

Abstract
'Nonstop' mutations are single base-pair substitutions that occur within translational termination (stop) codons and which can lead to the continued and inappropriate translation of the mRNA into the 3'-untranslated region. We have performed a meta-analysis of the 119 nonstop mutations (in 87 different genes) known to cause human inherited disease, examining the sequence context of the mutated stop codons and the average distance to the next alternative in-frame stop codon downstream, in comparison with their counterparts from control (non-mutated) gene sequences. A paucity of alternative in-frame stop codons was noted in the immediate vicinity (0-49 nucleotides downstream) of the mutated stop codons as compared with their control counterparts (p = 7.81 × 10-4). This implies that at least some nonstop mutations with alternative stop codons in close proximity will not have come to clinical attention, possibly because they will have given rise to stable mRNAs (not subject to nonstop mRNA decay) that are translatable into proteins of near-normal length and biological function. A significant excess of downstream in-frame stop codons was, however, noted in the range 150-199 nucleotides from the mutated stop codon (p = 8.55 × 10-4). We speculate that recruitment of an alternative stop codon at greater distance from the mutated stop codon may trigger nonstop mRNA decay, thereby decreasing the amount of protein product and yielding a readily discernible clinical phenotype. Confirmation or otherwise of this postulate must await the emergence of a clearer understanding of the mechanism of nonstop mRNA decay in mammalian cells.
Keywords: human inherited disease, stop codon, 3'-untranslated region, nonstop mutation, nonstop mRNA decay
Introduction
There are currently in excess of 60,000 missense and nonsense mutations (in nearly 4,000 different genes) listed in the Human Gene Mutation Database (HGMD) that are known to cause, or to be associated with, human inherited disease [1]. In addition, there are 119 examples of mutations (in 87 different genes) that occur within stop codons, a category of mutation which therefore constitutes ~0.2% per cent of codon-changing mutations [1]. Such lesions have been termed 'nonstop', 'nostop' or 'readthrough' mutations on the basis that the loss of the normal translational termination (stop) codon is likely to lead to continued translation of the mRNA further downstream into the 3'-untranslated region (UTR).
Although many authors tacitly assume that the normal open reading frame will simply be extended until the next in-frame stop codon is encountered, too few human nonstop mutations have so far been characterised to allow any general conclusions to be drawn as to their likely phenotypic consequences at either the mRNA or the protein level. In three reported cases, however (namely, those nonstop mutations in the gene encoding ribosomal protein S19 [RPS19], causing Diamond-Blackfan anaemia,[2] the F10 gene causing factor X deficiency [3] and the foxhead box E3 [FOXE3] gene causing anterior segment dysgenesis [4]), the levels of the mutant mRNA transcripts were found to be dramatically lower than those of their wild-type counterparts. By contrast, the mRNA level associated with a nonstop mutation in the 3-beta-hydroxy-delta-5-steroid dehyrogenase (HSD3B2) gene causing adrenal hyperplasia was found to be near normal, although both HSD3B2 enzymatic activity and antigen (associated with a predicted 467 amino-acid protein, extended by 95 residues beyond the wild-type length) were found to be dramatically reduced [5]. Similarly, in the case of a nonstop mutation in the thymidine phosphorylase (TYMP) gene responsible for mitochondrial neurogastrointestinal encephalomyopathy, the mRNA level was not found to be reduced, even although the thymine phosphorylase protein product it encoded was undetectable [6].
In yeast, nonstop mRNAs generated from mRNAs lacking translational termination codons are recognised, by the protein Ski7, on ribosomes that have become stalled at the 3' ends of the mRNAs; these RNAs are then targeted for exosome-mediated degradation [7-9]. While this process of 'nonstop mRNA decay' is fairly effective at removing nonstop mRNAs, any protein products generated by translation of residual nonstop mRNAs are degraded by the proteasome [10,11] Although few such studies have so far been attempted in mammalian cells, the expression level of nonstop mRNAs generally appears unaltered while ribosome stalling at the 3' end of the elongated nonstop mRNA blocks translation before the completion of synthesis of full-length polypeptides [12-14].
Precisely how nonstop mRNA decay impacts upon naturally occurring human nonstop mutations is unknown but, as is clear from the five disease-associated examples mentioned above, the evidence acquired to date suggests that this may be a gene- and mutation-dependent process [15]. Thus, although not uncommon, remarkably little is as yet known about the nature and consequences of this type of mutation. In this paper, we report a first meta-analysis of naturally occurring nonstop mutations causing human inherited disease. With a view to exploring the various possible factors that could impact upon the likelihood of a given nonstop mutation coming to clinical attention, we have performed an analysis of the sequence context of the mutated stop codons and the average distance to the next in-frame downstream stop codon in comparison with control (non-mutated) gene sequences.
Methods
Mutation and control datasets
A total of 119 naturally occurring nonstop mutations from 87 human genes (Supplementary Table S1 (Table 4)) were identified from the HGMD [1]. The majority of these nonstop mutations were single examples identified in specific genes but 18 genes harboured a total of 50 examples of this type of lesion. Since the multiple inclusion of identical sequences flanking mutated stop codons would have introduced considerable bias into the subsequent analysis, only one mutation per gene was considered in the analysis of the sequence context.
Table S1.
