

Abstract
Polymorphisms in drug transporter genes and/or drug-metabolising enzyme genes may contribute to inter-individual variability in rosiglitazone pharmacokinetics in humans. We sought to determine the joint effects of polymorphisms in the SLCO1B1 drug transporter gene and the cytochrome P450 (CYP) 2C8-metabolising enzyme gene on rosiglitazone pharmacokinetics in healthy volunteers. Healthy Caucasian subjects were prospectively enrolled on the basis of SLCO1B1 521 T > C genotype. Additionally, subjects were genotyped for SLCO1B1 11187 G > A, - 10499 A > C and 388 A > G polymorphisms, and the CYP2C8*3 polymorphism. SLCO1B1 haplotypes and diplotypes were computationally assigned. Rosiglitazone plasma concentrations were determined by high-performance liquid chromatography and analysed using non-compartmental methods. The study population consisted of 26 subjects, with a mean age of 33 ± 9 years, and a mean weight of 66.6 ± 11.7 kg. There were no significant differences in rosiglitazone pharmacokinetic parameters between SLCO1B1 diplotype groups. Subjects with the CYP2C8*1/*3 genotype (n = 7), however, had significantly lower rosiglitazone area under the plasma concentration-time curve (AUC) and significantly higher rosiglitazone oral clearance, compared with CYP2C8 wild-type homozygotes (n = 19). Stepwise linear regression analysis revealed that CYP2C8 genotype (p = 0.006) and weight (p = 0.022) were significant predictors of rosiglitazone AUC (overall p = 0.002; R2 = 41.6 per cent). We concluded that polymorphisms in the CYP2C8 drug-metabolising enzyme gene, but not the SLCO1B1 drug transporter gene, significantly influence rosiglitazone disposition in humans. Future studies examining the influence of CYP2C8 genotypes and haplotypes on thiazolidinedione disposition and response in patients with type 2 diabetes are warranted.
Keywords: rosiglitazone, thiazolidinedione, pharmacokinetic, pharmacogenetic, CYP2C8, SLCO1B1
Introduction
Rosiglitazone is an insulin sensitiser in the thiazolidinedione (TZD) drug class. TZDs stimulate the peroxisome proliferator-activated receptor- γ (PPAR-γ) in the cell nucleus and result in increased transcription of numerous genes involved in glucose and lipid metabolism and adipocyte differentiation [1]. Due to their glucose-lowering and insulin sensitising effects, TZDs are indicated for the treatment of type 2 diabetes [2,3]. Although TZDs are effective anti-diabetic agents, significant inter-individual variability exists in their disposition (ie pharmacokinetics) and response (ie pharmacodynamics) in the body. For example, pharmacokinetic data show that rosiglitazone area under the plasma concentration-time curve (AUC), a measure of plasma drug exposure, varies 2.4-fold between healthy individuals [4]. Rosiglitazone clinical response data show that 55 per cent of patients with type 2 diabetes fail to achieve a 30 mg/dl reduction in fasting plasma glucose following 26 weeks of therapy [3]. It is thought that the field of pharmacogenetics may help to provide insight regarding the extent to which genetic differences between people explain the observed variability in rosiglitazone pharmacokinetics and pharmacodynamics.
To date, many TZD pharmacogenetic investigations have focused on polymorphisms in drug target genes (eg PPAR-γ) and effector protein genes (eg the adiponectin gene) in hopes of explaining drug response variability [5-13]. Although these genes are certainly important, one must also consider the extent to which polymorphisms in drug-metabolising enzyme- and drug transporter genes affect rosiglitazone disposition in the body. Pharmacokinetic differences in plasma drug exposure, as a result of polymorphisms in drug disposition genes, may contribute to inter-individual variability in the glucose-lowering and insulin sensitising effects of the TZDs, and the risk of concentration-dependent side effects.
In terms of drug disposition, rosiglitazone is principally metabolised in the liver by the cytochrome P450 (CYP) 2C8 enzyme, and to a lesser extent by CYP2C9 [3,14]. The most frequently studied variant allele in the CYP2C8 gene is CYP2C8*3, which comprises two linked polymorphisms at codon 139 and codon 399 (Arg139Lys; Lys399Arg). In vitro, the CYP2C8*3 allele has been associated with decreased metabolic activity of the CYP2C8 enzyme [15-17]. In vivo, however, the effects of CYP2C8*3 appear to be substrate dependent, with reports of both increased and decreased metabolism of different CYP2C8 substrates. One clinical study showed that the CYP2C8*3 allele is associated with decreased rosiglitazone plasma drug exposure in healthy volunteers; however, two other studies have shown no significant associations between the CYP2C8*3 allele and rosiglitazone pharmacokinetics in humans [18-20].
