

Abstract
The hereditary hyperferritinaemia-cataract syndrome (HHCS) is characterised by an autosomal dominant cataract and high levels of serum ferritin without iron overload. The cataract develops due to L-ferritin deposits in the lens and its pulverulent aspect is pathognomonic. The syndrome is caused by mutations within the iron-responsive element of L-ferritin. These mutations prevent efficient binding of iron regulatory proteins 1 and 2 to the IRE in L-ferritin mRNA, resulting in an unleashed ferritin translation. This paper reviews all 31 mutations (27 single nucleotide transitions and four deletions) that have been described since 1995. Laboratory test showing hyperferritinaemia, normal serum iron and normal transferrin saturation are indicative for HHCS after exclusion of other causes of increased ferritin levels (inflammation, malignancy, alcoholic liver disease) and should prompt an ophthalmological consultation for diagnostic confirmation. Invasive diagnostics such as liver biopsy are not indicated. HHCS is an important differential diagnosis of hyperferritinaemia. Haematologists, gastroenterologists and ophthalmologists should be aware of this syndrome to spare patients from further invasive diagnosis (liver biopsy), and also from a false diagnosis of hereditary haemochromatosis followed by venesections. Patients diagnosed with HHCS should be counselled regarding the relative harmlessness of this genetic disease, with early cataract surgery as the only clinical consequence.
Keywords: ferritin, hyperferritinaemia, cataract, iron overload, genetic, eye lens
Cellular and body iron regulation
Iron is essential for a variety of biological functions at the cellular and systemic level, such as oxygen transport and the catalytic activity of many enzymes;[1] however, the specific chemical properties of iron as a transition metal also render it potentially toxic for cells and tissues. In the presence of reactive oxygen species (ROS), iron catalyses the generation of highly reactive hydroxyl radicals (Fenton/Haber-Weiss reactions) that damage membrane lipids, proteins and nucleic acids[2]. Considering that ROS are inevitable by-products of aerobiosis, organisms have to control iron concentrations tightly at the systemic and at the cellular level to satisfy metabolic needs but minimise iron toxicity. A complex network has evolved in mammals, assuring safe and balanced iron trafficking. Efficient absorption mechanisms exist for various forms of nutritional iron in the proximal small intestine (1 mg/day in humans)[3]. While the reticuloendothelial system is crucial for iron recycling and redistribution, the liver serves as an iron storage organ. Figure 1 schematically depicts important pathways in iron trafficking in a virtual cell. Iron can enter the cell only in the reduced form (ferrous iron, Fe2+), but is stored or bound to carrier proteins only in the ferric iron state (Fe3+). Several enzymes have been identified to oxidise/reduce iron, such as the ferrireductase Dcytb and hephaestin in intestinal cells or ceruloplasmin at the basolateral site of most cells.
Figure 1.
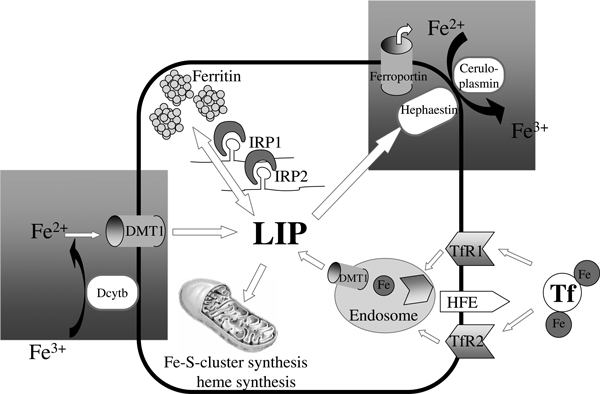
The role of ferritin in iron homeostasis. A virtual cell is shown, demonstrating the major cellular functions of iron uptake (TfR1 and -2, DMT1), export (ferroportin), storage (ferritin) and utilisation (eg haem synthesis). The LIP represents a chelated form of cytosolic iron that undergoes redistribution but also toxic side reactions. Hepatic hepcidin - the major systemic regulator - blocks iron export via ferroportin, while cytoplasmic IRPs coordinate cellular iron homeostasis by regulating proteins such as TfR1 and ferritin at the post-transcriptional level. Ferritin is the major iron storage protein and is mainly localised in hepatocytes and cells of the reticuloendothelial system. Connections between cellular and systemic iron metabolism are highlighted by hatched grey boxes. TfR1, transferrin receptor 1. DMT1, divalent metal transporter 1. LIP, labile iron pool. IRP, iron-regulatory protein.
Iron circulates in higher organisms bound to transferrin (Tf). It is internalised by cells via the ubiquitous transferrin receptor 1 (TfR1) - the major iron uptake protein. Expression of TfR2 - a second transferrin receptor - is limited to erythroid, hepatocellular and duodenal cells. The TfR-Tf complex is internalised by endocytosis, iron is then released, reduced and eventually transferred to the cytoplasm by the divalent metal transporter 1 (DMT1). An impaired interaction of TfR1 and/or TfR2 with a mutated human haemochromatosis (HFE) protein causes the most common monogenic hereditary disease in humans (hereditary haemochromatosis), which eventually leads to iron overload and tissue damage in several organs[4,5]. The central interchangeable form of iron is called the labile iron pool (LIP), which represents a loosely chelated form of iron that can undergo toxic redox reactions[6]. Cellular iron can be safely stored in ferritin, a multi-subunit protein of heavy- (H) and light- (L) chains, or it can be redistributed within the cells for major utilisation pathways like Fe-S-cluster or haem synthesis.
At the systemic level, iron can be exported from macrophages, duodenal cells, hepatocytes and some cells of the central nervous system via ferroportin to undergo systemic circulation. This export is regulated by the antimicrobial peptide hepcidin, which inhibits iron efflux by interacting with ferroportin, leading to ferroportin internalisation and degradation[7,8]. At present, hepcidin that is secreted from liver cells is generally considered as the 'systemic iron sensor' in higher organisms[3,9].