Nonstop mutations recorded in the Human Gene Mutation Database
| Entrez Gene ID | Gene | Base change | Amino acid change | Codon | Chromosome | Gene | Ref_Seq mRNA Acc Num (Longest) | CDS | Next STOP codon | polyA signals AATAAA ATTAAA | Flanking nucleotide sequence Terminal amino-acids | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transcript size | Number of Exons | |||||||||||
| 58 | ACTA1 | cTAG-CAG | Term-Gln | 376 | 1q42.13 | ACTA1 | NM_001100.3 | 1509 bp 7 exons | 106-1239 TAG |
1378-1380 TAA | 1465..1470 ATTAAA | tcgtccaccgcaaatgcttctagcacactccacctccagcacg tgc ttc tag = C F * |
| 58 | ACTA1 | TAG-TGG | Term-Trp | |||||||||
| 58 | ACTA1 | TAGa-TAT | Term-Tyr | |||||||||
| 326 | AIRE | TGAc-TGT | Term-Cys | 546 | 21q22.3 | AIRE | NM_000383.2 | 2257 bp 15 exons |
128-1765 TGA |
1943-1945 TAA |
1941..1946 ATTAAA |
cggcggcccccttcccctcctgaccccagatggccgggacatg ccc tcc tga = P S * |
| 336 | APOA2 | gTGA-AGA | Term-Arg | 78 | 1q21-q23 | APOA2 | NM_001643.1 | 473 bp 4 exons |
59-361 TGA |
422-424 TAA | 454..459 AATAAA |
gaacacagcctgccacccagtgaagtgtccagaccattgtctt acc cag tga = T Q * |
| 336 | APOA2 | gTGA-CGA | Term-Arg | |||||||||
| 336 | APOA2 | gTGA-GGA | Term-Gly | |||||||||
| 336 | APOA2 | TGA-TCA | Term-Ser | |||||||||
| 353 | APRT | TGA-CGA | Term-Arg | 181 | 16q24 | APRT | NM_000485.2 | 807 bp 5 exons |
36-578 TGA |
790-792 TAA | Not Identified |
tctctctcctgcagtatgagtgaccacagggcctcccagccca tat gag tga = Y E * |
| 353 | APRT | TGA-TCA | Term-Ser | |||||||||
| 411 | ARSB | gTAG-CAG | Term-Gln | 534 | 5q11-q13 | ARSB | NM_000046.2 | 6076 bp 9 exons |
1287-2888 TAG |
3036-3038 TAA |
3485..3490 AATAAA 4564-4569 AATAAA 5804-5809 AATAAA 6039..6044 AATAAA 6043..6048 AATAAA |
gggtgtggggcccttggatgtaggatttcagggaggctagaaa tgg atg tag = W M * |
| 435 | ASL | TAGg-TAC | Term-Tyr | 465 | 7cen-q11.2 | ASL | NM_000048.3 | 1937 bp 16 exons |
112-1506 TAG |
1654-1656 TAA |
1528-1533 AATAAA 1752-1757 AATAAA 1913-1918 AATAAA 1921-1926 AATAAA 1932-1937 AATAAA |
tactgcaggcacagcaggcctaggtcctcccacacctgccccc cag gcc tag = Q A * |
| 443 | ASPA | TAG-TGG | Term-Trp | 314 | 17pter-p13 | ASPA | NM_000049.2 | 1435 bp 6 exons |
159-1100 TAG |
1233-1235 TAA |
1364-1369 AATAAA |
gtattcgctgctgtttacattagaaatcacttccagcttacat tta cat tag = L H * |
| 472 | ATM | gTGA-GGA | Term-Gly | 3057 | 11q22-q23 | ATM | NM_000051.3 | 13147 bp 63 exons |
386-9556 TGA |
9641-9643 TAG |
10215-10220 ATTAAA 10514-10519 ATTAAA 13129-13134 AATAAA |
caggatggaaagcttgggtgtgatcttcagtatatgaattacc tgg gtg tga = W Y * |
| 472 | ATM | TGA-TCA | Term-Ser | |||||||||
| 477 | ATP1A2 | cTGA-CGA | Term-Arg | 1021 | 1q21-q23 | ATP1A2 | NM_000702.2 | 5496 bp 23 exons |
133-3195 TGA |
3277-3280 TAA |
5195-5200 AATAAA 5434..5439 AATAAA |
tggagaaggagacatactactgaccccattggaagaagaacca tac tac tga = Y Y * |
| 50617 | ATP6V0A4 | gTAG-CAG | Term-Gln | 841 | 7q33-q34 | ATP6V0A4 | NM_020632.2 | 3152 bp 23 exons |
284-2806 TAG |
2963-2965 TGA |
3039-3044 AATAAA 3116-3121 AATAAA |
tggatggcacagccgaggagtaggctgagggctgcacctccca gag gag tag = E E * |
| 540 | ATP7B | cTGA-CGA | Term-Arg | 1466 | 13q14.3 | ATP7B | NM_000053.2 | 6644 bp 21 exons |
158-4555 TGA |
4556-4558 TGA |
3788-3793 ATTAAA 4831-4836 ATTAAA 4892-4897 AATAAA |
gggatgaggagcagtacatctgatgacttcaggcaggcgggcc tac atc tga = Y I * |
| 166379 | BBS12 | TAGt-TAC | Term-Tyr | 711 | 4q27 | BBS12 | NM_152618.2 | 3260 bp 2 exons |
194-2326 TAG |
2375-2377 TAA |
2379-2384 AATAAA 3220-3225 AATAAA |
taacgggctttctatttttgtagtgttactggctaagtctttg ttt ttg tag = T L * |
| 120329 | CASP12 | gTGA-CGA | Term-Arg | 125 | 11q22.3 | CASP12 | AY358222.1 | 3-1057 TAA |
1064-1066 TAA |
1227..1232 AATAAA |
ctatctctttcctgggaattaaactcataagaagcaactca ggg aat taa = G N |
|
| 846 | CASR | aTAA-CAA | Term-Gln | 1079 | 3q13 | CASR | NM_000388.2 | ??bp ? exons |
439-3609 TAA |
3631-3633 TAG |
5831-5836 ATTAAA 6126-6131 AATAAA 6615..6620 AATAAA |
agaaaacgtagtgaattcataaaatggaaggagaagactg aat tca taa = N S * |
| 1027 | CDKN1B | gTAA-CAA | Term-Gln | 199 | 12p13.1-p12 | CDKN1B | NM_004064.2 | 2422 bp 3 exons |
466-1062 TAA |
1240-1242 TGA |
