

An abbreviated history of innovation in regulatory science
The mission of the US Food and Drug Administration (FDA) makes the FDA, in many ways, unique among federal agencies. As both a regulatory and a science agency, the FDA is charged with both protecting public health through regulation and enforcement and advancing public health by catalysing innovations in product (eg drug) development. To that end, the FDA's history has been hallmarked with patient-centred programmes and policies meant to coalesce innovation and scientific rigour in a way that is translatable to individual patient populations.
The Center for Drug Evaluation and Research (CDER) is the wing of the FDA that regulates over-the-counter and prescription drugs to ensure that they are safe and effective. CDER's recent initiatives are indicative of its commitment to innovation and improved patient outcomes. These include such programmes as the Critical Path, Safety First, Safe Use and Sentinel Initiatives. In the early period after the completion of the Human Genome Project, CDER embarked on one of its most forward-thinking infrastructure building projects to date. As pharmaceutical companies began to conduct exploratory genomics and pharmacogenetics studies in the context of their drug development programmes, it became clear to leaders in CDER and the Office of Clinical Pharmacology (OCP) within CDER that the Center needed to: 1) encourage companies to continue to evolve the science, with the ultimate goal of enhancing drug product development; and 2) develop internal FDA expertise to deal with the myriad complexities of analysing and interpreting pharmacogenetic, genomic, proteomic and like data. To that end, the Voluntary Genomic Data Submissions (VGDS) programme was developed in the early- to mid-2000s, to allow drug companies, consortia, academic researchers and individuals to engage in scientific exchange with the FDA, and to allow FDA regulatory scientists to gain experience with data in this then nascent field [1].
In its early days, VGDS were largely focused on technical aspects of platforms used to generate genomic-era data. Since the first VGDS in 2004, however, the FDA has received over 40 submissions, which have become more applied in nature. That is, more recent VGDS have been concerned with the practical application of genetic or biomarker information to drug development. Over the past five years, there has been significant growth in the use of pharmacogenetic principles in drug development. This has established the need for regulatory scientists with a keen understanding of pharmacogenetics, clinical pharmacology, population science/epidemiology, clinical trial design and pharmacotherapy (ie clinical practice). The Genomics Group within the OCP in CDER is a prototype regulatory review group within which such skill sets are being integrated and further developed.
The OCP Genomics Group: The clinical pharmacogenetics prototype
The stated mission of the OCP Genomics Group is: 'to advance personalised health for the benefit of patients and society'. The group's vision within the drug development and public health enterprises is the integration of pharmacogenetics and genomics in the discovery, development, regulation and safe and effective use of medications. To achieve this vision, the group has as its objectives to: 1) develop an integrated, translational regulatory review paradigm to enhance drug and biologic product development; 2) train regulatory scientists in pharmacogenetics and related disciplines; 3) conduct and disseminate the results of high-quality research to optimise clinical knowledge of the risk/benefit of products throughout their life cycles; and 4) develop regulatory policies and procedures to enhance the utilisation of genomics and related disciplines in drug discovery, development, regulation and utilisation.
Currently, the Genomics Group reviewers are integral members of the CDER cross-disciplinary review teams that regulate drug products in the neurology, cardiovascular, rheumatology, gastrointestinal, antiviral and oncology therapeutic areas. Opportunities for growth in the future are expected to include psychiatry, metabolic and endocrine diseases, pulmonology/allergy and biologics across all therapeutic areas.
With respect to enhancing drug product development in the context of regulatory review work, the Genomics Group is focused on two functions. First, reviewers work with drug companies and researchers in the investigational new drug (IND) application stage, to minimise the likelihood that experiments and research protocols will result in imperfect, ambiguous and uninterpretable data related to pharmacogenetics and applied biomarkers. At this stage, Genomics Group reviewers provide regulatory advice on everything from sample collection to the appropriateness of biomarker selection, to study design methodology. Secondly, the reviewers analyse and interpret data in regulatory submissions (eg new drug applications [NDAs] and therapeutic biologic applications [BLAs]) to support regulatory decisions as part of the cross-disciplinary review team. This includes medical officers, clinical pharmacologists, statisticians, preclinical pharmacologists/toxicologists, and other regulatory scientists.
In order to provide useful advice to the developers of therapeutic products at the IND stage and interpret data to support regulatory decisions on NDA and BLA submissions, Genomics Group reviewers essentially engage in five exercises to create biologically meaningful contexts for the evaluation of data: 1) consideration of all products of the genome; 2) consideration of all genomes; 3) integrative biology; 4) constructive pharmacology; and 5) translational analyses. These are briefly described below.