Ferritin as an iron storage molecule
Excess non-haem iron is stored in ferritin. Ferritin is a protein with a molecular mass of approximately 900 kDa which is present in virtually every cell. All human ferritins consist of 24 subunits of H and L chain ferritin. H-ferritin (21 kDa) is encoded on chromosome 11, while the L-ferritin sequence (19 kDa) is localised on chromosome 19. Both subunits together form a spherical shell around a cavity. Within this cavity, up to 4,500 iron atoms can be safely stored (ie without the possibility of reacting with ROS and generating hydroxyl radicals). Fe2+ is oxidised by the ferroxidase centre to insoluble hydroxyferric complexes through consumption of O2 or hydrogen peroxide (H2O2). Iron storage by ferritin is a reversible process, and during periods of iron starvation, ferritin readily releases iron whenever needed. Although ferritin always consists of 24 subunits, the ratio of H- to L-ferritin is variable (depending on the type of tissue), creating several isoferritins. H- and L-ferritin differ in terms of their function: while H-ferritin is responsible for iron storage and carries the ferroxidase activity needed to sequester iron, it is L-ferritin that exerts a regulatory function, owing to the presence of acidic groups that facilitate iron oxidation and hydrolysis. Ferritins containing exclusively H-chains oxidise iron approximately ten-fold faster than ferritins rich in L-ferritin[10]. By contrast, addition of even a small number of L-subunits to the ferritin molecule accelerates the transfer of iron from the ferroxidase centre to the iron core. This step is rate limiting and L-ferritin therefore improves the overall iron sequestering process[11]. Ferritins rich in H-ferritin have been found in the heart and brain and are believed to exert iron-segregating and antioxidant functions, whereas spleen and liver ferritins contain a larger proportion of L-ferritin and thus serve as iron stores, releasing iron on demand[12].
Serum ferritin as a marker for body iron stores
Small amounts of ferritin are also present in the serum. A large body of evidence suggests that circulating ferritin levels parallel the amount of iron stored in the liver. This is only true for the physiological state and true iron overload, however, since other pathological conditions--such as inflammatory, autoimmune and malignant diseases--can lead to a disproportionate elevation of serum ferritin[13,14]. Although clinicians have been using ferritin as a marker for body iron storage for more than three decades, little is known about the source of serum ferritin. Serum ferritin is iron poor, resembles L-ferritin immunologically and is thought to contain glycosylated (G) ferritin[15]. Reticuloendothelial cells (ie macrophages[16]) and hepatocytes[17] are the major iron-storing cell types in the body, and both contribute to extracellular ferritin production. Initially, it was thought that serum ferritin results from cellular leakage, but in vitro studies have demonstrated that L-ferritin can be targeted for glycosylation via the regular secretory pathway. This is an active process during translation, despite the lack of a conventional signal sequence[17].
Post-transcriptional control of ferritin synthesis is iron dependent
Iron availability is the main regulator of cellular ferritin synthesis. After ferritin was cloned in 1986[18], it was quickly learnt that it is mainly regulated at the translational level, controlled by a 157-nucleotide-containing leader sequence in the 5′-untranslated region of ferritin mRNA[19]. This leader sequence contains the so-called iron-responsive element (IRE), a sequence of approximately 50 nucleotides which forms a hairpin-like secondary structure and allows the iron-regulatory protein (IRP) to bind. There are two IRPs in the human body, IRP-1 and IRP-2. They are both able to sense iron, although via distinct mechanisms[20-22]. In the presence of iron, IRP-1 changes its conformation by assembling a cubane iron-sulphur cluster [4Fe-4S], loses its RNA-binding activity and acts a cytosolic aconitase[20,23,24]. By contrast, IRP-2 undergoes iron-dependent proteasomal degradation[25,26]. Under conditions of intracellular iron depletion, both IRPs can bind IRE with high affinity, prevent binding of the translation initiation complex and thus block ferritin translation (Figure 2). Both types of ferritin are regulated by an IRE, although data from heterozygous H-ferritin knockout mice suggest that there might be different iron thresholds at which L- and H-ferritin respond[27]. IREs have not only been identified in ferritin mRNA. They are also products of other genes, such as TfR1, which contains a series of five IREs in the 3′-untranslated region. IRP/IRE binding to the TfR1 IREs in iron deficiency increases the stability of TfR1 mRNA and, finally, iron uptake[28]. Interestingly, IRP-1 is not only affected by iron levels but also by various reactive oxygen and nitrogen species, such as H2O2, superoxide and hypochlorous acid and NO[29]. Comparable with iron depletion, low levels of H2O2 rapidly activate IRP-1 for binding to IRE, even at the whole-organ level[22,30].
Figure 2.
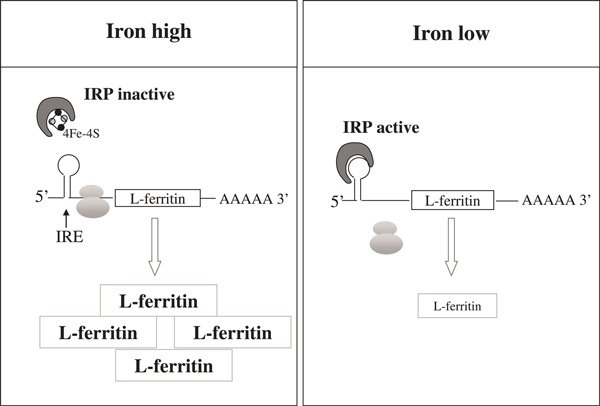
Post-transcriptional control of ferritin synthesis by the IRP/IRE network. Iron depletion activates IRP-1 and -2 for binding to the IRE in the 5′-untranslated region of ferritin mRNA. This IRP-IRE interaction prevents translation of ferritin and eventually decreases ferritin protein levels. IRP, iron-regulatory protein.
Hyperferritinemia Cataract Syndrome (HHCS) is due to uncontrolled translation of L-ferritin
Until the mid-1990s, hyperferritinaemia was thought primarily to be associated with iron overload, inflammation and malignancy. In 1995, two groups in Italy and France independently discovered families with hyperferritinaemia of unexplained aetiology that co-segregated with autosomal-dominant cataract at an unusually young age[31,32]. These patients had been initially misdiagnosed with hereditary haemochromatosis, although no iron overload had been found in liver biopsies and they developed iron-deficiency anaemia after repeated venesections. The novel clinical entity was eventually named as hyperferritinaemia-cataract syndrome (HHCS). Table 1 lists the typical clinical criteria for HHCS diagnosis. The chromosomal location of this disorder was found to be on chromosome 19, close to L-ferritin[31], and Bonneau et al. first discussed a potential involvement of the IRE in L-ferritin[32]. Shortly thereafter, both groups identified causative point mutations within the IRE of L-ferritin, although the mutations were not identical but located on neighbouring nucleotides within the same region (Paris-1 mutation: 40A > G[33], Verona-1 mutation: 40G > C)[34]. Protein-RNA interaction assays finally confirmed that the mutated IRE does indeed have a drastically reduced affinity for IRP. The disrupted IRE-IRP interaction uncouples L-ferritin synthesis from iron availability and leads to uncontrolled and sustained L-ferritin translation[33].