1836-1841 ATTAAA 1948-1953 ATTAAA 2382-2387 AATAAA |
ctcagaagacgtcaaacgtaaacagctcgaattaagaatatg caa acg taa = Q T * |
| 120329 | CFTR | TAG-TGG | Term-Trp | 1481 | 7q31.2 | CFTR | NM_000492.3 | 6132 bp 27 exons |
133-4575 TAG |
4585-4587 TAA |
6108..6113 AATAAA |
aggtgcaagatacaaggctttagagagcagcataaatgttgac agg ctt tag = R L * |
| 1080 | COL1A2 | aTAA-CAA | Term-Gln | 1277 | 7q22.1 | COL1A2 | NM_000089.3 | 5411 bp 52 exons |
472-4572 TAA |
4585-4587 TAA |
4848-4853 AATAAA 4861-4866 AATAAA 5357-5362 AATAAA 5378..5383 AATAAA |
ttggcccagtctgtttcaaataaatgaactcaatctaaattaa ttc aaa taa = F K * |
| 1378 | CRYBB1 | gTGA-CGA | Term-Arg | 253 | 22q12.1 | CRYBB1 | NM_001887.3 | 921 bp 6 exons |
71-829 TGA |
905..907 TAA |
903..908 AATAAA |
tggccacagagccccccaagtgagtccacacctcactctgcta ccc aag tga = P K * |
| 1414 | CRYM | TAAa-TAT | Term-Tyr | 315 | 16p13.11-p12.3 | CRYM | NM_001888.2 | 1303 bp 9 exons |
86-1030 TAA |
1043-1045 TGA |
1267..1272 AATAAA |
attcctggtcatctggtaaataaaacaaaggaacttgatgttg ggt aaa taa = G K * |
| 1428 | CTSK | TGAc-TGG | Term-Trp | 330 | 1q21 | CTSK | NM_000396.2 | 1702 bp 8 exons |
125-1114 TGA |
1169-1171 TAA |
1650-1655 AATAAA 1680-1685 AATAAA |
tggccagcttccccaagatgtgactccagccagccaaatccat aag atg tga = K M * |
| 1513 | CYP2C19 | TGAa-TGC | Term-Cys | 491 | 10q24.1-q24.3 | CYP2C19 | NM_000769.1 | 1473 bp 9 exons |
1-1473 TGA |
1549-1551 TGA |
1617-1622 ATTAAA 1733-1738 ATTAAA |
agctgtgcttcattcctgtctgaagaagcacagatggtctggc cct gtc tga = P V * |
| 1557 | DBT | TGA-TTA | Term-Leu | 422 | 1p31 | DBT | NM_001918.2 | 10831 bp 11 exons |
34-1482 TGA |
1501-1503 TGA |
Multiple polyA sites 10794-10799 AATAAA |
ttatgctactagatctgaaatgaagactgataagacattcttg ctg aaa tga = L K * |
| 1629 | DHCR7 | cTAA-CAA | Term-Gln | 476 | 11q13.2-q13.5 | DHCR7 | NM_001360.2 | 2665 bp 8 exons |
274-1701 TAA |
1852-1854 TAA |
2099-2105 AATAAA 2642-2648 ATTAAA |
gcctgctgcctggaatcttctaagggcacgccctagggagaag atc ttc taa = I F * |
| 1717 | DOK7 | tTGA-CGA | Term-Arg | 505 | 4p16.2 | DOK7 | NM_173660.3 | 2566 bp 7 exons |
71-1585 TGA |
2130-2132 TGA |
2547-2553 AATAAA |
tcaaggtaaacccccctccttgagagccgcagatcccgccccg cct cct tga = P P * |
| 285489 | EDA | cTAG-CAG | Term-Gln | 392 | Xq12 | EDA | NM_001399.4 | 5296 bp 10 exons |
243-1418 TAG |
1503-1505 TGA |
5251-5256 AATAAA |
tgggtgaagcccctgcatcctagattccccccattttgcctct gca tcc tag = A S * |
| 2110 | ETFDH | gTAA-CAA | Term-Gln | 618 | 4q32-q35 | ETFDH | NM_004453.2 | 2349 bp 13 exons |
333-2186 TAA |
2223-2225 TAA |
2307-2312 AATAAA |
acctgcttacaatggaatgtaaactgcagctagccagtttct gga atg taa = G M * |
| 1896 | EYA1 | TAAc-TAC | Term-Tyr | 593 | 8q13.3 | EYA1 | NM_000503.3 | 4326 bp 18 exons |
641-2419 TAA |
2435-2437 TGA |
3014-3020 ATTAAA 3585-3591 AATAAA 3849-3855 AATAAA 4299-4304 AATAAA |
ccttggaactggagtacctgtaacagcgctcggcactttgaca tac ctg taa = Y L * |
| 2138 | F8 | cTGA-CGA | Term-Arg | 2333 | Xq28 | F8 | NM_000132.2 | 9030 bp 27 exons |
172-7227 TGA |
7327-7329 TAG |
7637-7643 AATAAA 8004-8010 AATAAA 8048-8054 AATAAA 9010-9015 AATAAA |
gcgaggcacaggacctctactgagggtggccactgcagcacct ctc tac tga = L Y * |
| 2157 | FGB | aTAG-AAG | Term-Lys | 462 | 4q28 | FGB | NM_005141.2 | 1949 bp 8 exons |
26-1501 TAG |
1535-1537 TGA |
1649-1655 AATAAA 1913-1918 AATAAA |
ggcccttcttcccacagcaatagtccccaatacgtagattttt cag caa tag = Q Q * |
| 2244 | FGFR3 | gTGA-AGA | Term-Arg | 807 | 4p16.3 | FGFR3 | NM_000142.2 | 4093 bp 18 exons |
40-2460 TGA |
2759-2761 TAA |
4238-4243 AATAAA |
gcagtgggggctcgcggacgtgaagggccactggtccccaaca cgg acg tga = R T * |
| 2261 | FGFR3 | gTGA-GGA | Term-Gly | |||||||||
| 2261 | FGFR3 | TGA-TCA | Term-Ser | |||||||||
| 2261 | FGFR3 | TGA-TTA | Term-Leu | |||||||||
| 2261 | FGFR3 | TGAa-TGC | Term-Cys | |||||||||
| 2261 | FGFR3 | TGAa-TGG | Term-Trp | |||||||||
| 2261 | FGFR3 | TGAa-TGT | Term-Cys | |||||||||
| 2273 | FHL1 | gTAA-GAA | Term-Glu | 281 | Xq26 | FHL1 | NM_001449.3 | 2398 bp 8 exons |
209-1051 TAA |
1205-1207 TAA |
2360..2365 AATAAA |
cactgcaaaaaatgctccgtgaatctggccaacaagcgcttt gct ccg tga = A P * |
| 2261 | FKRP | cTGA-AGA | Term-Arg | 496 | 19q13.32 | FKRP | NM_024301.3 | 3349 bp 4 exons |
