concerned with experiments involving inbred animals.
Genotype data are usually in the form of large matrices of genotypes of marker loci (columns) from different persons (rows) [3]. Without loss of information, we can transform this person-marker matrix into a genotype-person matrix. For each marker, all of the genotypes appearing in the data are listed, one genotype per row, with a value of one for the cell if a person (column) has this genotype, and zero otherwise. Using this reformatting we now have a large 0-1 matrix. We can view this genotype-person matrix as a frequency matrix, with each cell denoting the frequency of the person (its column) who has the genotype that is denoted by its row. Such frequency matrices are commonly used in LSA and are called 'word-document matrices' or 'term-document matrices' [14,18,33]. Because we can fully reconstruct the original person-marker matrix from this genotype-person matrix, there is no information loss in this transformation. We impute the missing values on the basis of this genotype-person matrix. In this study, we used an imputation method which is similar to the 'K nearest-neighbour' (KNN)-based method used in Troyanskaya et al. [34] The rationale underlying this method is that where data points are clustered together (similar) in the lower dimension, we can expect them to be clustered together (similar) in the higher dimension as well. In this way, the missing dimensions of a data point (individual in our case) can be estimated by its neighbours (those which are very similar to the data points under study), with no missing data in these dimensions. Details of this method are provided in Appendix 2. In this study, we did not iterate to impute the missing values (ie we only use KNN once).
Results
Data
To evaluate fully the performance of the proposed strategy, we applied it to two real datasets and two simulated datasets. The first real dataset was that reported in Rosenberg et al. ,[5] which has genotypes at 377 autosomal microsatellite markers in 1,056 individuals from 52 populations. Here, we considered the whole dataset, as well as two American populations, the Pima and Surui populations, with 25 and 21 individuals, respectively, to demonstrate our methods. The other real dataset was from the HapMap project;[35] we used genotype data from 45 Chinese and 44 Japanese on chromosome 17 which was released in October 2005. A dataset is formed by the 500 most informative single nucleotide polymorphisms (SNPs) using the methods proposed in Rosenberg et al. [36]
One simulated dataset was generated under the coalescent model using MS, a program developed by Hudson [37]. A progenitor population gave rise to two subpopulations 3,000 generations ago. Subpopulation 1 had a constant size of 10,000, began to grow exponentially 1,000 generations ago and has now reached 40,000. Subpopulation 2 had a constant size of 2,000 before 2,000 generations, and then instantaneously expanded to 10,000 and has remained at that size until the present. We also assume that the mutation rate per site per generation is 10-8 and that we are interested in a segment of 10 kilobases. No recombination is set. In this fashion, we have generated 100 such chromosomal segments, each segment harbouring 27-77 SNPs. The chromosomal segments are pooled together to produce more genotype data. A dataset is formed by randomly sampling 400 haplotypes (200 individuals) from subpopulation 1 and 200 haplotypes (100 individuals) from subpopulation 2. The second simulated dataset was taken from Tang et al. [12] (http://www.fhcrc.org/science/labs/tang/). This dataset contains 50 individuals from each of the two ancestral populations and 200 individuals from the admixed population. The true individual admixture values of the admixed individuals are also available.
Population structure identification
We used STRUCTURE 2.0[10] and our SVD-based procedure on the datasets. In the second stage (the clustering stage) of our procedure, we used both the mixture model and K-means methods for clustering. For STRUCTURE, we tried the four available models. If not stated explicitly, we used the default model with admixture and correlated allele frequencies and set both burnin length and number of MCMC replications after burnin to be 20,000.
For Rosenberg et al.'s full dataset,[5] we followed their procedure and ran analyses (STRUCTURE 2.0 and mixture 4) multiple times for the number of populations (clusters) from two to six. Table 1 shows the Pearson correlation coefficients between the results of the STRUCTURE and mixture 4 analyses using the first five principal components. For number of populations (clusters) K = 5 and 6, the relatively small correlation coefficients of two clusters (cluster 5 for K = 5 and cluster 1 for K = 6) are caused by the accumulated differences between the estimates of the two methods (the probabilities of belonging to one cluster, or membership coefficients, must sum to 1 across clusters).
Table 1.