Table 1.
Clinical criteria of hyperferritinaemia-cataract syndrome.
| • High and constant levels of serum ferritin unresponsive to iron depletion |
|---|
| • No signs of iron overload (normal soluble serum iron and transferrin saturation) |
| • Normal blood cell counts and normal transaminases |
| • Familial cataract with phenotypic pulverulent appearance |
| • No relevant clinical symptoms apart from visual impairment by cataract |
Spectrum of mutations in the HHCS
Since the first description of HHCS (OMIM #600886), a total of 31 mutations have been described within the IRE region of L-ferritin in various countries. The geographical distribution of mutations described to date primarily suggests a stronger co-localisation with iron metabolism/haemochromatosis research centres than with prevalence of HHCS. A complete list (updated in September 2009) of all underlying mutations and patients published to date is given in Table 2. Table 2 also shows the different nomenclatures for each of the mutation sites, the patients' countries of origin, and the numbers of families and patients per publication. Twenty-seven of these mutations are single nucleotide transitions and four are deletions of 2-29 base pairs. The single nucleotide transitions in particular indicate which part of the IRE is critical for an efficient and functional IRE-IRP interaction. The most important sequences are a highly conserved hexanucleotide at the terminal loop, a bulge with an unpaired cytidin, and the upper stem of the hairpin (Figure 3). The lower stem seems to be less prone to mutations that could cause HHCS. Molecular models published by Lachlan et al. showed how point mutations in the L-ferritin IRE can distort the IRE hairpin structure[46]. An even more detailed analysis of IRP-IRE binding disturbances by various mutations was published by Allerson et al. in 1999, where they analysed thermodynamic changes in IRE structure and IRP-IRE interactions in different mutations[75] and were able to correlate the clinical severity of HHCS with the severity of IRE-IRP disturbance.
Table 2.
Single nucleotide transitions and deletions causing HHSC (as of September 2009).
| Type of mutation | Location FTL cDNA nomenclature | Location based on transcription start site | Nomenclature EMBL HSAFL12 | Trivial name | Author | Year of publication | No. of families/patients | Patients' country of origin |
|---|---|---|---|---|---|---|---|---|
| SNT | c.-186C > T | 14C > G | Cremonesi et al.[35] | 2001 | 1/1 | Italy | ||
| SNT | c.-182C > T+ c.-178T > G | 18C > T + 22T > G | Pavia-2 | Cazzola et al.[36] | 1997 | 1/3 | Italy | |
| SNT | c.-171C > G | 29C > G | Bosio et al.[37] | 2004 | 1/2 | Italy | ||
| SNT | c.-168G > A | 32G > A | Pavia-1 | Cazzola et al.[36] | 1997 | 1/7 | Italy | |
| Cicilano et al.[38] | 1999 | 1/4 | Italy | |||||
| Volkmann et al.[39] | 2000 | 1/5 | Italy | |||||
| Vanita et al.[40] | 2006 | 1/13 | India | |||||
| Simsek et al.[41] | 2003 | 1/10 | Netherlands | |||||
| Millonig et al.[42] | 2009 | 1/17 | Germany | |||||
| SNT | c.-168G > C | 32G > C | Baltimore | Kato and Casella[43] | 1999 | - | Italy | |
| Campagnoli et al.[44] | 2002 | 1/7 | Italy | |||||
| Craig et al.[45] | 2003 | 1/4 | UK | |||||
| Simsek et al.[41] | 2003 | 1/6 | Netherlands | |||||
| Lachlan et al.[46] | 2004 | 2/6 | UK | |||||
| SNT | c.-168G > T | 32G > T | Paris-2 | Martin et al.[47] | 1998 | 2/7 | France | |
| Craig et al.[45] | 2003 | 1/1 | Australia | |||||
| Hetet et al.[48] | 2003 | 5/- | France | |||||
| Lachlan et al.[46] | 2004 | 1/1 | UK | |||||
| SNT | c.-167C > A | 33C > A | Giansily et al.[49] | 2001 | - | Italy | ||
| Hetet et al.[48] | 2003 | 1/1 | France | |||||
| SNT | c.-167C > T | 33C > T | Madrid | Balas et al.[50] | 1999 | 1/6 | Spain | |
| Brooks et al.[51] | 2002 | 1/5 | USA | |||||
| Hetet et al.[48] | 2003 | 3/- | France | |||||
| Ladero et al.[52] | 2004 | 1/7 | Spain | |||||
| Lachlan et al.[46] | 2004 | 1/1 | UK | |||||
| SNT | c.-166T > C | 34T > C | Hetet et al.[48] | 2003 | 1/3 | France | ||
| SNT | c.-164C > A | 36C > A | London-2 | Mumford et al.[53] | 1998 | 1/8 | UK | |
| Hetet et al.[48] | 2003 | 1/1 | France | |||||
| Lachlan et al.[46] | 2004 | 1/1 | UK | |||||
| SNT | c.-164C > G | 36C > G | Cremonesi et al.[54] | 2003 | - | Italy | ||
| SNT | c.-163A > T | 37A > T | Zaragoza | Garcia Erce et al.[55] | 2006 | 2/10 | Spain | |
| SNT | c.-163A > C | 37A > C | Cremonesi | Personal communication | Italy | |||
| SNT | c.-163A > G | 37A > G | Cremonesi et al.[54] | 2003 | - | Italy | ||