Consideration of all products of the genome
'Genomics' review is essentially a misnomer, as it implies that Genomics Group reviewers limit their purview to data such as those generated from microarray or other expression platforms. Rather, we use the term 'genomics' expansively to include all products of the human genome. We consider the impact of genetic variations on disease heterogeneity, drug exposure variability and differential therapeutic or toxic responses, we also consider gene and protein expression as prognosticators of clinical outcomes, predictors of drug response or measures of drug activity important for demonstrating proof-of-concept or dose selection. Proteomic and metabolomic data in the context of a drug or biologic submission would also be reviewed where appropriate.
Consideration of all genomes
There is complex interplay between multiple genomes in certain therapeutic areas. For example, in areas such as HIV, hepatitis C virus (HCV) and oncology drug development, we are not only interested in whether variations in host/germline DNA are important correlates of drug response, we are also interested in whether viral genotypes or somatic mutations modify responses to therapeutic interventions. As has been recently demonstrated, HIV tropism, HCV genotype and tumour mutations (eg KRAS) are important determinants of responses to such drugs as HIV entry inhibitors (eg maraviroc), anti-hepatitis agents (eg ribavirin/interferon) and oncology agents (eg panitumumab, cetuximab). It would be inappropriate singly to consider host/germline DNA variants in drug development programmes where variability in viral or tumour genomes may also be important in modulating responses. Failure to consider all genomes of relevance jointly may lead to failure to demonstrate adequate benefit-over-risk and failure to optimise therapy for a given patient population.
Integrative biology
Sound regulatory advice on selection of appropriate pre-pivotal study designs (eg pharmacokinetics [PK], proof-of-concept, dose-selection) through the use of pharmacogenetic and biomarker information is highly reliant on expansive knowledge of the drug (to the extent possible at that time in its development) and of disease biology. As such, our reviewers constantly survey and critically evaluate the biomedical literature and internal FDA datasets with respect to genotype-phenotype relationships, disease pathogenesis and drug/biological pathways. The clear advantage of this approach to institutionalising an expansive knowledge management system is to allow for the consideration of 'priors' in regulatory review. For example, prior understanding of biopathways, the relationship between candidate genes/variants and drug response phenotypes, or mechanistic insights into biological heterogeneity can allow for more informed study designs and interpretation of NDA/BLA data.
Constructive pharmacology
Early phase clinical studies in drug development are largely designed to define the PK, pharmacodynamics (PD) and exposure-response relationship for a new drug, and are generally considered exploratory in nature. Furthermore, in registration studies meant to provide definitive evidence in support of a drug approval, pharmacogenetics and biomarker studies are predominantly also exploratory (eg no pre-specified statistical analysis plan). Therefore, when pharmacogenetic associations are uncovered in these contexts, it becomes critical to ensure that the findings are not spurious. For example, if an exploratory analysis demonstrates that therapeutic benefit is limited to a specific genetic subgroup, we must be utterly convinced that this is not a chance finding because of the public health implications of 'getting it wrong'.
Constructive pharmacology refers to the practice of Genomics Group reviewers to use the totality of what is known for the drug being developed--from a pre-clinical and clinical pharmacology standpoint--to establish or refute biological plausibility for the observed pharmacogenetic association, PD response, differential response by race/ethnicity, or safety signal in the studied population. It becomes critical to apply what is observed directly from clinical pharmacology, toxicology or drug interaction studies, or that can be expected through pharmacometric modelling and simulation, to the interpretation of the observed phenotype uncovered in these so-called exploratory studies. Exploratory studies may ultimately be used to support regulatory decisions related to approvability. Therefore, constructive pharmacology is necessary to provide pharmacological context to observed drug response phenotypes in both early exploratory and later phase clinical trials.
Translational analyses
The above exercises serve as a framework for either provision of regulatory advice during drug product development (eg through IND reviews) or for data interpretation in regulatory submissions (NDAs and BLAs). We coined the term 'translational analyses' to mean the synthesis of information into a regulatory point of view to be shared with the CDER review team and sponsors of therapeutic submissions. Translational analyses go beyond traditional statistical analyses, although statistical and other quantitative methodologies may be employed. Translational analyses integrate information from in vitro, in silico, pre-clinical, ex vivo and PK/PD studies, late phase clinical trials and the curated literature to provide clear, pragmatic advice or interpretation of data in the form of a review work product that becomes part of the regulatory history of the submission. Translational analyses may also be conducted for drugs already approved by the FDA when evaluating whether or not a label should be updated to include pharmacogenetic data. In this case, structured question-based review frameworks, such as our previously described 'pharmacogenetic pyramid' may be useful [2].