Correlation coefficients between the estimates of STRUCTURE and the singular value decomposition (SVD)-based method from the full data of Rosenberg et al. [5]
| Cluster/population | ||||||
|---|---|---|---|---|---|---|
| K a | 1 | 2 | 3 | 4 | 5 | 6 |
| 2 | 0.9602 | 0.9602 | ||||
| 3 | 0.9756 | 0.9695 | 0.9826 | |||
| 4 | 0.9824 | 0.9853 | 0.9667 | 0.9708 | ||
| 5 | 0.9602 | 0.9836 | 0.9470 | 0.9719 | 0.5715 | |
| 6 | 0.5688 | 0.9473 | 0.9473 | 0.9719 | 0.9642 | 0.9596 |
aK is the number of clusters/populations.
Note: For the SVD-based method, the first five principal components were used. The mixture model was used for clustering.
We tried the four models available in STRUCTURE 2.0 on the Pima-Surui data subset with 100 randomly chosen markers. The model assuming independent allele frequencies among populations with no admixture yielded the best results. Burnin length and number of MCMC replications after burnin were all 10,000 in the analyses. The results are summarised in Table 2. Clearly, it is difficult to draw the conclusion that there are two populations in the dataset from the above results, but once we set K = 2, STRUCTURE 2.0 can assign each individual correctly to the population it belongs to.
Table 2.
Results from STRUCTURE 2.0 on the Pima-Surui data with 100 randomly selected markers without missing genotypes.
| Ka | Run 1b | Run 2b | Run 3b | Run 4b | Run 5b |
|---|---|---|---|---|---|
| 1 | -10849.6 | -10847.8 | -10850.1 | -10848.9 | -10849.5 |
| 2 | -9616.9 | -9619.3 | -9619.4 | -9629.2 | -9614.2 |
| 3 | -9417 | -9418.3 | -9412.9 | -9529.3 | -9498.3 |
| 4 | -9648.8 | -9369.8 | -9557 | -9397.9 | -9445.7 |
| 5 | -9303.7 | -9472.3 | -9346.6 | -9405.1 | -9317 |
| 6 | -9541.6 | -10457.2 | -9315.6 | -9484.1 | -11348.8 |
| 7 | -9408.4 | -10576.9 | -10151.4 | -9443.4 | -10938.6 |
| 8 | -9451.9 | -9369.1 | -10715.4 | -9393.1 | -10304.5 |
| 9 | -10403.1 | -9450.7 | -9489.1 | -10100.8 | -9205.1 |
aK is the number of clusters/subpopulations; bDifferent runs of STRUCTURE 2.0.
Before performing SVD, we first transformed the data into the genotype-person format. Wall et al. reported that pre-processing is critical in SVD/PCA,[38] which is well known in LSA [14,18,19,39]. We applied the so-called tf-idf transformation[18,39] on the genotype-person matrix. For the Pima-Surui sub-dataset with 100 randomly chosen markers, on the reduced two-dimensional space, mixture 4 finds two clusters. The mixture 4 and K-means methods assign individuals correctly to their populations with two clusters.
Figure 1 plots pairwise cosine similarities between individuals in the reduced two-dimensional space. Figure 2 shows the pairwise cosine similarities between individuals using the original data without reduction. It seems that the original data are noisier and that SVD not only reduces the dimension but also reduces noise.
Figure 1.

Pairwise cosine similarities between individuals in the reduced two-dimensional space of the Pima-Surui data with 100 randomly selected markers without missing genotypes.
Figure 2.

Pairwise cosine similarities between individuals using the original Pima - Surui data with the same 100 markers as in Figure 1.
To evaluate our method's performance, we reduced the number of markers. When the numbers of markers were 80, 60, 40 and 20, both methods performed equally well (data not shown). When the number of markers was reduced to ten, both methods still performed well, with STRUCTURE slightly better when the marker information was limited (data not shown).
To compare the performance of the methods fully, we conducted a simulation study using population genetics models. Table 3 shows the results for three datasets from the simulation study. It has been reported that STRUCTURE provides very stable estimates when the model assumptions hold [5]. In the presence of tightly linked SNPs, STRUCTURE not only performed worse but was also not very stable. For example, Table 3 shows that when 494 SNPs were used, the best performance of STRUCTURE only misclassified three individuals; however, the median number of misclassified individuals by STRUCTURE was 33. For the same dataset, the time taken for the analyses (using a laptop with Intel Pentium M 1.80 GHz CPU, 512 MB RAM and Windows XP) was about 30 minutes for STRUCTURE and about 28 seconds for the proposed method computing 100 largest singular values in MatLab software (The Mathworks, Inc.).
Table 3.