| SNT | c.-161C > A | 39C > A | Craig et al.[45] | 2003 | 1/2 | Italy | ||
| SNT | c.-161C > G | 39C > G | Garderet et al.[56] | 2004 | 1/3 | France | ||
| Papanikolaou et al.[57] | 2006 | 3/19 | Greece | |||||
| SNT | c.-161C > T | 39C > T | London-1 | Mumford et al.[53] | 1998 | 1/2 | UK | |
| Arosio et al.[58] | 1999 | 1/1 | Italy | |||||
| Cicilano et al.[38] | 1999 | 1/8 | Italy | |||||
| Hernandez Martin et al.[59] | 2008 | 1/1 | Spain | |||||
| Hetet et al.[48] | 2003 | 3/5 | France | |||||
| Lachlan et al.[46] | 2004 | 1/1 | UK | |||||
| Ferrante et al.[60] | 2005 | 2/ | Belgium | |||||
| SNT | c.-160A > G | 40A > G | 146A > G | Paris-1 Montpellier-1 | Beaumont et al.[33] | 1996 | 1/5 | France |
| Aguilar-Martinez et al.[61] | 1996 | 1/1 | France | |||||
| Arnold et al.[62] | 1997 | 1/6 | UK | |||||
| Barton et al.[63] | 1998 | 1/1 | European | |||||
| Cicilano et al.[38] | 1999 | 1/8 | Italy | |||||
| Perez de Nanclares et al.[64] | 2001 | 1/- | Spain (Basque) | |||||
| Craig et al.[45] | 2003 | 1/11 | Australia | |||||
| Hetet et al.[48] | 2003 | 4/- | France | |||||
| Lachlan et al.[46] | 2004 | 1/1 | UK | |||||
| Del Castillo Rueda et al.[65] | 2007 | 1/3 | Spain | |||||
| Hughes and Vosylius[66] | 2006 | 1/1 | UK | |||||
| Mumford et al.[53] | 2000 | 1/6 | UK | |||||
| Ferrante et al.[60] | 2005 | 2/ | Belgium | |||||
| Mohn et al.[67] | 2005 | 1/1 | Italy | |||||
| 40A > G + 41G > C | Cremonesi et al.[35] | 2001 | Italy | |||||
| SNT | c.-159G > C | 41G > C | 147G > C | Verona-1 | Girelli et al.[34] | 1995 | 1/3 | Italy |
| SNT | c.-157G > A | 43G > A | Phillips et al.[68] | 2005 | 1/14 | USA | ||
| SNT | c.-153G > A | 47G > A | Hetet et al.[48] | 2003 | 1/3 | France | ||
| SNT | c.-150C > A | 50C > A | Gonzalez-Huerta et al.[69] | 2008 | 1/11 | Mexico | ||
| SNT | c.-149G > C | 51G > C | Camaschella et al.[70] | 2000 | 1/2 | Italy | ||
| SNT | c.-144A > T | 56A > T | Cremonesi | Personal communication | France | |||
| Deletion | c.-220_-196del25 | Burdon et al.[71] | 2007 | 1/6 | Australia | |||
| Deletion | c.-190-162del29 | 10_38del29 | Verona-2 | Girelli et al.[72] | 1997 | 1/6 | Italy | |
| Deletion | c.-178-173del30 | 22_27del6 | Cazzola et al.[73] | 2002 | 1/- | Italy | ||
| Deletion | c.-162-161delAC | 38_39delAC | Giansily et al.[49] | 2001 | - | |||
| Hetet et al.[48] | 2003 | 1/1 | Canada | |||||
| Deletion | c.-158-143del16 | 42_57del16 | Feys et al.[74] | 2001 | 1/3 | France | ||
| Hetet et al.[48] | 2003 | Same family | ||||||
Key
SNT: single nucleotide transition.
Bold text: first description of a new mutation.
Normal text: further publications with a known mutation.
FTL, ferritin-light.
Figure 3.
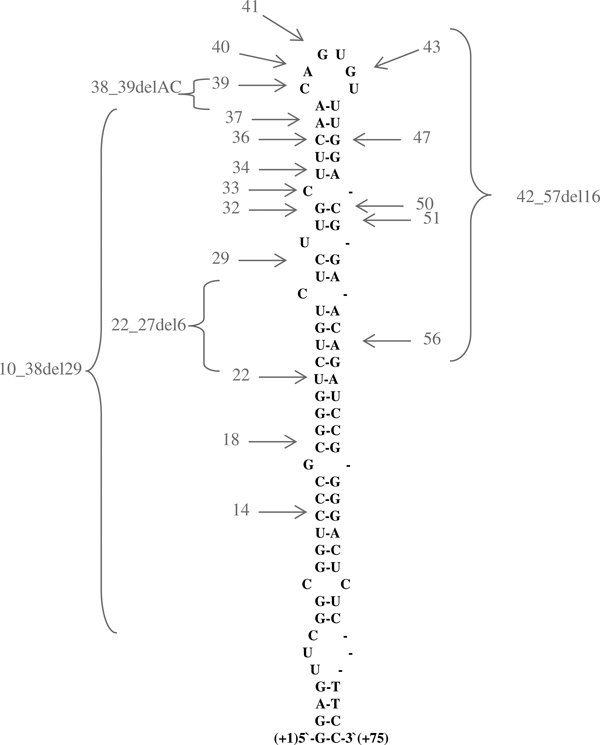
Mutations causing hyperferritinaemia-cataract syndrome (HHCS) in the iron-responsive element (IRE) of L-ferritin. The IRE of L-ferritin forms a hairpin-like structure. Most of the mutations that cause HHCS discovered so far are located in the upper stem and the conserved hexanucleotide of the IRE. Single nucleotide transitions are depicted by grey arrows, and the number of the nucleotide deletions are represented by brackets. An extensive overview of all mutations can be found in Table 2.
Ophthalmological presentation of the cataract in HHCS
Although the cataract is the only clinical manifestation in HHCS, it is still unknown to many ophthalmologists and remains undiagnosed even after ophthalmological consultation for cataract surgery. The descriptions of HHCS cataract vary considerably. A comprehensive study of cataract features in several families affected by HHCS using the slit-lamp and retro-illumination technique has been performed by Craig and colleagues[45]. According to this study, cataract morphology in HHCS families is highly distinctive and consistent but varies with age at onset and severity. The primary characteristics are the appearance of axial and peripheral white flecks and small crystalline aggregates. Other opacities are small cortical vacuoles which are translucent but easily visible on retro-illumination. Craig and colleagues even concluded that cataract morphology should be sufficient to suspect HHCS[45]. Figure 4 shows a typical cataract in a patient with a 32G > A mutation representing all of the above-mentioned features. Due to the lack of larger studies, it remains unclear whether or not the cataract in HHCS patients is congenital or develops in early infanthood. Girelli et al. described a child with hyperferritinaemia at birth, in whom no cataract appeared during follow-up for one year[76]. Similar observations have been reported by others[64].