Shortening the lag on the critical path to personalised medicine
'Personalised medicine' (PM) means different things to different people. From our standpoint, PM is largely the development and delivery of safe and effective treatments to the right patient subgroups, at the right dose, at the right time in their disease process. Unfortunately, there are significant bottlenecks on the critical path to PM. At the front end, a well-recognised bottleneck is the 'valley of death' that companies must traverse in successfully bringing a new drug to market. Over the past decade, there has been a decline in the number of novel drugs approved each year by the FDA [3]. This reduction in the number of approvals is not due to a changing evidentiary standard at the FDA, however, but rather to excessively high attrition rates (~90 per cent) across the phases of drug development due to lack of efficacy or differentiation, or the presence of a notable safety signal [4]. Rational use of pharmacogenetics principles and application of biomarkers may reduce attrition rates through maximising knowledge about the compound being tested throughout its development life. At the back end, a second bottleneck lies in the translation of pharmacogenetics into clinical practice. We have previously described what we believe to be some of the barriers to the translation of pharmacogenetics into clinical practice [2]. A major obstacle is conflicting points of view on how to generate evidence of clinical utility for pharmacogenetics. While clinical trialists promulgate the randomised controlled trial (RCT) as the gold standard for evidence generation, it should be noted that for some pharmacogenetics questions (eg related to safety), RCTs may not be feasible. Furthermore, the current research infrastructure does not explicitly incentivise the conduct of genotype-stratified, prospective RCTs for pharmacogenetics. In many cases, a 'totality of evidence' standard can be applied that synthesises mechanistic biological data and clinical sources. For example, the utility of certain pharmacogenetic tests is likely to be evaluable in large observational studies conducted in such settings as pharmacy benefits management systems or integrated managed care organisations. This is especially probable in the dawning era of the electronic medical record. Willingness to accept data from well-conducted observational studies with rigorous methodology, combined with an understanding of the underlying biology, will be a prerequisite to integration of PM into clinical practice in a meaningful way. The critical path to PM is cyclical (Figure 1). Each phase of this cycle informs the next. As a translator of information across this pathway, the clinical pharmacogeneticist will be an integral part of this process. The FDA remains committed to the development of such translators in the regulatory science space, with the ultimate expectation of improving efficiency in the development of safe and effective medications for individuals.
Figure 1.
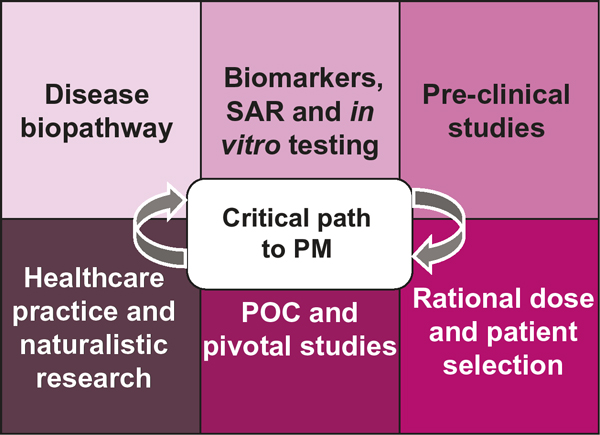
The critical path to personalised medicine. Key: PM, personalised medicine; POC, proof-of-concept; SAR, structure-activity relationship.
References
- Orr MS, Goodsaidm F, Amur S, Rudman A. et al. The experience with voluntary genomic data submissions at the FDA and a vision for the future of the voluntary data submission program. Clin Pharmacol Ther. 2007;81:294–297. doi: 10.1038/sj.clpt.6100053. [DOI] [PubMed] [Google Scholar]
- Zineh I, Lesko LJ. Pharmacogenetics in medicine: Barriers, critical factors and a framework for dialogue. Per Med. 2009;6:359–361. doi: 10.2217/pme.09.27. [DOI] [PubMed] [Google Scholar]
- Hughes B. FDA drug approvals. Nat Rev Drug Discov. 2008;8:93–96. doi: 10.1038/nrd2813. [DOI] [PubMed] [Google Scholar]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–5. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]