2980-1785 TGA |
1846-1848 TGA |
2489-2494 AATAAA 2540-2545 AATAAA |
tgagtctgacgggaagcggctgaagccctgataacctcgcctt agc ggc tga = S G * |
| 79147 | FMO2 | tTAG-CAG | Term-Gln | 472 | 1q23-q25 | FMO2 | NM_001460.2 | 5181 bp 10 exons |
118-1533 TAG |
1723-1725 TAG |
Multiple polyA sites ?????? AATAAA |
tcggaccctgcaactcctattagtatcgcctggttgggcctgg tcc tat tag = S Y * |
| 2301 | FOXE3 | gTGA-CGA | Term-Arg | 320 | 1p32 | FOXE3 | NM_012186.2 | 2000 bp SINGLE Exon |
245-1204 TGA |
1418-1420 TGA |
1939-1944 ATTAAA 1954-1959 ATTAAA |
cggggctggagcgctacctgtgagcctgcgccgcgcgggcag tac ctg tga = Y L |
| 2301 | FOXE3 | TGA-TCA | Term-Ser | |||||||||
| 2294 | FOXF1 | gTGA-CGA | Term-Arg | 380 | 16q24 | FOXF1 | NM_001451.2 | 2579 bp 3 exons |
44-1183 TGA |
1400-1402 TAG |
3218-3223 AATAAA 3301-3306 AATAAA |
acatcaagccttgcgtgatgtgaggctgccgccgcaggccct gtg atg tga = V M * |
| 2327 | FOXH1 | gTGA-CGA | Term-Arg | 366 | 8q24.3 | FOXH1 | NM_003923.1 | 1793 bp 3 exons |
580-1677 TGA |
1684-1686 TAA |
Not found | tgctctcctggtgcagcctgtgaggctcttaagacaggggcca agc ctg tga = S L * |
| 8928 | FUCA1 | gTAA-AAA | Term-Lys | 462 | 1p34 | FUCA1 | NM_000147.3 | 2095 bp 8 exons |
46-1446 TAA |
1681-1683 TGA |
1575-1581 ATTAAA 2044-2049 AATAAA |
taaagctgacaggagtgaagtaatcatttgagtgcaagaagaa gtg aag taa = V K * |
| 2517 | GALT | cTGA-CGA | Term-Arg | 380 | 9p13 | GALT | NM_000155.2 | 1347 bp 11 exons |
68-1207 TGA |
1352-1354 TAA |
1315-1320 AATAAA |
gggagacagcaaccatcgcctgaccacgccgaccacagggcct atc gcc tga = I A * |
| 2592 | GALT | TGAc-TGC | Term-Cys | |||||||||
| 2623 | GATA1 | aTGA-CGA | Term-Arg | 414 | Xp11.23 | GATA1 | NM_002049.2 | 1522 bp 6 exons |
113-1354 TGA |
1475-1477 TAA |
1478-1484 AATAAA |
gtggctccgctcagctcatgagggcacagagcatggcct agc tca tga = S S * |
| 2592 | GCDH | TGAg-TGG | Term-Trp | 439 | 19p13.2 | GCDH | NM_000159.2 | 1839 bp 12 exons |
78-1394 TGA |
1473-1475 TGA |
1802..1807 AATAAA |
aggcgttcacggccagcaagtgagccgctccatcaggggcccg agc aag tga = S K * |
| 2639 | GCH1 | cTGA-CGA | Term-Arg | 251 | 14q22.1-q22.2 | GCH1 | NM_000161.2 | 2941 bp 6 exons |
162-914 TGA |
1017-1019 TGA |
Multiple polyA sites 2896-2901 ATTAAA |
tcctgactctcattaggagctgagcttcattcagtgtgtgtgc agg agc tga = R S * |
| 2645 | GCK | gTGA-CGA | Term-Arg | 466 | 7p15.3-p15.1 | GCK | NM_000162.2 | 2759 bp 10 exons |
487-1884 TGA |
2314-2316 TGA |
2724-2729 ATTAAA |
aggcctgtatgctgggccagtgagagcagtggccgcaagcgcag ggc cag tga = G Q * |
| 2645 | GCK | TGA-TTA | Term-Leu | |||||||||
| 55806 | HR | cTAG-CAG | Term-Gln | 35 | 8p21.2 | HR | NM_005144.3 | 4981 bp 19 exons |
131-3700 TAG |
4151-4153 TGA |
4311-4316 ATTAAA 4952-4957 ATTAAA 4956-4961 AATAAA |
caggaggccaaatagagggatgctaggtg gcc aaa tag = A K * |
| HR | TAG-TGG | Term-Trp | ||||||||||
| 2643 | HBA2 | TAAg-TAT | Term-Tyr | 142 | 16p13.3 | HBA2 | NM_000517.3 | 575 bp 3 exons |
38-466 TAA |
557-559 TAA |
555-560 AATAAA |
tgctgacctccaaataccgttaagctggagcctcggtagccgt tac cgt taa = Y R * |
| 3040 | HBA2 | tTAA-AAA | Term-Lys | |||||||||
| 3040 | HBA2 | tTAA-CAA | Term-Gln | |||||||||
| 3040 | HBA2 | tTAA-GAA | Term-Glu | |||||||||
| 3040 | HBA2 | tTAA-TCA | Term-Ser | |||||||||
| 3081 | HGD | tTGA-CGA | Term-Arg | 446 | 3q13.33 | HGD | NM_000187.2 | 1920 bp 14 exons |
371-1708 TGA |
1778-1780 TAG |
1892..1898 AATAAA |
ccagcagaacctaattgagactggaacattgctaccataa cct aat tga = P N * |
| 3284 | HSD3B2 | TGAt-TGC | Term-Cys | 373 | 1p13.1 | HSD3B2 | NM_000198.2 | 1669 bp 4 exons |
143-1261 TGA |
1544-1546 TGA |
1649..1654 AATAAA |
ccctgaagtccaagactcagtgatttaaggatgacagagatgt act cag tga = T Q * |
| 3425 | IDUA | aTGA-GGA | Term-Gly | 654 | 4p16.3 | IDUA | NM_000203.3 | 2203 bp 14 exons |
89-2050 TGA |
2231-2233 TGA |
2145-2150 AATATA |
ccccatccccgggcaatccatgagcctgtgctgagccccagtg aat cca tga = N P * |
| 3425 | IDUA | TGAg-TGT | Term-Cys | |||||||||
| 8517 | IKBKG | TAG-TGG | Term-Trp | 420 | Xq28 | IKBKG | NM_001099856.1 | 2073 bp 10 exons |
225-1483 TAG |
1563-1565 TAG |
2049-2054 AGTAAA |
atgtcatggagtgcattgagtagggccggccagtgcaaggcca att gag tag = I E * |