Figure 4.
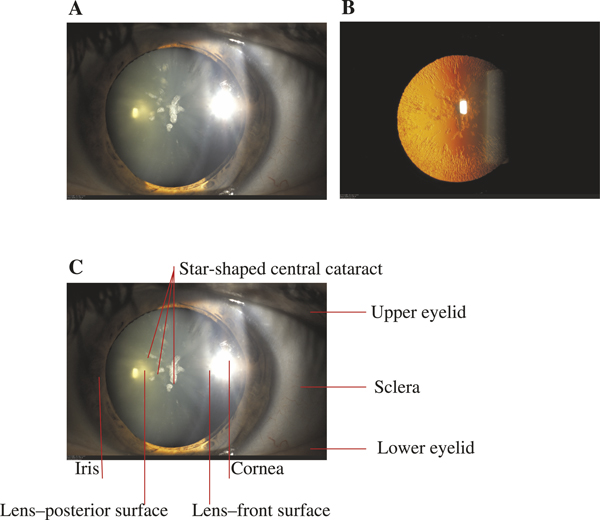
Pathognomonic aspect of cataract in HHCS. Slit-lamp examination (a) and the retro-illumination technique (b) show the typical aspect of a star-shaped central cataract and peripheral flecks in the lens. From Millonig et al. 2009[42]. The slit-lamp is a low-power microscope combined with a high-intensity light source that can be focused to shine a narrow beam, thus allowing examination of the front parts of the eye (cornea, iris, lens). Panel (c) shows the anatomical details of (a).
After unravelling the pathomechanism of HHCS and collecting increasing numbers of HHCS families, it became clear that the clinical severity with regard to serum ferritin levels, and visual impairment due to cataract correlated with the type of mutation. Cazzola and co-workers showed that mutations in the highly conserved hexanucleotide region (eg at position 40 or 41) caused a severe cataract, while mutations around the bulge with unpaired cytidine (eg position 32) caused mild cataract. In addition, mutations in the lower stem of IRE caused only asymptomatic cataract (double mutation at positions 18 and 22)[36]. Molecular models published by Lachlan et al. showed how point mutations in the L-ferritin IRE can distort the IRE hairpin structure[46].
It should be noted that not only the extent of the cataract, but also the local distribution of the nuclear and cortical opacities affect visual impairment. In the case of the first German family to be added to the database, recently published by the present authors, the degree of impairment of the central visual axis determined the timing of cataract surgery, which varied considerably from the age of 14 to 57 years, although all family members had an identical single point mutation[42].
Clinical relevance of HHCS
Thus far, most patients with HHCS have been identified in the course of a check-up for high serum ferritin. Many patients are initially misdiagnosed with hereditary haemochromatosis and treated with venesections that lead to rapid iron depletion and iron-deficiency anaemia. Haematologists, gastroenterologists and ophthalmologists should therefore be aware of HHCS to prevent false diagnosis and needless invasive diagnostics, such as liver biopsy, but also harmful therapeutic approaches such as venesection. The laboratory triad of hyperferritinaemia, normal serum iron and normal transferrin saturation is indicative of HHCS after exclusion of other causes of increased ferritin levels (inflammation, malignancy, alcoholic liver disease) and should prompt an ophthalmological consultation. HHCS patients do not show increased transaminases or any signs of inflammation (elevated C-reactive protein or increased white blood cell count). After diagnosing HHCS, patient counselling should focus on the relative harmlessness of this genetic disease, with early cataract surgery as the only clinical consequence.
Acknowledgements
This work was supported by the Dietmar Hopp Foundation, the Manfred Lautenschläger Foundation and an Olympia Morata Fellowship from the Medical School of Heidelberg to G.M.
References
- Frausto da Silva JJR, Williams RJP. The Biological Chemistry of the Elements: The Inorganic Chemistry of Life. Clarendon Press, Oxford, UK; 1991. [Google Scholar]
- Halliwell B, Gutteridge JMC. 'Role of free radicals and catalytic metal ions in human disease: An overview'. Methods Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. 'Balancing acts: Molecular control of mammalian iron metabolism'. Cell. 2004;117:285–297. doi: 10.1016/S0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Gao J, Chen J, Kramer M, Tsukamoto H. et al. 'Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression'. Cell Metab. 2009;9:217–227. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z. et al. 'A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis'. Nat. Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- Breuer W, Epsztejn S, Cabantchik ZI. 'Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron(II)'. J. Biol. Chem. 1995;270:24209–24215. doi: 10.1074/jbc.270.41.24209. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB. et al. 'Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization'. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Knutson MD, Oukka M, Koss LM, Aydemir F. et al. 'Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin'. Proc. Natl. Acad. Sci. USA. 2005;102:1324–1328. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T. 'Hepcidin: A key regulator of iron metabolism and mediator of anemia of inflammation'. Blood. 2003;102:783–788. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- Levi S, Luzzago A, Cesareni G, Cozzi A. et al. 'Mechanism of ferritin iron uptake: Activity of the H-chain and deletion mapping of the ferro-oxidase site. A study of iron uptake and ferro-oxidase activity of human liver, recombinant H-chain ferritins, and of two H-chain deletion mutants'. J. Biol. Chem. 1988;263:18086–18092. [PubMed] [Google Scholar]
- Santambrogio P, Levi S, Cozzi A, Rovida E. et al. 'Production and characterization of recombinant heteropolymers of human ferritin H and L chains'. J. Biol. Chem. 1993;268:12744–12748. [PubMed] [Google Scholar]