In addition to metabolism, recent computational modelling data suggest that rosiglitazone disposition in the body may be influenced by the hepatic drug transporter organic anion-transporting polypeptide 1B1 (OATP1B1), which is responsible for the active uptake of a diverse range of substrates from the blood into the liver [21-23]. OATP1B1 is encoded by the solute carrier organic anion transporter family, member 1B1 (SLCO1B1) gene [24,25]. The most widely studied SLCO1B1 polymorphism is a T to C substitution at nucleotide position 521 (Val174Ala). In vitro, this polymorphism has been associated with decreased function of the OATP1B1 transporter [24,26]. Clinical studies have demonstrated that subjects who carry a variant SLCO1B1 521C allele have increased plasma concentrations of certain OATP1B1 substrates (eg pravastatin and repaglinide), compared with subjects who are wild-type homozygotes [27-30]. These findings speak to the hypothesis that decreased OATP1B1 transporter function, as a result of functional SLCO1B1 polymorphisms, causes decreased substrate uptake in the liver, subsequently leading to higher concentrations of the substrate in plasma. Only one study has been published evaluating the impact of SLCO1B1 polymorphisms on TZD pharmacokinetics, and no significant associations were found [4]. This lack of an association has yet to be replicated in other clinical studies, however, and clinical studies examining the joint effects of SLCO1B1 and CYP2C8 polymorphisms on TZD pharmacokinetics in the same study cohort have not yet been performed.
Taken together, functional polymorphisms in drug metabolism and/or drug transporter genes may explain, in part, the observed pharmacokinetic variability of rosiglitazone in humans. As such, we sought to determine the joint effects of SLCO1B1 and CYP2C8 polymorphisms on inter-individual variability in rosiglitazone pharmacokinetics in healthy volunteers.
Materials and methods
The study was conducted as an open-label, single dose, 24-hour pharmacokinetic study in healthy Caucasian men and women between 18 and 55 years of age. Race was determined by patient self-report. Subjects were required to be of the same race as their parents and grandparents. Subjects were prospectively screened, enrolled and stratified in the study based on SLCO1B1 521 T > C genotype. The clinical study was approved by the University of Colorado Multiple Institutional Review Board, and all subjects provided written, informed consent. Subjects were excluded from the study if they had a body mass index ≥ 35 kg/m2 and/or a current or past clinical history of liver, kidney, cardiovascular, gastrointestinal, endocrine, neurological or haematological disease. Patients with active malignancy, self-reported human immunodeficiency virus positivity, active alcohol abuse, or pregnancy and/or lactation were also excluded from the study. In terms of laboratory tests, subjects were excluded from the study if they had metabolic, renal, hepatic or haematological laboratory tests outside of normal limits. Exclusionary medications included: gemfibrozil, trimethoprim, phenobarbital, primidone, carbamazepine, rifampin, cyclosporine, oral anti-diabetic agents and/or insulin.
Study design
The intensive 24-hour pharmacokinetic study took place at the University of Colorado General Clinical Research Center Inpatient Unit. After an overnight fast, subjects were administered a single, 4 mg oral dose of rosiglitazone with 150 ml of water at 08.00. A standardised meal (50 per cent carbohydrate, 30 per cent protein and 20 per cent fat) was administered two hours after rosiglitazone ingestion. Additional warm meals were provided six, ten and 24 hours after rosiglitazone ingestion. All meals were caffeine free, and subjects were required to abstain from smoking during the 24-hour period. Blood samples (10 ml in ethylene-diaminetetraacetic acid [EDTA] vacuum tubes) were collected pre-rosiglitazone dose and 0.5, 1, 1.5, 2, 3, 4, 5, 7, 9, 12, 16, 20 and 24 hours post-rosiglitazone dose. Plasma was harvested within 30 minutes of sample collection, and plasma samples were stored at -80°C until high-performance liquid chromatography (HPLC) analysis was performed.
Genotyping
After obtaining informed consent, a pre-screening genetic sample was collected from each potential subject via a mouthwash collection method [31]. Subjects were asked vigorously to swish 15 ml of Scope mouthwash (Procter & Gamble, Cincinnati, OH, USA) for one minute and then expectorate into a sterile collection tube. Genomic DNA was isolated from buccal cells in the mouthwash expectorate using a commercially available kit (QIAamp® DNA Mini Kit; Qiagen, Valencia, CA, USA). Subjects were prospectively genotyped and enrolled in the study based on SLCO1B1 521 T > C genotype. The goal was to enrol equal numbers of SLCO1B1 521 T/T homozygotes and SLCO1B1 521C allele carriers (ie 521 T/C and C/C genotypes). Because the SLCO1B1 521 T > C polymorphism is commonly studied in haplotype form, subjects were also genotyped for the following SLCO1B1 polymorphisms: -11187 G > A, -10499 A > C and 388 A > G (Asn130Asp). In terms of CYP2C8, subjects were genotyped for the CYP2C8*3 (Arg139Lys) polymorphism.