The performance of STRUCTURE 2.0 and the singular value decomposition (SVD)-based method on the simulated data.
| Number of single nucleotide polymorphisms | Number of misclassified individuals | |
|---|---|---|
| STRUCTURE | SVD-based | |
| 401 | 36 (12%) | 3 (1%) |
| 453 | 34 (11.3%) | 1 (0.3%) |
| 494 | 3 (1%) | 0 (0%) |
Note: The numbers in the table are the numbers of misclassified individuals and the numbers in parentheses are the misclassification rates. For the SVD-based method, the K-means method was employed for clustering using 30 principal components. The three datasets in the table correspond to the combinations of the first eight, nine and ten chromosomal segments from the original simulated data, respectively.
For the dataset from the HapMap project, STRUCTURE best performance's misclassified two of the 45 Chinese individuals and two of the 44 Japanese individuals, whereas the best performance of the proposed strategy classified all individuals correctly. The inferior performance of STRUCTURE was partly due to the fact that some of the markers in the dataset were tightly linked. For example, the distances between some SNPs were less than 100 base pairs. It is well known that Chinese and Japanese populations are closely related and very difficult to distinguish in genetics studies. It is not too difficult, however, to distinguish between informative markers. This also confirms that SNPs can be very informative in inferring population structures [40].
We used the simulated data with admixed individuals to evaluate the performance of the proposed strategy on data exhibiting population admixture. Figure 3 shows the results. The membership coefficients were calculated following the method of Nascimento et al. [41] We found that STRUCTURE performed slightly better than the proposed strategy. This is expected because when the model assumptions hold, parametric methods with correct assumptions should always perform better.
Figure 3.
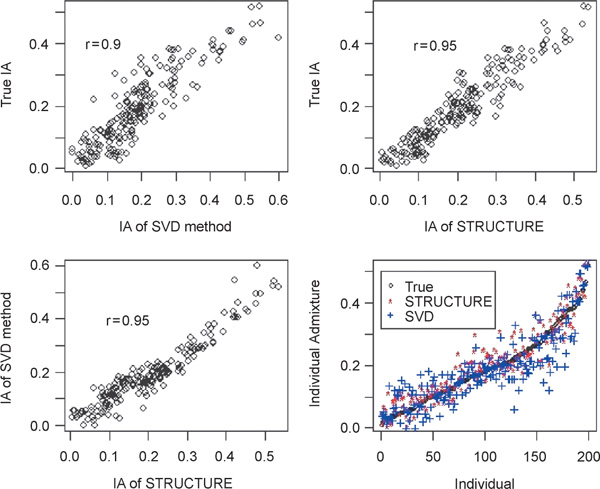
The results of STRUCTURE and the singular value decomposition (SVD)-based method based on the simulated data set of Tang et al. [12]For the SVD-based method, ten principal components were used. The K-means method was used for clustering. The individual admixture (IA) values were calculated using the method of Nascimento et al. [41].
Evaluation of DBMC
We used our DBMC method on the reduced data to evaluate its performance. DBMC performed well in different reduced dimensions. Figure 4 shows the formation of the initial partitioning by DBMC. There were four points (blue circles without other symbols superimposed) left ungrouped by Gap statistics. They were classified into the second cluster by the initialisation procedure. Therefore, after the density-based initialisation, all but one individual (number 42 in the figures with coordinates - 5.95710, - 0.19782) were correctly classified. Only one iteration was required to finish the clustering correctly.
Figure 4.
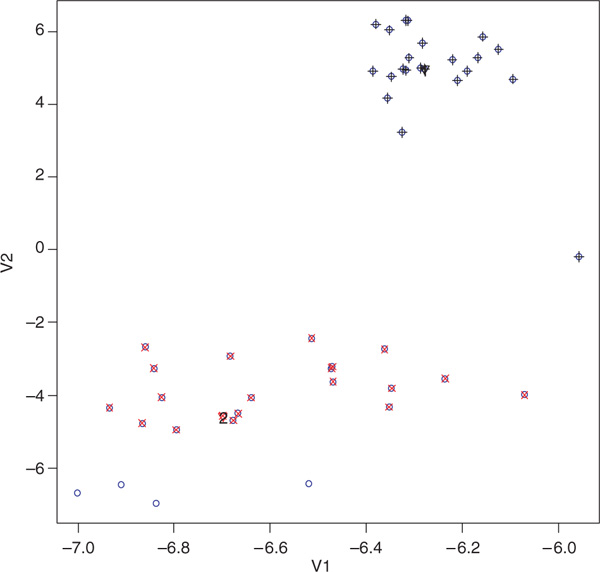
Illustration of the formation of the initial partitioning by density-based mean clustering on the first two principal components of the Pima-Surui data with 100 randomly selected markers without missing genotypes. The numbers (1 and 2 here) over the triangles indicate the order of cluster starting points identified. Points with black plus signs indicate the points identified as the first cluster by Gap statistics. Points with red crosses indicate the points identified by Gap statistics as the second cluster. Blue circles represent the original points in the two-dimensional space after singular value decomposition. Downward triangles indicate the starting points for each cluster. Points with red crosses indicate the points identified by Gap statistics as the second cluster.