- Harrison PM, Arosio P. 'The ferritins: Molecular properties, iron storage function and cellular regulation'. Biochim. Biophys. Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- Lipschitz DA, Cook JD, Finch CA. 'A clinical evaluation of serum ferritin as an index of iron stores'. N. Engl. J. Med. 1974;290:1213–1216. doi: 10.1056/NEJM197405302902201. [DOI] [PubMed] [Google Scholar]
- Torti SV, Torti FM. 'Iron and ferritin in inflammation and cancer'. Adv. Inorg. Biochem. 1994;10:119–137. [PubMed] [Google Scholar]
- Santambrogio P, Cozzi A, Levi S, Arosio P. 'Human serum ferritin G-peptide is recognized by anti-L ferritin subunit antibodies and concanavalin-A. Br. J. Haematol. 1987;65:235–237. doi: 10.1111/j.1365-2141.1987.tb02271.x. [DOI] [PubMed] [Google Scholar]
- Yuan XM, Li W, Baird SK, Carlsson M, Melefors O. 'Secretion of ferritin by iron-laden macrophages and influence of lipoproteins'. Free Radic. Res. 2004;38:1133–1142. doi: 10.1080/10715760400011692. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Hevi S, Chuck SL. 'Regulated secretion of glycosylated human ferritin from hepatocytes'. Blood. 2004;103:2369–2376. doi: 10.1182/blood-2003-09-3050. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Keim S, Papadopoulos P, O'Brien S. et al. 'Cloning, characterization, expression, and chromosomal localization of a human ferritin heavy-chain gene'. Proc. Natl. Acad. Sci. USA. 1986;83:7226–7230. doi: 10.1073/pnas.83.19.7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Rouault TA, Caughman SW, Dancis A. et al. 'A cis-acting element is necessary and sufficient for translational regulation of human ferritin expression in response to iron'. Proc. Natl. Acad. Sci USA. 1987;84:6730–6734. doi: 10.1073/pnas.84.19.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Kühn LC. 'Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide and oxidative stress'. Proc. Natl. Acad. Sci. USA. 1996;93:8175–8182. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairo G, Pietrangelo A. 'Iron regulatory proteins in pathobiology'. Biochem. J. 2000;352:241–250. doi: 10.1042/0264-6021:3520241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantopoulos K. 'Iron metabolism and the IRE/IRP regulatory system: An update'. Ann. N. Y. Acad. Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- Haile DJ, Rouault TA, Harford JB, Kennedy MC. et al. 'Cellular regulation of the iron-responsive element binding protein: Disassembly of the cubane iron-sulfur cluster results in high-affinity RNA binding'. Proc. Natl. Acad. Sci. USA. 1992;89:11735–11739. doi: 10.1073/pnas.89.24.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault T, Klausner R. 'Regulation of iron metabolism in eukaryotes'. Curr. Top. Cell. Regul. 1997;35:1–19. doi: 10.1016/s0070-2137(97)80001-5. [DOI] [PubMed] [Google Scholar]
- Salahudeen AA, Thompson JW, Ruiz JC, Ma HW. et al. 'An E3 ligase possessing an iron responsive hemerythrin domain is a regulator of iron homeostasis'. Science. 2009;326:722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashisht AA, Zumbrennen KB, Huang X, Powers DN. et al. 'Control of iron homeostasis by an iron-regulated ubiquitin ligase'. Science. 2009;326:718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira C, Santambrogio P, Martin ME, Andrieu V. et al. 'H ferritin knockout mice: A model of hyperferritinemia in the absence of iron overload'. Blood. 2001;98:525–532. doi: 10.1182/blood.V98.3.525. [DOI] [PubMed] [Google Scholar]
- Koeller DM, Casey JL, Hentze MW, Gerhardt EM. et al. 'A cytosolic protein binds to structural elements within the iron regulatory region of the transferrin receptor mRNA'. Proc. Natl. Acad. Sci. USA. 1989;86:3574–3578. doi: 10.1073/pnas.86.10.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S. 'Iron regulatory protein 1 as a sensor of reactive oxygen species'. Biofactors. 2005;24:171–181. doi: 10.1002/biof.5520240121. [DOI] [PubMed] [Google Scholar]
- Mueller S, Pantopoulos K, Hübner CA, Stremmel W. et al. 'IRP1 activation by extracellular oxidative stress in the perfused rat liver'. J. Biol. Chem. 2001;276:23192–23196. doi: 10.1074/jbc.M100654200. [DOI] [PubMed] [Google Scholar]
- Girelli D, Olivieri O, De Franceschi L, Corrocher R. et al. 'A linkage between hereditary hyperferritinaemia not related to iron overload and autosomal dominant congenital cataract'. Br. J. Haematol. 1995;90:931–934. doi: 10.1111/j.1365-2141.1995.tb05218.x. [DOI] [PubMed] [Google Scholar]
- Bonneau D, Winter-Fuseau I, Loiseau MN, Amati P. et al. 'Bilateral cataract and high serum ferritin: A new dominant genetic disorder?'. J. Med. Genet. 1995;32:778–779. doi: 10.1136/jmg.32.10.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont C, Leneuve P, Devaux I, Scoazec JY. et al. 'Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract'. Nat. Genet. 1995;11:444–446. doi: 10.1038/ng1295-444. [DOI] [PubMed] [Google Scholar]
- Girelli D, Corrocher R, Bisceglia L, Olivieri O. et al. 'Molecular basis for the recently described hereditary hyperferritinemia-cataract syndrome: A mutation in the iron-responsive element of ferritin L-subunit gene (the "Verona mutation")'. Blood. 1995;86:4050–4053. [PubMed] [Google Scholar]
- Cremonesi L, Fumagalli A, Soriani N, Ferrari M. et al. 'Double-gradient denaturing gradient gel electrophoresis assay for identification of L-ferritin iron-responsive element mutations responsible for hereditary hyperferritinemia-cataract syndrome: Identification of the new mutation C14G'. Clin. Chem. 2001;47:491–497. [PubMed] [Google Scholar]