All genotyping was performed with polymerase chain reaction (PCR) pyrosequencing analysis. PCR primers, pyrosequencing primers and PCR annealing temperatures used for the genotyping procedures are shown in Table 1. PCR reactions were performed in a final volume of 25 μl that consisted of: 12.5 μl HotStarTaq™ Master Mix (Qiagen), 10 pmol of each primer, 8.5 μl water and 50-100 ng genomic DNA. The standard PCR cycling conditions were: initial denaturation at 95°C for 15 minutes, 40 cycles of denaturation at 95°C for 30 seconds, annealing at the designated temperature for 30 seconds and extension at 72°C for one minute, followed by a final extension step at 72°C for seven minutes. Samples were genotyped using a PSQ 96MA genotyping system (Biotage, AB, Uppsala, Sweden) according to standard manufacturer protocol. Genotype determinations were made using automated pyrosequencing software (PSQ™ 96MA SNP software v. 2.0; Biotage).
Table 1.
PCR primers, pyrosequencing primers and PCR annealing temperatures used for genotyping
| Polymorphism | Primer sequence (5' to 3') | Annealing temperature |
|---|---|---|
| SLCO1B1 - 11187 G > A | PCR-forward: TGCTGCAACCATATCAACAAA PCR-reverse: Biotin-TGGACATTAAGCTCTCTTCTGAAA Sequencing: ATATATATGTGCATATGTG |
60°C |
| SLCO1B1 -10499 A > C | PCR-forward: Biotin-GTAAAAGCCATTCCCAAAAGTAAG PCR-reverse: CCACCTTATTATGTCATTGCTCTA Sequencing: GTTTTAAGAGTTCACTCCAG |
60°C |
| SLCO1B1 388 A > G | PCR-forward: CATTTCACTTTTACCCATC PCR-reverse: Biotin-TACCTTTTCCCACTATCTCA Sequencing: ATTCTAAAGAAACTAAT |
55°C |
| SLCO1B1 521 T > C | PCR-forward: Biotin-AGGAATCTGGGTCATACATGTGG PCR-reverse: CCCCTATTCCACGAAGCATATT Sequencing: AAGCATATTACCCATGAAC |
60°C |
| CYP2C8*3, Argl39Lys | PCR-forward: Biotin-GGCGTTTCTCCCTCACAACCT PCR-reverse: GTCACCCACCCTTGGTTTTTCT Sequencing: CGGTCCTCAATGCTC |
60°C |
Following genotyping, SLCO1B1 haplotypes were computationally assigned using HelixTree® Genetics Analysis Software (Golden Helix, Inc., Bozeman, MT, USA). SLCO1B1 haplotypes were assigned as follows:
*1A (-11187G/ - 10499A/388A/521T);
*1B (-11187G/ - 10499A/388G/521T);
*5 (-11187G/ - 10499A/388A/521C);
*15 (-11187G/ -10499A/388G/521C);
*16 (-11187G/ - 10499C/388G/521C);
*17 (-11187A/ - 10499A/388G/521C); and
*21 (-11187A/ - 10499A/388G/521T).
Rosiglitazone plasma concentration analysis
Rosiglitazone plasma concentrations were determined by HPLC with fluorescence detection using an adaptation of a previously published method [32]. The HPLC system comprised a Beckman 127 binary gradient HPLC pump (Beckman Coulter, Inc., Fullerton, CA, USA), Agilent 1100 series autosampler (G1313A; Agilent Technologies, Inc., Santa Clara, CA, USA) and fluorescence detector (G1321A; Agilent Technologies). Chromatographic data analysis was conducted using ChromPerfect Data Collection and Analysis software (version 3.52; Justice Innovations, Mountain view, CA, USA). Betaxolol (United States Pharmacopeia, Rockville, MD, USA) was used as the internal standard and was dissolved in methanol to make a 25 μg/ml stock solution. Separation was performed using a Hypersil-phenyl column (100 mm × 3 mm, 5 μm; Thermo Scientific, Waltham, MA, USA). The mobile phase consisted of 20 mM sodium acetate buffer (pH = 4.9) and acetonitrile (60:40, v:v) and was run through the system at a flow rate of 0.5 ml/min. Rosiglitazone fluorescence was detected at an excitation wavelength of 235 nm and an emission wavelength of 310 nm. Betaxolol fluorescence was detected at an excitation wavelength of 247 nm and an emission wavelength of 367 nm. Sample preparation was conducted using a liquid-liquid extraction procedure in the following manner: 10 μl betaxolol (250 ng) and 20 μl methanol were added to 200 μl EDTA plasma and were vortexed. Methyl-tert butyl ether (2 ml) was added to each tube and vortexed at high speed for ten seconds. The samples were centrifuged at 2000 × g for five minutes at room temperature. After centrifugation, the aqueous layer was frozen in a dry ice/isopropanol bath for ten minutes. The organic layer was decanted into a glass tube, allowed to equilibrate to room temperature and then dried down for ten minutes at 37°C in a water bath using nitrogen gas. The dried samples were reconstituted with 100 μl mobile phase, transferred into vials, and a 35 μl aliquot was injected onto the column. The lower limit of quantification of the assay was 5 ng/ml.