| 9445 | ITM2B | tTGA-AGA | Term-Arg | 267 | 13q14.3 | ITM2B | NM_021999.3 | 1870 bp 6 exons |
1874-987 TGA |
1018-1020 TAA |
1131-1136 ATTAAA 1440-1445 ATTAAA 1664-1669 AATAAA 1785-1790 AATAAA 1834-1839 ATTAAA |
tggaaactttaatttgttcttgaacagtcaagaaaaacattat tgt tct tga = C K * |
| 169522 | KCNV2 | tTAG-TAT | Term-Tyr | 546 | 9p24.2 | KCNV2 | NM_133497.2 | 1882 bp 2 exons |
215-1852 TAG |
2031-2033 TAG |
2142-2147 AATAAA |
tcaccccaagacaagagaattagtattttataggacatgtggc gag aat tag = E N * |
| 169522 | KCNV2 | tTAG-CAG | Term-Gln | |||||||||
| 84634 | KISS1R | cTGA-AGA | Term-Arg | 399 | 19p13.3 | KISS1R | NM_032551.4 | 1607 bp 5 exons |
146-1342 TGA |
1839-1841 TAA |
1554-1559 ATTAAA |
gggaggacaacgcccctctctgagcggacccggtgggaatccg cct ctc tga = P L * |
| 3914 | LAMB3 | TGAt-TGG | Term-Trp | 1173 | 1q32 | LAMB3 | NM_000228.2 | 4093 bp 23 exons |
145-3663 TGA |
3829-3831 TGA |
4008-4013 AATGAA 4020-4025 AATAAA |
tctactatgccacctgcaagtgatgctacagcttccagcccgt tgc aag tga = C K * |
| 9388 | LIPG | cTGA-CGA | Term-Arg | 501 | 18q21.1 | LIPG | NM_006033.2 | 4143 bp ? exons |
253-1755 TGA |
1900-1902 TGA |
4094-4099 ATTAAA 4118-4123 AATAAA |
actgtggagcttccctgagggtgcccgggcaagtcttg ctt ccc tga = L P * |
| 4143 | MAT1A | TAGa-TAT | Term-Tyr | 396 | 10q22 | MAT1A | NM_000429.2 | 3419 bp 9 exons |
256-1443 TAG |
1645-1647 TAA |
3382-3387 AATAAA |
ttcccaggaagcttgtattttagagccagggggagctgggcct gta ttt tag = V F * |
| 4159 | MC3R | TAG-TCG | Term-Ser | 361 | 20q13.2-q13.3 | MC3R | NM_019888.2 | 1112 bp 1 exon |
1-1083 TAG |
1102-1104 TGA |
Not found | gcaacggcatgaacttgggataggatgcagggccatggaaatg ttg gga tag = L G * |
| 64087 | MCCC2 | gTAA-CAA | Term-Gln | 564 | 5q12-q13 | MCCC2 | NM_022132.3 | 2329 bp 17 exons |
100-1791 TAA |
17987-1800 TAA |
1796-1801 AATAAA |
acttcggtatcttcaggatgtaactggaataaaggatgttttc agg atg taa = R M * |
| 5080 | MECP2 | cTGA-TGG | Term-Trp | 487 | Xq28 | MECP2 | NM_004992.2 | 10241 bp 4 exons |
227-1687 TGA |
1766-1768 TGA |
1790-1795 AATAAA 7191-7196 TATAAA 7300-7305 AATAAA 9490-9495 AATAAA |
ccgtgaccgagagagttagctgactttacacggagcggattgc gtt agc tga = V S * |
| 5080 | MECP2 | cTGA-CGA | Term-Arg | |||||||||
| 5080 | MECP2 | cTGA-TTA | Term-Leu | |||||||||
| 5080 | MECP2 | cTGA-TGC | Term-Cys | |||||||||
| 4338 | MOCS2 | TAAt-TAC | Term-Tyr | 189 | 5q11 | MOCS2 | NM_004531.3 | 1347 bp 8 exons |
40-793 TAA |
845-847 TGA |
1238-1243 ATTAAA 1289-1294 ATTAAA 1299-1304 AATAAA |
gcttttgggcatccaacagttaatcacttatgtttttagagca aac agt taa = N S * |
| 4524 | MTHFR | TGA-TCA | Term-Ser | 657 | 1p36.3 | MTHFR | NM_005957.3 | 7105 bp 12 exons |
185-2155 TGA |
2303-2305 TGA |
3833-3838 ?????? 7086-7091 AATAAA |
cgagagaaacggaggctccatgaccctgcgtcctgacgccctg gct cca tga = A P * |
| 55651 | NHP2 | aTGA-AGA | Term-Arg | 154 | 5q35.3 | NHP2 | NM_017838.3 | 867 bp 4 exons |
144-605 TGA |
756-758 TGA |
802-805 ACTAAA 836-841 AGTAAA |
agtccctgcccctacccctatgaggggctccggtagcacctgg ccc cta tga = P L * |
| 4878 | NPPA | cTGA-CGA | Term-Arg | 152 | 1p36.21 | NPPA | NM_006172.2 | 840 bp 3 exons |
95-550 TGA |
554-556 TAA |
768-773 ATTAAA 819-824 AATAAA |
ctgtgttctctttgcagtactgaagataacagccagggaggac cag tac tga = Q Y * |
| 190 | NR0B1 | aTAA-GAA | Term-Glu | 471 | Xp21.3-p21.2 | NR0B1 | NM_000475.3 | 1555 bp 2 exons |
13-1424 TAA |
1447-1479 TAA |
1475-1480 AA TAAA 1514-1519 AATAAA |
aaatgctctgtacaaagatataaagtcatgtgggccacacaag aag ata taa = K I * |
| 4939 | OAS2 | TAG-TGG | Term-Trp | 720 | 12q24.2 | OAS2 | NM_016817.2 | 3539 bp 11 exons |
141-2300 TAG |
2322-2324 TAA |
3015-3020 AATAAA 3340-3345 AATAAA 3513-3518 AATAAA |
ataattctaaaagaaacttctagagatcatctggcaatcgctt aac ttc tag = N F * |
| 4976 | OPA1 | TAAa-TAC | Term-Tyr | 961 | 3q28-q29 | OPA1 | NM_015560.1 | 5864 bp 31 exons |
56-2938 TGA |
2975-29771 TGA |
3046-3051 AATAAA |
aagctcttcatcaggagaaataaattaagtgagtaaaaattct gag aaa taa = E K * |
| 5009 | OTC | TGAt-TGG | Term-Trp | 355 | Xp21.1 | OTC | NM_000531.4 | 1647 bp 10 exons |
215..1279 TGA |
1319-1321 TAA |
1365-1370 AATAAA 1622-1627 AATAAA |
agctccagaagcctaaattttgatgttgtgttacttgtcaaga aaa ttt tga = K F * |
| 5080 | PAX6 | TAA-TTA | Term-Leu | 423 | 11p13 | PAX6 | NM_000280.2 | 2816 bp 15 exons |