Discussion
We have described a two-stage strategy for using multilocus genotype data to examine population structure and to assign individuals to populations. We prefer to call this approach a strategy, instead of a method, because it provides a framework, not just one method. One can choose different dimension reduction and clustering methods to fit into the framework.
Our strategy does not rely on any population genetic assumptions, such as Hardy-Weinberg equilibrium and linkage equilibrium between loci within populations. This means that violation of the assumptions does not invalidate our strategy. For model-based methods, the violation of assumptions makes these methods invalid, at least theoretically, although some methods may be robust to certain departures from assumptions. We have shown, through simulation and real data analyses, that the proposed approach is not affected by departure from the linkage equilibrium assumption for markers in the data; however, tightly linked markers may provide redundant information, so more markers are usually needed. In this situation, the validity of our strategy is not affected, but the validity of model-based methods becomes questionable.
It is reported that pre-transformation is critical in SVD [18,38,39]. We choose to use the tf-idf transformation, which is widely used in information retrieval, but other pre-transformations are possible. Before the pre-transformation, it would be helpful to eliminate the non-informative rows and columns [19]. In our experiment, we only eliminated the markers in situations where just one genotype appears in the whole sample. It is possible to use some criteria (such as entropy) to filter out the non-informative data. This would make the analysis faster (because the matrix becomes smaller) and more efficient (because the remaining matrix is more informative).
There are many dimension reduction techniques besides SVD, such as PCA and its variants (probabilistic PCA, non-linear PCA etc), correspondence analysis, multidimensional scaling, independent component analysis, projection pursuit and projection pursuit regression, principal curves and methods based on topologically continuous maps and neural networks [42]. The reason we chose SVD is that many efficient algorithms exist for this method. Because we only need a few principal components, even more efficient algorithms are available for this purpose. Although the person-marker matrix can be large if there are many markers or individuals, the SVD procedure can be performed very quickly. In addition, SVD seems to reduce noise, as shown in Figures 1 and 2. It is likely that other dimension reduction methods may also yield good results.
It would be more statistically sound to view our genotype-person matrix as a binary matrix. Several methods have been proposed for PCA on binary data,[43-45] but they implicitly assume that the observations are independent across the dimensions, which does not apply in our case--the genotypes are by no means independent at each marker. Nevertheless, we have tried the logistic PCA[44] on our data, but the results were not as good as those achieved by treating the genotype-person matrix as a real matrix. Figure 5 shows the first three principal components from logistic PCA using the criterion that the log likelihood change is less than 0.1. It is obvious that the two clusters are not clearly separated. Although we could perform dimension reduction using probabilistic PCA on the original genotype data matrix, which is a categorical matrix, no methods are available for such analyses. One PCA method for categorical data is implemented in SPSS software (SPSS Inc.) through the use of the optimal scaling (or optimal scoring) approach to turn the categorical problem into a quantitative one,[46] and applies PCA to the numeric matrix. The coding scheme of the categorical data in the original matrix may affect the resulting numeric matrix, however; moreover, one needs to set the optimal scaling level for analysis variables subjectively, whereas we intend to avoid subjective choices in our strategy.
Figure 5.
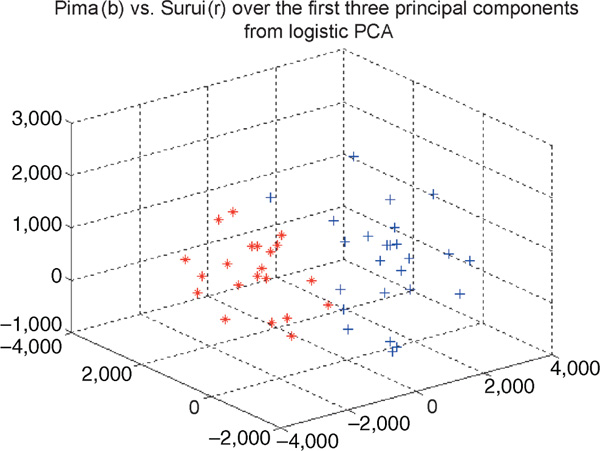
The first three principal components from logistic principal component analysis using the criterion that the log likelihood change is less than 0.1 in the Pima-Surui data.