Pharmacokinetic analysis
Rosiglitazone pharmacokinetic parameters were determined by non-compartmental methods using WinNonlin software 5.0.1 (Pharsight Corp., Mountain View, CA, USA). Rosiglitazone plasma concentration-time curves were generated, and maximum plasma concentration (Cmax) and time to maximum plasma concentration (Tmax) were read from these curves. The linear trapezoidal method was used to calculate the area under the plasma concentration-time curve from 0 to ∞ (AUC0-∞). The half-life (t1/2) was calculated from the following equation: t1/2 = ln2/ke, where ke was calculated using linear regression analysis of the log-linear portion of the concentration-time curve. Total apparent oral clearance (CL/F) of rosiglitazone was calculated as dose (mg)/AUC, and weight-adjusted oral clearance (CL/F/kg) was calculated as [dose (mg)/AUC]/weight (kg).
Statistical analysis
A one-way analysis of variance was used to compare baseline demographics, AUC0-∞, CL/F, CL/F/kg, t1/2, Cmax and Tmax between the three SLCO1B1 diplotype groups. Unpaired Student's t-tests were used to compare baseline demographics and pharmacokinetic parameters between the CYP2C8*3 genotype groups (wild-type homozygotes versus CYP2C8*3 heterozygotes). A stepwise linear regression analysis was used to determine the joint effects of CYP2C8*3 genotype, SLCO1B1 diplotype, age, sex and body weight on rosiglitazone AUC0-∞. All statistical analyses were conducted using SPSS (version 14.0 for Windows; SPSS Inc., Chicago, IL, USA). A p value <0.05 was considered statistically significant.
Results
The study population consisted of 26 healthy Caucasian volunteers (22 women, four men), mean age 33 ± 9 years (range 19 to 55 years), and mean weight 66.6 ± 11.7 kg (range 45.7 to 93.3 kg). In terms of the SLCO1B1 521 T > C screening genotype, 13 subjects were T/T homozygotes, 11 subjects were heterozygotes and two subjects were C/C homozygotes. Subjects were assigned to the following groups based on SLCO1B1 diplotype: Group 1 (*1A/*1A); Group 2 (*1A/*1B or *1B/*1B); and Group 3 (subjects with at least one copy of the *5, *15, *16 or *17 haplotype). The number of subjects in each diplotype group, the specific diplotypes and corresponding baseline characteristics are shown in Table 2. Baseline continuous variables did not differ significantly between the three SLCO1B1 diplotype groups. In the study population, 19 subjects possessed the CYP2C8*1/*1 genotype and seven subjects possessed the CYP2C8*1/*3 genotype. There were no subjects with the CYP2C8*3/*3 genotype in the study population. Average age, average weight and sex distribution were 32 ± 8 years, 65.9 ± 12.4 kg and 16 women, respectively, in the CYP2C8*1/*1 genotype group; and 35 ± 12 years, 68.2 ± 10.4 kg and six women, respectively, in the CYP2C8*1/*3 genotype group. Baseline continuous variables did not differ significantly between CYP2C8 genotype groups. Rosiglitazone plasma concentration-time curves by SLCO1B1 diplotype group and CYP2C8 genotype group are shown in Figures 1 and 2, respectively.
Table 2.
Baseline characteristics by SLCO1B1 diplotype group
| Group 1 n = 6 | Group 2 n = 7 | Group n = 13 | |
|---|---|---|---|
| SLCO1B1 diplotypes | *1A/*1A, n = 6 | *1A/*1B, n = 4 | *1A/*15, n = 2 |
| *1B/*1B, n = 3 | *1A/*16, n = 2 | ||
| *1A/*17, n = 2 | |||
| *1B/*15, n = 1 | |||
| *1B/*16, n = 1 | |||
| *1B/*17, n = 2 | |||
| *5/*15, n = 1 | |||
| *15/*16, n = 1 | |||
| *17/*21, n = 1 | |||
| Age (years) | 33.8 ± 10.8 | 34.9 ± 12.1 | 31.3 ± 7.3 |
| Weight (kg) | 65.9 ± 14 | 65.3 ± 9.6 | 67.5 ± 12.5 |
| Women, n | 5 | 7 | 10 |
Data are presented as mean ± SD or number.
Figure 1.
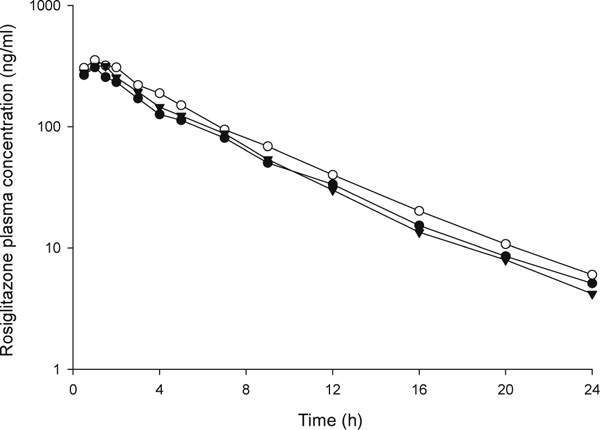
Rosiglitazone plasma concentration-time curves by SLCO1B1 diplotype group. Closed circles represent SLCO1B1 diplotype Group 1 (subjects with the *1A/*1A diplotype). Open circles represent SLCO1B1 diplotype Group 2 (subjects with the *1A/*1B or *1B/*1B diplotype). Closed triangles represent SLCO1B1 diplotype Group 3 (subjects with at least one copy of the *5, *15, *16, or *17 haplotype).