513-1781 TAA |
1821-1823 TAA |
2269-2274 ATTAAA 2495-2500 AATAAA |
aatactggccaagattacagtaa aaaaaaaaaaaaaaaaaaaaaggaaaggaaa tta cag taa = L Q * |
| 5080 | PAX6 | TAA-TAT | Term-Tyr | |||||||||
| 5189 | PEX1 | aTAA-CAA | Term-Gln | 1284 | 7q21.2 | PEX1 | NM_000466.2 | 4390 bp 24 exons |
97-3948 TAA |
4030-4032 TGA |
4261-4266 AATAAA 4356-4361 AATAAA |
gacagaaagtaactttagcataaaatatacttctttttgattt tta gca taa = L A * |
| 8929 | PHOX2B | TGAt-TGG | Term-Trp | 315 | 4p12 | PHOX2B | NM_003924.2 | 3033 bp 3 exons |
361-1305 TGA |
1426-1428 TGA |
1452-1457 AATAAA 1766-1771 AATAAA 1798-1803 ATTAAA 1861-1866 ATTAAA |
tagtgaagagcagtatgttctgatctggaatcctgcggcggcg atg ttc tga = M F * |
| 8929 | PHOX2B | TGAt-TGC | Term-Cys | |||||||||
| 55163 | PNPO | tTAA-CAA | Term-Gln | 262 | 17q21.32 | PNPO | NM_018129.2 | 3482 bp 7 exons |
154-939 TAA |
1021-1023 TAA |
1405-1410 ATTAAA 2412-2417 ATTAAA 3438-3443 AATAAA |
tctatgagagacttgcaccttaactctgggacctgctggccca gca cct taa = A P * |
| 5627 | PROS1 | TAAg-TAT | Term-Tyr | 636 | 3q11.2 | PROS1 | NM_000313.1 | 3309 bp 15 exons |
147-2177 TAA |
2217-2219 TAA |
2636-2641 ATTAAA 2735-2740 ATTAAA 3289-3294 AATAAA |
ggaaaaagacaaagaattcttaaggcatcttttctctgcttat aat tct taa = N A * |
| 10594 | PRPF8 | cTGA-CGA | Term-Arg | 2336 | 17p13.3 | PRPF8 | NM_006445.3 | 7311 bp 43 exons |
115-7122 TGA |
7243-7245 TGA |
7261-7266 AATAAA 7274-7279 AATAAA |
atcgggaggacctgtatgcctgaccgtttccctgcctcctgct tat gcc tga = Y A * |
| 5744 | PTHLH | TGAa-TGG | Term-Trp | 178 | 12p12.1-p11.2 | PTHLH | NM_198965.1 | 1331 bp 5 exons |
323-856 TGA |
1013-1015 TGA |
1304-1309 AATAAA |
ttcacggaggcattgaaattttcagcagagaccttc agg cat tga = R H |
| 10111 | RAD50 | TAAa-TAT | Term-Tyr | 1313 | 5q31 | RAD50 | NM_005732.2 | 5891 bp 25 exons |
388-4326 TAA |
4522-4524 TGA |
5836-5841 AATAAA |
tgggattcaatgttcattaaaaatatccaagatttaaatg gtt cat taa = V H * |
| 6066 | RHCE | TAAg-TAC | Term-Tyr | 418 | 1p36.11 | RHCE | NM_020485.3 | 1635 bp 9 exons |
87-1340 TAA |
1416-1418 TGA |
1482-1487 ATTAAA 1490-1495 ATTAAA 1536-1541 AATAAA 1596-1601 ATTAAA |
atttggctgttggattttaagcaaaagcatccaagaaaaa gga ttt taa = G F * |
| 6010 | RHO | cTAA-CAA | Term-Gln | 349 | 3q21-q24 | RHO | NM_000539.2 | 2768 bp 5 exons |
96-1142 TAA |
1293-1295 TAA |
1239-1244 AATGAA 1506-1511 AATAAA 1659-1664 TATAAA 2563-2568 ATTAAA |
cgagccaggtggccccggcctaagacctgcctaggactctgtg ccg gcc taa = P A * |
| 6010 | RHO | cTAA-GAA | Term-Glu | |||||||||
| 860 | RUNX2 | TGA-TCA | Term-Ser | 522 | 6p21 | RUNX2 | NM_001024630.2 | 5572 bp 9 exons |
7-1776 TGA |
1853-1855 TAG |
2761-2666 ATTAAA 3073-3078 ATTAAA 3892-3897 AATAAA 4183-4188 ATTAAA 4448-4453 ATTAAA 4591-4596 ATTAAA |
aatctgtttggcgaccatattgaaattcctcagcagtggccca cca tat tga = P Y |
| 710 | SERPING1 | cTGA-AGA | Term-Arg | 479 | 11q12-q13.1 | SERPING1 | NM_000062.2 | 1984 bp 8 exons |
192-1694 TGA |
1830-1832 TGA |
1940-1945 AATAAA |
gagtatatgaccccagggcctgagacctgcaggatcaggttag agg gcc tga = R A * |
| 4068 | SH2D1A | aTGA-AGA | Term-Arg | 129 | Xq25-q26 | SH2D1A | NM_002351.2 | 2507 bp 4 exons |
346-732 TGA |
766-768 TAA |
738-743 AATAAA 1036-1041 AATAAA |
atgtctgcctgaaagccccatgaagaaaaataaaacaccttgt gcc cca tga = A P * |
| 6473 | SHOX | cTGA-CGA | Term-Arg | 293 | Xp22.33 | SHOXa | NM_000451.3 | 3757 bp 6 exons |
692-1570 TGA |
1712-1715 TAG |
2486-2491 ATTAAA |
gcggaggccctggggctctgacccgccgcgcagccc ggg ctc tga = G L * |
| 6473 | SHOX | aTGA-CGA | Term-Arg | 226 | SHOXb | NM_006883.2 | 1951 bp 6 exons |
692-1369 TGA |
1433-1436 TAG |
Not found | ||
| 5172 | SLC26A4 | TGAa-TGG | Term-Trp | 781 | 7q31 | SLC26A4 | NM_000441.1 | 4930 bp 21 exons |
225-2567 TGA |
2691-2693 TAA |
2719-2724 AATAAA 3014-3019 AATAAA 3038-3043 AATAAA 3066-3071 AATAAA 3229-3234 AATAAA |
ctatgcgtacacttgcatcctgaaagtgggttcgggaggtctc gca tcc tga = A S * |
| 54977 | SLC25A38 | cTGA-CGA | Term-Arg | 305 | 3p22.1 | SLC25A38 | NM_017875.2 | 2124 bp 7 exons |
402-1316 TGA |
1398-1400 TAA |
1897-1902 AATAAA 1965-1670 ATTAAA 2092-2097 AATAAA |
gggcctgaagtcctgaccaagagaggactgg aag tcc tga = K S * |