It is an open question as to how to choose dimensionality for the reduced space when using SVD or PCA. Much work has been done on this topic, including likelihood ratio, Minimum Description Length (MDL), Akaike information criterion (AIC), BIC,[47] Laplace's method,[48] and probabilistic PCA model [26]. All of these methods are based on some probabilistic model, however -- usually the normality assumption. In our case, the assumption is obviously not appropriate. Laplace's method seems to give a more reasonable choice than others, but can only serve as a guideline. Because our purpose is clustering, one possible way for choosing the optimal dimension is by clustering results. For each given dimension, we can perform cluster analysis on the reduced space and evaluate the resulting clusters -- for example, between to within-cluster variation. We can then select the optimal dimension as the one with the best clustering evaluation. Methods based on appropriate models (perhaps binary or categorical models) or non-parametric (empirical) approaches should be more appropriate for our problem -- and we are planning to investigate this in the future.
Ando observed that, in LSA, using SVD, the topics underlying outlier documents (ie those documents that are very different from other documents) tend to be lost as lower numbers of dimensions are chosen [33]. A general explanation of the good performance of LSI is that when eigenvectors with smaller eigenvalues are left out, noise is eliminated, and, as a result, the similarities among the linguistic units are measured more accurately than in the original space. According to the mathematical formulation of SVD, dimensional reduction comes from two sources: outlier documents and minor terms. These two types of noise are mathematically equivalent and are inseparable under SVD. However, people do not want to consider the outlier documents as 'noise', when their interest is in characterising the relationships among the documents while all the documents are assumed to be equal. In our case, fine structure (small numbers of individuals who are very different from others) may be lost, especially when the sample size is small. Hastie et al. noted that finer structure can be lost with any dimension reduction method [49]. Ando proposed an algorithm which differs from SVD in that terms and documents are treated in a non-symmetrical way [33]. By scaling the vectors in each computation of eigenvectors, his algorithm tries to eliminate noise from the minor terms without eliminating the influence of the outlier documents. Further analyses are needed to evaluate this method.
In this paper, we chose to use mixture models as our clustering methods. The advantages of the mixture models include their readiness for use, their ability to choose the number of clusters automatically and their computational efficiency. These are by no means the only choice, however, and we have also considered K-means methods here. In fact, both mixture models and DBMC perform well. Conventional K-means performs a little worse (given the number of clusters as a priori). In our analysis it happened that some initial values produced very different (worse) clustering results by the conventional K-means. In general, when the sample size is large and the model provides a reasonable description of the data, mixture models (model-based methods in general) perform well. When the clusters are restricted to globular regions, K-means should work well. In our analysis of the Pima-Surui dataset, our sample size was not small (25 and 21 for two clusters) and the cluster shapes were convex (data not shown), so it is not surprising that both mixture models and K-means performed well.
We used cosine similarity to measure 'similarity' between individuals. Because cosine similarity is easy to interpret and simple to compute, it is widely used in text mining and information retrieval [14,18,19]. It is natural to measure 'similarity' between vectors by their inner product. Cosine is closely related to inner product and correlation. If the vectors have unit length, cosine is equivalent to inner product. If the vectors are centred, cosine is the same as correlation.
Our strategy can be used for identifying populations and assigning individuals in situations where there is little information about population structure. It should also be useful in situations where cryptic population structure is a concern, such as in case-control studies in association mapping.
In summary, we find that the strategy we have described in this paper has the ability to identify population structure, make correct inferences of the number of subpopulations and assign individuals to their corresponding subpopulation. Most of all, it is model free and does not depend on any genetics assumptions. Although it has several advantages over its parametric counterpart, as pointed out by Tang et al.: [12] 'no one method is universally preferable'; however, it provides a useful alternative to analyse genetic data for population structure inferences.
Appendix 1
A variant of the K-means method
K-means is a commonly used non-parametric clustering method, but it has the following drawbacks:
(1) The initial partition may affect the results. Randomisation is often used but has limited success [13].
(2) The procedure may not converge. If the procedure is not well defined, it is quite possible for the procedure to oscillate indefinitely between two or more partitions and never converge. This defect was recognised by the developer of the K-means method [50].
(3) It cannot determine the number of clusters, which is either preset or visually determined.