Figure 2.
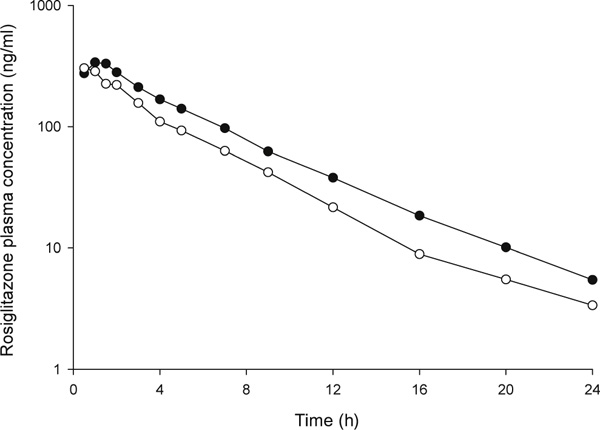
Rosiglitazone plasma concentration-time curves by CYP2C8 genotype. Closed circles represent the CYP2C8*1/*1 genotype group. Open circles represent the CYP2C8*1/*3 genotype group.
In univariate analysis, rosiglitazone pharmacokinetic parameters did not differ significantly between SLCO1B1 diplotype groups (Table 3). By contrast, rosiglitazone pharmacokinetic parameters were significantly different by CYP2C8*3 genotype (Table 4). Specifically, rosiglitazone AUC0-∞ was 29.4 per cent lower, and oral clearance was 38.9 per cent higher, in CYP2C8 heterozygotes compared with CYP2C8*1 homozygotes. After adjustment for weight, rosiglitazone oral clearance remained significantly higher (35.4 per cent) in CYP2C8 heterozygotes compared with CYP2C8*1 homozygotes. Rosiglitazone t1/2 was shorter in heterozygotes versus CYP2C8*1 homozygotes; however, this difference did not reach statistical significance.
Table 3.
Rosiglitazone pharmacokinetic parameters by SLCO1B1 diplotype group
| Pharmacokinetic parameter | Group 1 (*1A/*1A) n = 6 | Group 2 (*1A/*1B or *1B/*1B) n = 7 | Group 3 (*5, *15, *16, or *17 haplotype carriers) n = 13 | p value |
|---|---|---|---|---|
| AUC0-∞ (ng*h/ml) | 1601 ± 451 (1128, 2074) | 2019 ± 624 (1442, 2595) | 1699 ± 417 (1447, 1951) | 0.26 |
| CL/F (ml/h) | 2673 ± 766 (1869, 3477) | 2153 ± 673 (1530, 2775) | 2495 ± 633 (2112, 2877) | 0.37 |
| CL/F/kg (ml/h/kg) | 40.9 ± 9.4 (31, 50.8) | 33 ± 9.2 (24.5, 41.5) | 37.5 ± 10 (31.5, 43.6) | 0.35 |
| t1/2 (h) | 4.1 ± 0.9 (3.2, 5) | 3.9 ± 0.8 (3.2, 4.7) | 3.8 ± 0.8 (3.3, 4.3) | 0.78 |
| Cmax (ng/ml) | 337 ± 125 (206, 469) | 409 ± 86 (329, 489) | 383 ± 113 (315, 451) | 0.50 |
| Tmax (hr) | 1.0 ± 0.6 (0.4, 1.6) | 0.8 ± 0.4 (0.4, 1.2) | 0.9 ± 0.5 (0.7, 1.2) | 0.68 |
Data are presented as mean ± SD, along with 95% confidence intervals. Abbreviations: AUC, area under the plasma concentration-time curve; CL/F, apparent oral clearance; CL/F/kg, weight-adjusted oral clearance; t1/2, half-life; Cmax, maximum plasma concentration; Tmax, time to maximum plasma concentration.
Table 4.
Rosiglitazone pharmacokinetic parameters by CYP2C8 genotype
| Pharmacokinetic parameter | CYP2C8*1/*1 n = 19 | CYP2C8*1/*3 n = 7 | p value |
|---|---|---|---|
| AUC0-∞ (ng*h/ml) | 1914 ± 469 (1688, 2140) | 1352 ± 300 (1075, 1629) | 0.002 |
| C1/F (ml/h) | 2212 ± 550 (1947, 2477) | 3072 ± 602 (2516, 3629) | 0.008 |
| CL/F/kg (ml/h/kg) | 33.9 ± 7.1 (30.4, 37.3) | 45.9 ± 11 (35.7, 56) | 0.03 |
| t1/2 (h) | 4.0 ± 0.8 (3.7, 4.4) | 3.5 ± 0.7 (2.9, 4.1) | 0.09 |
| Cmax (ng/ml) | 402 ± 115 (347, 458) | 317 ± 56 (266, 369) | 0.02 |
| Tmax (h) | 1.0 ± 0.5 (0.7, 1.2) | 0.7 ± 0.3 (0.5, 1.0) | 0.13 |
Data are presented as mean ± SD, along with 95% confidence intervals. Abbreviations: AUC, area under the plasma concentration-time curve; CL/F, apparent oral clearance; CL/F/kg, weight-adjusted oral clearance; t1/2, half-life; Cmax, maximum plasma concentration; Tmax, time to maximum plasma concentration.