| 6663 | SOX10 | cTAA-TAC | Term-Tyr | 467 | 22q13.1 | SOX10 | NM_006941.3 | 2882 bp 4 exons |
279-1679 TAA |
1935-1937 TGA |
2840-2845 AATAAA 2846-2851 ATTAAA |
atacgacactgtcccggccctaaagggggccctgtcgccacca cgg ccc taa = R P * |
| 6663 | SOX10 | cTAA-AAA | Term-Lys | |||||||||
| 6716 | SRD5A2 | TAA-TCA | Term-Ser | 255 | 2p23 | SRD5A2 | NM_000348.3 | 2446 bp 5 exons |
72..836 TAA |
918-920 TAA |
846-851 ATTAAA 1235-1240 ATTAAA 2426-2431 ATTAAA |
cccttattccattcatcttttaaaggaaccaaattaaaaagga atc ttt taa = I F * |
| 7170 | TPM3 | TAA-TCA | Term-Ser | 286 | 1q21.2 | TPM3 | NM_152263.2 | 7116 bp 12 exons |
116-973 TAA |
1142-1144 TAA |
1140-1145 ATTAAA 1414-1419 ATTAAA 1894-1899 ATTAAA |
tccttactttttcatacagataattatcaccgtttctgctctg tac aga taa = Y R * |
| 7454 | WAS | cTGA-AGA | Term-Arg | 503 | Xp11.4-p11.21 | WAS | NM_000377.1 | 1806 bp 12 exons |
35-1543 TGA |
1805-1807 TAA |
1777-1782 AATAAA |
aagatgatgaatgggatgactgagtggctgagttacttgctgc gat gac tga = D D * |
| 7454 | WAS | cTGA-CGA | Term-Arg | |||||||||
| 7454 | WAS | TGA-TCA | Term-Ser | |||||||||
| 7490 | WT1 | TGAg-TGG | Term-Trp | 450 | 11p13 | WT1 | NM_024424.2 | 3020 bp 10 exons |
197-1741 TGA |
1805-1807 TGA |
2206-2211 AATAAA 3002-3007 ATTAAA |
ccaaactccagctggcgctttgaggggtctccctcggggaccg gcg ctt tga = A L * |
A control dataset was established which comprised 1,692 genes listed in the HGMD (for which both coding and 3'-UTRs were obtainable from Ensembl [Build 37] but for which no termination codon [nonstop] mutations have so far been recorded). Data from the Transterm database http://uther.otago.ac.nz/Transterm.html, [16] representing a total of 29,210 stop codons associated with annotated human genes, were used as genome-wide controls.
Analysis of nonstop mutations
The relative frequency of each type of stop codon (ie TAG, TAA and TGA) in the mutated (nonstop mutation-bearing) sequences and non-mutated wild-type control gene sequences was assessed. Stop codons harbouring single and multiple mutations were examined separately.
To detect any bias in the pattern of stop codon mutability, the mutability of the dinucleotides within a pentanucleotide spanning the stop codon and including one flanking nucleotide on either side was assessed. The number of mutations occurring in each of the 12 possible dinucleotides (note that four dinucleotides [CC, CA, CG and TC] cannot occur in conjunction with any stop codon-spanning pentanucleotide and were therefore omitted) was counted. In the HGMD control dataset, one nucleotide position within each stop codon was randomly mutated and the numbers of mutations in each possible dinucleotide were then counted. Statistical significance was determined using Fisher's exact test with a Bonferroni correction being applied to allow for multiple testing.
Since the identity of the nucleotides immediately flanking the stop codon may influence the susceptibility of the stop codon to mutation, the frequencies of each DNA base in each of the six positions upstream and downstream of the normally used stop codon were obtained for both the mutated sequences and the controls. The expected frequency E of the DNA bases at each position was calculated based on the probability of observing this nucleotide in the HGMD control sequences:
where Eij is the expected frequency of the base I = {A, C, G, T} at position j, Fij is the observed frequency of base i at position j in the HGMD control dataset, Nm is the total number of mutated sequences and Nc is the number of sequences in the HGMD control dataset. Under the assumption that the data follow a binomial distribution, we considered that an increase or decrease in the observed frequency of a particular nucleotide in a specified position was statistically significant if the corresponding p value was < 0.01. In addition, to investigate whether any particular stop codon (ie TGA, TAG or TAA) was associated with any specific flanking nucleotides, we placed both the mutated and control sequences into separate datasets for each of the three stop codons and repeated the above analysis for each of the new datasets.