We propose a clustering method which is based on K-means and can avoid the above drawbacks. The basic idea of our method is that to identify a cluster starting from the point with the highest density around it in the current dataset. To be more specific, suppose we are given n data points X1, X2,..., Xn. Let π1, π2,..., πk denote a partitioning of the data into k disjoint clusters such that
The algorithm of this method (density-based mean clustering [DBMC]) is as follows. Vary the total number of clusters from k = 1, 2,..., K. For each k, perform the following procedure:
1. For every data point in the sample, calculate the distance to its mth (usually three or four) nearest neighbour and identify the point that has the smallest value (highest density). Choose this point as the starting point.
2. Find the point nearest to the starting point and merge these two points to form a cluster. Repeat until all the points are in the cluster. This results in a sequence of nested clusters.
3. Use the Gap statistic[49] to obtain the cluster size and form one cluster. The Gap statistic uses the criterion of between-to-total variance for the goodness of a cluster. The Gap statistic selects the optimal cluster among the nested clusters as the one with the biggest difference (Gap) between the observed and the expected variance by permutation.
4. For the remaining data, repeat steps 1 to 3, until k clusters are found or all points are included (no point left). This leads to the initial partitioning, namely Calculate the centroids for each cluster, denoted as , and set the index of iteration to t = 0.
5. For each data point, find the nearest centroid and assign the point to the cluster represented by this centroid. This results in a new partitioning:
Compute the new centroids and repeat this updating procedure until a certain stopping criterion described below is met.
6. Evaluate the resulting clusters as described below. Among all of the k values studied, select the best clustering according to the quality evaluated in step 6.
For step 5, a stopping criterion is needed, an example is:
where one choice for E() is the objective function discussed by Dhillon and Modha[19] and an alternative candidate is the between-to total (between-cluster plus within-cluster) sums of squares [51]. To ensure the convergence of when we use the between-to total sums of squares as the stopping criterion at each iteration, we choose the new partitioning to have a larger value of the between-to within-sums of squares, or else the iteration stops. Therefore, the algorithm outlined above never results in a decrease in the E(.) value, which is bounded from above by some constant [19]. Therefore, if DBMC is iterated indefinitely, then the value of E(.) will eventually converge. Note that this only means that the algorithm procedure will converge, but it does not imply that the underlying partitioning converges [19,52].
There are a number of methods for estimating the number of clusters [53]. Here, we chose the Gap statistic using the resampling method [54]. Suppose that the maximum possible number of clusters in the data is M. The basic idea of the Gap method for estimating the number of cluster K is to identify , , which provides the strongest significant evidence against the null hypothesis H0 of K = 1, that is, 'no cluster' in the data. The Gap method employs the so-called uniformity hypothesis, which states that the data are sampled from a uniform distribution in the d-dimensional space. It compares an observed internal index, such as the within-clusters sum of squares, to its expectation under a reference null distribution via resampling, and chooses the smallest k which maximises the Gap statistic as the number of clusters [53,54]. The basic idea of the Gap statistics for estimating cluster size[49] is similar to that of the Gap method for estimating the number of clusters.
Appendix 2
Missing genotype imputation
If we consider a person A who has a missing value in marker G, the KNN-based method would find K other persons, who have observed genotypes for marker G and are most similar to A in genotypes in markers other than G. A weighted average of frequencies of genotypes of marker G from the K closest persons is then used as an estimate for the missing value in person A. In the weighted average, the contribution of each person is weighted by the similarity of his/her genotypes to those of person A. We then use SVD on the imputed matrix to obtain the projections of each person onto a reduced space; choose the K nearest neighbours for person A in the reduced space; and repeat the KNN imputation. This iteration is repeated until some preset criteria are met. In summary, the algorithm works as follows.
1. Start with the genotype-person matrix X which has missing values.
2. Compute cosine similarity between x*, who has missing values, and all other persons, using only those coordinates not missing in x*. Identify the K nearest neighbours.
3. Impute the missing coordinates of x* by the weighted average of the corresponding coordinates of the K individuals closest to produce X0. Set i = 0.
4. Apply SVD to the complete matrix Xi to derive the reduced space and identify the K nearest neighbours in the reduced space.
Set i < -i + 1 and repeat steps 3 and 4 until a preset number of iterations is reached, or ||Mi - Mi+1||/||Mi|| is below some threshold, where Mi is the entire imputed matrix at the ith stage.