In stepwise linear regression analysis, both CYP2C8 genotype (coefficient = -0.479; p = 0.006) and weight (coefficient = -0.392; p = 0.022) were significantly associated with rosiglitazone AUC0-∞ (overall model significance = 0.002; R2 = 0.416).
Discussion
Our intensive 24-hour pharmacokinetic study revealed that variation in the CYP2C8 drug-metabolising enzyme gene, but not the SLCO1B1 drug transporter gene, was associated with inter-individual differences in the pharmacokinetics of rosiglitazone in healthy volunteers. Previous in vitro data show that the CYP2C8*3 allele is associated with decreased metabolic activity for some CYP2C8 substrates. Therefore, we would expect this to translate clinically into higher rosiglitazone AUC and decreased rosiglitazone clearance in subjects carrying a variant CYP2C8*3 allele. In a population of healthy volunteers, however, we found that the CYP2C8*3 allele was associated with lower rosiglitazone AUC and higher rosiglitazone oral clearance, compared with wild-type homozygotes.
Although these clinical findings contrast with in vitro data, our findings are consistent with recent data published by Kirchheiner and colleagues showing that the variant CYP2C8*3 allele is associated with decreased rosiglitazone plasma exposure, and increased rosiglitazone oral clearance, in healthy volunteers [18]. The most marked pharmacokinetic effects observed in that study were in CYP2C8*3 homozygotes, with intermediate effects in subjects with the CYP2C8*1/*3 genotype. Although we did not have any CYP2C8*3 homozygotes in our study population, our clearance results in heterozygotes were consistent with Kirchheiner et al.'s population pharmacokinetic analysis, which showed total clearance values of 2980 ml/h and 2420 ml/h in CYP2C8 heterozygotes and CYP2C8*1 homozygotes, respectively. Kirchheiner et al. also found that the significant predictors of rosiglitazone AUC in linear regression analysis were CYP2C8*3 genotype and body weight, which explained 48 per cent of the variability in rosiglitazone AUC. We observed the same independent predictors of rosiglitazone AUC in our study, and the R2 value was of similar magnitude.
The finding that the CYP2C8*3 allele is associated with increased metabolism in vivo has also been reported for pioglitazone, another member of the TZD class [33]. Like rosiglitazone, pioglitazone is principally metabolised in the liver by CYP2C8, and to a lesser extent by CYP2C9 and CYP3A4. Recently, Tornio and colleagues reported an association between CYP2C8*3 genotype and pioglitazone pharmacokinetics in healthy volunteers [33]. Specifically, plasma concentrations of pioglitazone were lower in CYP2C8*3 homozygotes and heterozygotes than in wild-type homozygotes. The weight-adjusted AUC was 26 per cent lower in the heterozygotes than in wild-type homozygotes, which is consistent with the magnitude of effect observed in our study of rosiglitazone.
While drug-metabolising enzymes are routinely studied in relation to substrate disposition, the contribution of drug transporters to substrate pharmacokinetics merits attention as well. In regard to rosiglitazone, a pharmacophore modelling study provided intriguing data suggesting that rosiglitazone may be a substrate for OATP1B1 [21]. The notion of rosiglitazone as a putative OATP1B1 substrate was further strengthened by existing drug-drug interaction data showing that rosiglitazone concentrations were increased following administration of gemfibrozil, which has both OATP1B1- and CYP2C8-inhibitory actions [34]. Polymorphisms in SLCO1B1, particularly 521 T > C, have been shown significantly to alter the pharmacokinetics of substrates such as pravastatin and repaglinide [28-30]. Therefore, it can be hypothesised that if rosiglitazone is indeed an OATP1B1 substrate, plasma drug exposure would be higher in subjects carrying a variant SLCO1B1 521C allele, as a result of diminished substrate uptake in the liver. In our study population, however, univariate and multivariate analysis revealed no significant associations between SLCO1B1 genotype and rosiglitazone pharmacokinetics. These results are consistent with a recent report by Kalliokoski et al. which showed no association between SLCO1B1 521 T > C genotype and rosiglitazone or pioglitazone pharmacokinetics in healthy volunteers [4]. Given our similar study designs and SLCO1B1 haplotype assignments, our work provides further evidence of a lack of association between SLCO1B1 polymorphisms and rosiglitazone pharmacokinetics, even after controlling for the potential confounding effects of the CYP2C8*3 polymorphism. It is important to note, however, that our small sample size and multiple statistical tests may have limited our power to detect differences in rosiglitazone pharmacokinetics between SLCO1B1 diplotype groups. Thus, additional studies, using a larger sample size, are needed conclusively to determine the joint effects of SLCO1B1 and CYP2C8 polymorphisms on rosiglitazone pharmacokinetics. Additionally, in-depth in vitro and in vivo mechanistic investigations are needed to clarify the role of OATP1B1 in thiazolidinedione disposition.