- Cazzola M, Bergamaschi G, Tonon L, Arbustini E. et al. 'Hereditary hyperferritinemia-cataract syndrome: Relationship between phenotypes and specific mutations in the iron-responsive element of ferritin light-chain mRNA'. Blood. 1997;90:814–821. [PubMed] [Google Scholar]
- Bosio S, Campanella A, Gramaglia E, Porporato P. et al. 'C29G in the iron-responsive element of L-ferritin: A new mutation associated with hyperferritinemia-cataract'. Blood Cells Mol. Dis. 2004;33:31–34. doi: 10.1016/j.bcmd.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Cicilano M, Zecchina G, Roetto A, Bosio S. et al. 'Recurrent mutations in the iron regulatory element of L-ferritin in hereditary hyperferritinemia-cataract syndrome'. Haematologica. 1999;84:489–492. [PubMed] [Google Scholar]
- Volkmann M, Schiff JH, Hor M, Hentze MW. et al. ['Pair of siblings of Italian ethnicity with hyperferritinemia and cataract'] Internist (Berl) 2000;41:381–384. doi: 10.1007/s001080050521. [DOI] [PubMed] [Google Scholar]
- Vanita V, Hejtmancik JF, Hennies HC, Guleria K. et al. 'Sutural cataract associated with a mutation in the ferritin light chain gene (FTL) in a family of Indian origin'. Mol. Vis. 2006;12:93–99. [PubMed] [Google Scholar]
- Simsek S, Nanayakkara PW, Keek JM, Faber LM. 'Two Dutch families with hereditary hyperferritinaemia-cataract syndrome and heterozygosity for an HFE-related haemochromatosis gene mutation'. Neth. J. Med. 2003;61:291–295. [PubMed] [Google Scholar]
- Millonig GH, Holzer MP, Tolle G, Auffarth GU. et al. 'Das hereditäre Hyperferritinämie-Katarakt-Syndrom - die erste Familie in Deutschland'. ('Hereditary Hyperferritinemia Caratact Syndrome -the first family in Germany') Z. Gastroenterol. 2009;47:1211–1215. doi: 10.1055/s-0028-1109523. [DOI] [PubMed] [Google Scholar]
- Kato GJ, Casella F. 'L-ferritin Baltimore-1: A novel mutation in the iron responsive element (C32G) as a cause of hyperferritinemia-cataract syndrome'. Blood. 1999;94:407a. [Google Scholar]
- Campagnoli MF, Pimazzoni R, Bosio S, Zecchina G. et al. 'Onset of cataract in early infancy associated with a 32G- >C transition in the iron responsive element of L-ferritin'. Eur. J. Pediatr. 2002;61:499–502. doi: 10.1007/s00431-002-1019-4. [DOI] [PubMed] [Google Scholar]
- Craig JE, Clark JB, McLeod JL, Kirkland MA. et al. 'Hereditary hyperferritinemia-cataract syndrome: Prevalence, lens morphology, spectrum of mutations, and clinical presentations'. Arch. Ophthalmol. 2003;121:1753–1761. doi: 10.1001/archopht.121.12.1753. [DOI] [PubMed] [Google Scholar]
- Lachlan KL, Temple IK, Mumford AD. 'Clinical features and molecular analysis of seven British kindreds with hereditary hyperferritinaemia cataract syndrome'. Eur. J. Hum. Genet. 2004;12:790–796. doi: 10.1038/sj.ejhg.5201252. [DOI] [PubMed] [Google Scholar]
- Martin ME, Fargion S, Brissot P, Pellat B, Beaumont C. 'A point mutation in the bulge of the iron-responsive element of the L ferritin gene in two families with the hereditary hyperferritinemia-cataract syndrome'. Blood. 1998;91:319–323. [PubMed] [Google Scholar]
- Hetet G, Devaux I, Soufir N, Grandchamp B. et al. 'Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations'. Blood. 2003;102:1904–1910. doi: 10.1182/blood-2003-02-0439. [DOI] [PubMed] [Google Scholar]
- Giansily M, Beaumont C, Desveaux C, Hetet G. et al. 'Denaturing gradient gel electrophoresis screening for mutations in the hereditary hyperferritinaemia cataract syndrome'. Br. J. Haematol. 2001;112:51–54. doi: 10.1046/j.1365-2141.2001.02513.x. [DOI] [PubMed] [Google Scholar]
- Balas A, Aviles MJ, Garcia-Sanchez F, Vicario JL. 'Description of a new mutation in the L-ferritin iron responsive element associated with hereditary hyperferritinemia-cataract syndrome in a spanish-family'. Blood. 1999;93:4020–4021. [PubMed] [Google Scholar]
- Brooks DG, Manova-Todorova K, Farmer J, Lobmayr L. et al. 'Ferritin crystal cataracts in hereditary hyperferritinemia cataract syndrome'. Invest. Ophthalmol. Vis. Sci. 2002;43:1121–1126. [PubMed] [Google Scholar]
- Ladero JM, Balas A, Garcia-Sanchez F, Vicario JL, Diaz-Rubio M. 'Hereditary hyperferritinemia-cataract syndrome. Study of a new family in Spain'. Rev. Esp. Enferm. Dig. 2004;96:507–509. doi: 10.4321/s1130-01082004000700008. 510-511. [DOI] [PubMed] [Google Scholar]
- Mumford AD, Vulliamy T, Lindsay J, Watson A. 'Hereditary hyperferritinemia-cataract syndrome: Two novel mutations in the L-ferritin iron-responsive element'. Blood. 1998;91:367–368. [PubMed] [Google Scholar]
- Cremonesi L, Paroni R, Foglieni B, Galbiati S. et al. 'Scanning mutations of the 5'UTR regulatory sequence of L-ferritin by denaturing high-performance liquid chromatography: Identification of new mutations'. Br. J. Haematol. 2003;121:173–179. doi: 10.1046/j.1365-2141.2003.04253.x. [DOI] [PubMed] [Google Scholar]
- Garcia Erce JA, Cortes T, Cremonesi L, Cazzola M. et al. ['Hyperferritinemia-cataract syndrome associated to the HFE gene mutation. Two new Spanish families and a new mutation (A37T: "Zaragoza")'] Med. Clin. (Barc.) 2006;127:55–58. doi: 10.1157/13089990. [DOI] [PubMed] [Google Scholar]