Acknowledgements
We thank Dr Mário Figueiredo for providing the source code for his mixture model and patiently answering questions; Mr Preslav Nakov for providing information about pre-processing; Dr Thomas Minka for providing the source code for the choice of dimensionality for PCA; Dr Michael Tipping for answering questions about dimension reduction for binary data; Dr Andrew Schein for providing the code for logistic PCA; Dr Noah Rosenberg for providing the data; Drs Rebecka Jörnsten and Hongyuan Zha for discussion on clustering number selection; Dr Andrew Pakstis for providing information about marker selection; and Dr Baolin Wu for general discussions. This work was supported in part by National Institutes of Health grants GM59507 and GM57672.
References
- Corander J, Waldmann P, Sillanpaa MJ. Bayesian analysis of genetic differentiation between populations. Genetics. 2003;163:367–374. doi: 10.1093/genetics/163.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Hattangadi N, Lane B. et al. Methods for high-density admixture mapping of disease genes. Am J Hum Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Burke T, Elo K. et al. Empirical evaluation of genetic clustering methods using multilocus genotypes from 20 chicken breeds. Genetics. 2001;159:699–713. doi: 10.1093/genetics/159.2.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Pritchard JK, Weber JL. et al. Genetic structure of human populations. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- Risch NJ. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–856. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Verdu P, Pakstis AJ. et al. Use of autosomal loci for clustering individuals and populations of East Asian origin. Hum Genet. 2005;117:511–519. doi: 10.1007/s00439-005-1334-8. [DOI] [PubMed] [Google Scholar]
- Dawson KJ, Belkhir K. A Bayesian approach to the identification of panmictic populations and the assignment of individuals. Genet Res. 2001;78:59–77. doi: 10.1017/s001667230100502x. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satten GA, Flanders WD, Yang Q. Accounting for unmeasured population substructure in case-control studies of genetic association using a novel latent-class model. Am J Hum Genet. 2001;68:466–477. doi: 10.1086/318195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Peng J, Wang P, Risch NJ. Estimation of individual admixture: Analytical and study design considerations. Genet Epidemiol. 2005;28:289–301. doi: 10.1002/gepi.20064. [DOI] [PubMed] [Google Scholar]
- Ding CH, X H, H Z, H S. Adaptive dimension reduction for clustering high dimensional data. Proc 2nd IEEE Intl Conf Data Mining. 2002. pp. 147–154.
- Landauer TK, Foltz PW, Laham D. Introduction to latent semantic analysis. Discourse Process. 1998;25:259–284. doi: 10.1080/01638539809545028. [DOI] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB. Maximum likelihood for incomplete data via the EM algorithm (with discussion) J Roy Stat Soc Ser B. 1977;39:1–38. [Google Scholar]
- Moore AW. In: Advances in Neural Information Processing Systems. Kearns M, Solla S, Cohn D, editor. MIT Press, Cambridge, MA; 1999. Very fast EM-based mixture model clustering using multiresolution kd-trees; pp. 543–549. [Google Scholar]
- Bartell BT, Cottrell GW, Belew RK. Latent semantic indexing is an optimal special case of multidimensional scaling. Proc SIGIR'92 Research and Development in Information Retrieval. 1992. pp. 161–167.
- Ding CH. A probabilistic model for dimensionality reduction in information retrieval and filtering. Proc 1st SIAM Computational Information Retrieval Workshop. 2000.
- Dhillon ID, Modha DS. Concept decomposition for large sparse text data using clustering. Machine Learning. 2001;42:143–175. doi: 10.1023/A:1007612920971. [DOI] [Google Scholar]
- Golub G, Van Loan C. Matrix Computations. Johns Hopkins University Press, Baltimore, MD; 1996. [Google Scholar]
- Figueiredo M, Jain AK. Unsupervised learning of finite mixture models. IEEE Trans Pattern Anal Mach Intell. 2002;24:381–396. [Google Scholar]
- Celeux G, Chrétien S, Forbes F, Mkhadri A. A component-wise EM algorithm for mixtures. J Comput Graph Stat. 2001;10:699–712. [Google Scholar]
- Zhu X, Zhang S, Zhao H, Cooper RS. Association mapping, using a mixture model for complex traits. Genet Epidemiol. 2002;23:181–196. doi: 10.1002/gepi.210. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Alter O, Brown PO, Botstein D. Singular value decomposition for genome-wide expression data processing and modeling. Proc Natl Acad Sci USA. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop CM. Variational principle components. Proc 9th International Conference on Artificial Neural Networks. 1999;1:509–514. [Google Scholar]
- Brand ME. Incremental singular value decomposition of uncertain data with missing values. European Conference on Computer Vision (ECCV) 2002;2350:707–720. [Google Scholar]
- Chan KL, Lee TW, Sejnowski TJ. In: Advances in Neural Information Processing Systems. Michael J, Kearns MN, Solla SA, editor. MIT Press, Cambridge, MA; 2003. Handling missing data with variational learning of ICA; pp. 415–420. [Google Scholar]
- Roweis S. In: Advances in Neural Information Processing Systems. Michael J, Kearns MN, Solla SA, editor. MIT Press, Cambridge, MA; 1998. EM algorithms for PCA, SPCA. [Google Scholar]
- Butte AJ, Ye J, Haring HU, Determining significant fold differences in gene expression analysis. Pac Symp Biocomput. 2001. pp. 6–17. [DOI] [PubMed]
- Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Ando RK. Latent semantic space: Iterative scaling improves precision of inter-document similarity measurement. Proc 23rd SIGIR. 2000. pp. 216–223.