The issue that remains to be determined is whether the CYP2C8*3 polymorphism will significantly influence rosiglitazone pharmacodynamics in patients with type 2 diabetes. To date, all of the rosiglitazone drug metabolism pharmacokinetic-pharmacogenetic studies have been conducted in healthy volunteers. As such, the drugs have only been given for a single dose, or for a short duration of multiple doses. Thus, it is unclear if the observed reductions in rosiglitazone plasma exposure in subjects carrying the variant CYP2C8*3 allele will have an impact on the pharmacodynamic (ie glucose-lowering and insulin sensitising) effects of rosiglitazone in patients with type 2 diabetes. It is also not known whether patients carrying a CYP2C8*3 allele will be at a lower risk of concentration-dependent side effects such as oedema and weight gain. Furthermore, the mechanism(s) by which the CYP2C8*3 polymorphism is associated with increased metabolism in humans is still unclear. Recent data by Rodriguez-Antona and colleagues, however, have shed some light on this subject, suggesting that two novel CYP2C8 haplotypes (ie haplotypes B and C), rather than the CYP2C8*3 allele alone, are important determinants of substrate disposition in vitro and in vivo[35].
In sum, our study shows that the CYP2C8*3 allele, but not polymorphisms in the SLCO1B1 drug transporter gene, is associated with inter-individual differences in the pharmacokinetics of rosiglitazone in healthy volunteers. Future studies examining the effects of the CYP2C8*3 polymorphism, and novel CYP2C8 haplotypes, on TZD disposition and response in healthy volunteers and patients with type 2 diabetes are warranted.
Acknowledgments
We would like to thank Courtney V. Fletcher, Pharm.D., for his support in terms of study design and rosiglitazone analytical methods. This work was supported, in part, by a University of Colorado Denver General Clinical Research Center Pilot and Feasibility Grant, and University of Colorado Denver General Clinical Research Center Grant M01 RR00051.
References
- Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- Actos Prescribing Information. Takeda Pharmaceuticals America, Inc.; 2007. [Google Scholar]
- Avandia Prescribing Information. GlaxoSmithKline; 2007. [Google Scholar]
- Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. No significant effect of SLCO1B1 polymorphism on the pharmacokinetics of rosiglitazone and pioglitazone. Br J Clin Pharmacol. 2008;65:78–86. doi: 10.1111/j.1365-2125.2007.02986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluher M, Lubben G, Paschke R. Analysis of the relationship between the Pro12Ala variant in the PPAR-gamma2 gene and the response rate to therapy with pioglitazone in patients with type 2 diabetes. Diabetes Care. 2003;26:825–831. doi: 10.2337/diacare.26.3.825. [DOI] [PubMed] [Google Scholar]
- Snitker S, Watanabe RM, Ani I. et al. Changes in insulin sensitivity in response to troglitazone do not differ between subjects with and without the common, functional Pro12Ala peroxisome proliferator-activated receptor-gamma2 gene variant: Results from the Troglitazone in Prevention of Diabetes (TRIPOD) study. Diabetes Care. 2004;27:1365–1368. doi: 10.2337/diacare.27.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang ES, Park SY, Kim HJ. et al. Effects of Pro12Ala polymorphism of peroxisome proliferator-activated receptor gamma2 gene on rosiglitazone response in type 2 diabetes. Clin Pharmacol Ther. 2005;78:202–208. doi: 10.1016/j.clpt.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Kang ES, Park SY, Kim HJ. et al. The influence of adiponectin gene polymorphism on the rosiglitazone response in patients with type 2 diabetes. Diabetes Care. 2005;28:1139–1144. doi: 10.2337/diacare.28.5.1139. [DOI] [PubMed] [Google Scholar]
- Kang ES, Cha BS, Kim HJ. et al. The 11482G > A polymorphism in the perilipin gene is associated with weight gain with rosiglitazone treatment in type 2 diabetes. Diabetes Care. 2006;29:1320–1324. doi: 10.2337/dc05-2466. [DOI] [PubMed] [Google Scholar]
- Florez JC, Jablonski KA, Sun MW. et al. Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab. 2007;92:1502–1509. doi: 10.1210/jc.2006-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolford JK, Yeatts KA, Dhanjal SK. et al. Sequence variation in PPARG may underlie differential response to troglitazone. Diabetes. 2005;54:3319–3325. doi: 10.2337/diabetes.54.11.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang X, Zhang Q, Ma Z. Response to pioglitazone treatment is associated with the lipoprotein lipase S447X variant in subjects with type 2 diabetes mellitus. Int J Clin Pract. 2007;61:552–557. doi: 10.1111/j.1742-1241.2006.01242.x. [DOI] [PubMed] [Google Scholar]