- Garderet L, Hermelin B, Gorin NC, Rosmorduc O. 'Hereditary hyperferritinemia-cataract syndrome: A novel mutation in the iron-responsive element of the L-ferritin gene in a French family'. Am. J. Med. 2004;117:138–139. doi: 10.1016/j.amjmed.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Papanikolaou G, Chandrinou H, Bouzas E, Contopoulos-Ioannidis D. et al. 'Hereditary hyperferritinemia cataract syndrome in three unrelated families of western Greek origin caused by the C39>G mutation of L-ferritin IRE'. Blood Cells Mol. Dis. 2006;36:33–40. doi: 10.1016/j.bcmd.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Arosio C, Fossati L, Vigano M, Trombini P. et al. 'Hereditary hyperferritinemia cataract syndrome: A de novo mutation in the iron responsive element of the L-ferritin gene'. Haematologica. 1999;84:560–561. [PubMed] [Google Scholar]
- Hernandez Martin D, Cervera Bravo A, Balas Perez A. ['Hereditary hyperferritinemia and cataract syndrome: A de novo mutation'] An. Pediatr. (Barc.) 2008;68:408–410. doi: 10.1157/13117721. [DOI] [PubMed] [Google Scholar]
- Ferrante M, Geubel AP, Fevery J, Marogy G. et al. 'Hereditary hyperferritinaemia-cataract syndrome: A challenging diagnosis for the hepatogastroenterologist'. Eur. J. Gastroenterol. Hepatol. 2005;17:1247–1253. doi: 10.1097/00042737-200511000-00016. [DOI] [PubMed] [Google Scholar]
- Aguilar-Martinez P, Biron C, Masmejean C, Jeanjean P, Schved JF. 'A novel mutation in the iron responsive element of ferritin L-subunit gene as a cause for hereditary hyperferritinemia-cataract syndrome'. Blood. 1996;88:1895. [PubMed] [Google Scholar]
- Arnold JD, Mumford AD, Lindsay JO, Hegde U. et al. 'Hyperferritinaemia in the absence of iron overload'. Gut. 1997;41:408–410. doi: 10.1136/gut.41.3.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton JC, Beutler E, Gelbart T. 'Coinheritance of alleles associated with hemochromatosis and hereditary hyperferritinemia-cataract syndrome'. Blood. 1998;92:4480. [PubMed] [Google Scholar]
- Perez de Nanclares G, Castano L, Martul P, Rica I. et al. 'Molecular analysis of hereditary hyperferritinemia-cataract syndrome in a large Basque family'. J. Pediatr. Endocrinol. Metab. 2001;14:295–300. doi: 10.1515/jpem.2001.14.3.295. [DOI] [PubMed] [Google Scholar]
- Del Castillo Rueda A, Fernandez Ruano ML. ['Hereditary hyperferritinemia cataracts syndrome in a Spanish family caused by the A40G mutation (Paris) in the L-ferritin (FTL) gene associated with the mutation H63D in the HFE gene'] Med. Clin. (Barc.) 2007;129:414–417. doi: 10.1016/S0025-7753(07)72873-7. [DOI] [PubMed] [Google Scholar]
- Hughes M, Vosylius P. 'Dual diagnoses of hereditary hyperferritinaemia-cataract syndrome and hereditary haemochromatosis'. Clin. Lab. Haematol. 2006;28:357–359. doi: 10.1111/j.1365-2257.2006.00824.x. [DOI] [PubMed] [Google Scholar]
- Mohn A, Capanna R, Chiarelli F. 'A girl with persistent hyperferritinaemia'. Lancet. 2005;365:1744. doi: 10.1016/S0140-6736(05)66549-X. [DOI] [PubMed] [Google Scholar]
- Phillips JD, Warby CA, Kushner JP. 'Identification of a novel mutation in the L-ferritin IRE leading to hereditary hyperferritinemia-cataract syndrome'. Am. J. Med. Genet. A. 2005;134A:77–79. doi: 10.1002/ajmg.a.30425. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Huerta L, Ramirez-Sanchez V, Rivera-Vega M, Messina-Baas O, Cuevas-Covarrubias S. 'A family with hereditary hyperferritinaemia cataract syndrome: Evidence of incomplete penetrance and clinical heterogeneity'. Br. J. Haematol. 2008;143:596–598. doi: 10.1111/j.1365-2141.2008.07345.x. [DOI] [PubMed] [Google Scholar]
- Camaschella C, Zecchina G, Lockitch G, Roetto A. et al. 'A new mutation (G51C) in the iron-responsive element (IRE) of L-ferritin associated with hyperferritinaemia-cataract syndrome decreases the binding affinity of the mutated IRE for iron-regulatory proteins'. Br. J. Haematol. 2000;108:480–482. doi: 10.1046/j.1365-2141.2000.01920.x. [DOI] [PubMed] [Google Scholar]
- Burdon KP, Sharma S, Chen CS, Dimasi DP. et al. 'A novel deletion in the FTL gene causes hereditary hyperferritinemia cataract syndrome (HHCS) by alteration of the transcription start site'. Hum. Mutat. 2007;28:742. doi: 10.1002/humu.9501. [DOI] [PubMed] [Google Scholar]
- Girelli D, Corrocher R, Bisceglia L, Olivieri O. et al. 'Hereditary hyperferritinemia-cataract syndrome caused by a 29-base pair deletion in the iron responsive element of ferritin L-subunit gene'. Blood. 1997;90:2084–2088. [PubMed] [Google Scholar]
- Cazzola M, Foglieni B, Bergamaschi G, Levi S. et al. 'A novel deletion of the L-ferritin iron-responsive element responsible for severe hereditary hyperferritinaemia-cataract syndrome'. Br. J. Haematol. 2002;116:667–670. doi: 10.1046/j.0007-1048.2001.03310.x. [DOI] [PubMed] [Google Scholar]
- Feys J, Nodarian M, Aygalenq P, Cattan D. et al. ['Hereditary hyperferritinemia syndrome and cataract'] J. Fr. Ophtalmol. 2001;24:847–850. [PubMed] [Google Scholar]
- Allerson CR, Cazzola M, Rouault TA. 'Clinical severity and thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia-cataract syndrome'. J. Biol. Chem. 1999;274:26439–26447. doi: 10.1074/jbc.274.37.26439. [DOI] [PubMed] [Google Scholar]
- Girelli D, Olivieri O, Gasparini P, Corrocher R. 'Molecular basis for the hereditary hyperferritinemia-cataract syndrome'. Blood. 1996;87:4912–4913. [PubMed] [Google Scholar]