- Troyanskaya O, Cantor M, Sherlock G. et al. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. The international HapMap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Li LM, Ward R, Pritchard JK. Informativeness of genetic markers for inference of ancestry. Am J Hum Genet. 2003;73:1402–1422. doi: 10.1086/380416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Wall ME, Rechtsteiner A, Rocha LM. In: A Practical Approach to Microarray Data Analysis. Berrar DP, Dubitzky W, Kluwer MG, editor. Norwell, MA; 2003. Singular value decomposition and principal component analysis. [Google Scholar]
- Nakov P, Popova A, Mateev P. Weight functions impact on LSA performance. Proc RANLP. 2001. pp. 187–193.
- Liu N, Chen L, Wang S. et al. Comparison of single-nucleotide polymorphisms and microsatellites in inference of population structure. BMC Genet. 2005;6:S26. doi: 10.1186/1471-2156-6-S1-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento S, Mirkin B, Moura-Pires F. Modeling proportional membership in fuzzy clustering. IEEE Trans Fuzzy Syst. 2003;11:173–186. doi: 10.1109/TFUZZ.2003.809889. [DOI] [Google Scholar]
- Carreira-Perpinan M. A review of dimension reduction techniques. Technical Report CS-96-09, Department of Computer Science, University of Sheffield, Sheffield, UK. 1997.
- Collins M, Dasgupta S, Schapire RE. A generalization of principal component analysis to the exponential family. Proc NIPS. 2001. pp. 617–624.
- Schein A, Saul L, Ungar L. A generalized linear model for principal component analysis of binary data. Proc of the Ninth International Workshops Artificial Intelligence and Statistics. 2003.
- Tipping M. In: Advances in Neural Information Processing Systems. Michael J, Kearns MN, Solla SA, editor. MIT Press, Cambridge, MA; 1999. Probabilistic visualisation of high-dimensional binary data; pp. 592–598. [Google Scholar]
- Jornsten R, Yu B. Simultaneous gene clustering and subset selection for sample classification via MDL. Bioinformatics. 2003;19:1100–1109. doi: 10.1093/bioinformatics/btg039. [DOI] [PubMed] [Google Scholar]
- Xu G, Zha H, Golub G, Kailath T. Fast algorithms for updating signal subspaces. IEEE Trans Circuits Syst II: Analog and Digital Signal Processing. 1994;41:537–549. doi: 10.1109/82.318942. [DOI] [Google Scholar]
- Minka T. Automatic choice of dimensionality for PCA. Proc of NIPS. 2000. pp. 598–604.
- Hastie T, Tibshirani R, Eisen MB. et al. "Gene shaving" as a method for identifying distinct sets of genes with similar expression patterns. Genome Biol. 2000;1:3.1–3.21. doi: 10.1186/gb-2000-1-2-research0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen J. Some methods for classification and analysis of multivariate observations. Proc 5th Berkeley Symp. 1967;1:281–297. [Google Scholar]
- Calinski RB, Harabasz J. A dendrite method for cluster analysis. Communications in Statistics. 1974;3:1–27. [Google Scholar]
- Pollard D. Quantization and the method of k-means. IEEE Trans Inform Theory. 1982;28:199–205. doi: 10.1109/TIT.1982.1056481. [DOI] [Google Scholar]
- Fridlyand J, Dudoit S. Application of resampling methods to estimate the number of clusters and to improve the accuracy of a clustering method. Technical Report 600, Department of Statistics, University of California, Berkeley, CA. 2001.
- Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. JR Stat Soc Ser B. 2001;63:411–423. doi: 10.1111/1467-9868.00293. [DOI] [Google Scholar]