- Hansen L, Ekstrom CT, Tabanera YPR. et al. The Pro12Ala variant of the PPARG gene is a risk factor for peroxisome proliferator-activated receptor-gamma/alpha agonist-induced edema in type 2 diabetic patients. J Clin Endocrinol Metab. 2006;91:3446–3450. doi: 10.1210/jc.2006-0590. [DOI] [PubMed] [Google Scholar]
- Baldwin SJ, Clarke SE, Chenery RJ. Characterization of the cytochrome P450 enzymes involved in the in vitro metabolism of rosiglitazone. Br J Clin Pharmacol. 1999;48:424–432. doi: 10.1046/j.1365-2125.1999.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahadur N, Leathart JB, Mutch E. et al. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem Pharmacol. 2002;64:1579–1589. doi: 10.1016/S0006-2952(02)01354-0. [DOI] [PubMed] [Google Scholar]
- Dai D, Zeldin DC, Blaisdell JA. et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11:597–607. doi: 10.1097/00008571-200110000-00006. [DOI] [PubMed] [Google Scholar]
- Soyama A, Saito Y, Hanioka N. et al. Non-synonymous single nucleotide alterations found in the CYP2C8 gene result in reduced in vitro paclitaxel metabolism. Biol Pharm Bull. 2001;24:1427–1430. doi: 10.1248/bpb.24.1427. [DOI] [PubMed] [Google Scholar]
- Kirchheiner J, Thomas S, Bauer S. et al. Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin Pharmacol Ther. 2006;80:657–667. doi: 10.1016/j.clpt.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Hruska MW, Cheong JA, Amico JA. et al. Effect of CYP2C8 genotype on rosiglitazone pharmacokinetics. Clin PharmacolTher. 2005;P36:PI–107. [Google Scholar]
- Pedersen RS, Damkier P, Brosen K. The effects of human CYP2C8 genotype and fluvoxamine on the pharmacokinetics of rosiglitazone in healthy subjects. Br J Clin Pharmacol. 2006;62:682–689. doi: 10.1111/j.1365-2125.2006.02706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Pang KS, Swaan PW, Ekins S. Comparative pharmacophore modeling of organic anion transporting polypeptides: A meta-analysis of rat Oatp1a1 and human OATP1B1. J Pharmacol ExpTher. 2005;314:533–541. doi: 10.1124/jpet.104.082370. [DOI] [PubMed] [Google Scholar]
- Ho RH, Kim RB. Transporters and drug therapy: Implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78:260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–446. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Tirona RG, Leake BF, Merino G, Kim RB. Polymorphisms in OATP-C: Identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J Biol Chem. 2001;276:35669–35675. doi: 10.1074/jbc.M103792200. [DOI] [PubMed] [Google Scholar]
- Konig J, Seithel A, Gradhand U, Fromm MF. Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:432–443. doi: 10.1007/s00210-006-0040-y. [DOI] [PubMed] [Google Scholar]
- Kameyama Y, Yamashita K, Kobayashi K. et al. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15 + C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005;15:513–522. doi: 10.1097/01.fpc.0000170913.73780.5f. [DOI] [PubMed] [Google Scholar]
- Kivisto KT, Niemi M. Influence of drug transporter polymorphisms on pravastatin pharmacokinetics in humans. Pharm Res. 2007;24:239–247. doi: 10.1007/s11095-006-9159-2. [DOI] [PubMed] [Google Scholar]
- Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics and pharmacodynamics of repaglinide and nateglinide. J Clin Pharmacol. 2008;48:311–321. doi: 10.1177/0091270007311569. [DOI] [PubMed] [Google Scholar]
- Niemi M, Schaeffeler E, Lang T. et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-CSLCO1B1) Pharmacogenetics. 2004;14:429–440. doi: 10.1097/01.fpc.0000114750.08559.32. [DOI] [PubMed] [Google Scholar]
- Niemi M, Backman JT, Kajosaari LI. et al. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther. 2005;77:468–478. doi: 10.1016/j.clpt.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Andrisin TE, Humma LM, Johnson JA. Collection of genomic DNA by the noninvasive mouthwash method for use in pharmacogenetic studies. Pharmacotherapy. 2002;22:954–960. doi: 10.1592/phco.22.12.954.33598. [DOI] [PubMed] [Google Scholar]
- Hruska MW, Frye RF. Simplified method for determination of rosiglitazone in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;803:317–320. doi: 10.1016/j.jchromb.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Tornio A, Niemi M, Neuvonen PJ, Backman JT. Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab Dispos. 2008;36:73–80. doi: 10.1124/dmd.107.018010. [DOI] [PubMed] [Google Scholar]
- Niemi M, Backman JT, Granfors M. et al. Gemfibrozil considerably increases the plasma concentrations of rosiglitazone. Diabetologia. 2003;46:1319–1323. doi: 10.1007/s00125-003-1181-x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Antona C, Niemi M, Backman JT. et al. Characterization of novel CYP2C8 haplotypes and their contribution to paclitaxel and repaglinide metabolism. Pharmacogenomics J. 2007;8:268–277. doi: 10.1038/sj.tpj.6500482. [DOI] [PubMed] [Google Scholar]