Determining the distance to the next downstream in-frame stop codon
The distance to the next downstream stop codon in the required reading frame is likely to determine the length of any extended protein product. For each mutated (nonstop mutation-bearing) DNA sequence and each sequence in the HGMD control dataset, we therefore determined the distance to the next in-frame stop codon downstream. Sequences in the HGMD control dataset, for which the next downstream stop codon was beyond the 3'-UTR sequence available from Ensembl, were not used in this analysis. Distances between 0 and 500 base pairs (bp) from the original stop codon were divided into 'bins', each 50 bp long, the final bin containing all sequences where the distance was greater than 500 bp. The number of sequences which fell into each bin was recorded for both the mutated sequences and the HGMD control sequences. The same procedure was repeated for those sequences with single mutations and for those sequences harbouring two or more mutations. To assess the statistical significance of our findings, we employed Fisher's exact test using a Bonferroni correction to allow for multiple testing. p values of < 0.05 were considered to be statistically significant.
Using the same method as for the original stop codons, we also investigated the frequency of occurrence of specific nucleotides surrounding the next in-frame stop codon downstream. It is possible that at least a proportion of these downstream in-frame stop codons are associated with naturally occurring splice isoforms of the gene,[17] and might therefore possess comparable sequence characteristics to the stop codons involved in the mutational events. The flanking sequence may also affect the likelihood of a mutation coming to clinical attention.
Results and discussion
Relative frequency of stop codon involvement in nonstop mutation
We have performed a meta-analysis of the 119 nonstop mutations (in 87 different genes) known to cause human inherited disease (Supplementary Table S1 (Table 4)) and recorded in the HGMD [1]. HGMD is a comprehensive collection of germline mutations causing (or associated with) human inherited disease and is an invaluable source of data for meta-analyses of human gene mutations.
The termination of synthesis of every human protein is effected by one of three stop codons, TAG, TAA and TGA, listed in increasing order of usage in human genes. We posed the question as to whether one of these stop codons might be more susceptible to mutation, or alternatively might be more likely to come to clinical attention once mutated, than the others. We noted that a majority of the nonstop mutations (57 per cent) in our dataset occurred within TGA codons (Table 1). Since 49.4 per cent and 48.6 per cent of stop codons in the HGMD control gene dataset and human genome dataset, respectively, were of this type, however, this finding did not attain statistical significance (Table 1; p values 0.107 and 0.066, respectively).
Table 1.
The proportion of nonstop mutations harboured by each type of stop codon in mutated gene sequences, HGMD control gene sequences and the human genome at large
| Stop codon type | Proportion of stop codons harbouring nonstop mutations causing human genetic disease (%)a | Proportion of stop codons in HGMD control gene sequences (%)b | Estimated proportion (number) of stop codons in the human genome (%)c |
|---|---|---|---|
| TAA | 26.05 | 28.60 | 27.8 (8106) |
| TAG | 16.81 | 21.99 | 23.6 (6901) |
| TGA | 57.14 | 49.40 | 48.6 (14203) |
aMutations and sequences were taken from the HGMD [1].
bThe control dataset comprises 1,692 genes listed in the HGMD but for which no nonstop mutations have been recorded to date.
cBased on a total of 29,210 stop codons associated with annotated human genes. Data from the Transterm database http://uther.otago.ac.nz/Transterm.html[16]
The proportion of mutations in the other two types of stop codon was also not significantly different from the corresponding proportions in the set of HGMD control gene sequences (p values, 0.674 for TAA and 0.201 for TAG) and in the human genome at large (p values, 0.753 for TAA and 0.88 for TAG).
The above notwithstanding, we speculated whether TAA codons flanked on the 3' side by A might be hypermutable, since this would in effect constitute a short polyadenine run. It has been reported that bases adjacent to mononucleotide runs in the human genome are characterised by an increased single nucleotide polymorphism frequency [18]. We therefore assessed whether the nucleotide A following the TAA stop codon might influence the mutability of this codon. In agreement with our postulate, the presence of an A adjacent to a TAA stop codon was indeed found to increase the mutability of this codon by 1.4 fold (p = 0.016).
Genes exhibiting an abundance of missense/ nonsense mutations do not harbour a disproportionate number of nonstop mutations
As we have noted above, a total of 18 human genes are known to harbour multiple nonstop mutations. We therefore sought to determine whether this was simply due to a particularly large number of mutations having been reported from these genes. At the time this analysis was performed (October 2010), the HGMD contained mutation data from a total of 2,249 human genes, for which a total of 55,813 missense or nonsense mutations had been reported. No correlation was found, however, between the probability of finding multiple nonstop mutations in a given gene and the total number of missense and nonsense mutations reported for that gene (Pearson's correlation -0.108; p = 0.67). Thus, for example, the largest number of missense/nonsense mutations was reported from the F8 gene (1,217) but only one nonstop F8 mutation has been reported. Conversely, no missense/nonsense mutations have been recorded for the HR gene, even though two nonstop mutations have been identified. Hence we may conclude that the observation that some genes harbour multiple nonstop mutations is unrelated to the number of reported missense and nonsense mutations for those genes.
Gene ontology analysis for genes harbouring nonstop mutations
The Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) was used to identify enriched biological themes within the group of 87 genes harbouring either multiple or single nonstop mutations [19]. A total of 13 terms were found to be significantly enriched (p < 0.001, without correction for multiple testing) for single mutations (see Supplementary Table S2 (Table 5)). One of the most significantly enriched terms was 'oxidoreductase' (p = 0.005 after Bonferroni correction), which was associated with 11 of the 67 nonstop mutation-harbouring genes identified in the DAVID database [20]. Six terms were found to be significantly enriched (p < 0.001 without correction for multiple testing) for genes harbouring multiple nonstop mutations (Supplementary Table S3 (Table 6)); however, no significant bias in gene function was noted for these genes after correction for multiple testing. A search using all nonstop mutation-containing genes revealed an association with the protein information resource (PIR) term 'deafness' (p = 0.0248), corresponding to six of 86 sequences, although the biological relevance of this observation remains unclear.
Table S2.
Major enriched (p < 0.001) categories for genes harbouring single mutations in stop codons
| Category | Term | Count | % | p value | Genes |
|---|---|---|---|---|---|
| SP_PIR_KEYWORDS | Oxidoreductase | 11 | 16.42 | 2.03E-05 |
HSD3B2, DBT, GCDH, MTHFR, CYP2C19, DHCR7, FMO2, ETFDH, HGD, PNPO, SRD5A2 |
| GOTERM_BP_FAT | GO:0044271 ~ nitrogen compound biosynthetic process | 9 | 13.43 | 1.40E-04 |
MOCS2, OTC, SLC25A38, ATP1A2, ASL, ATP6V0A4, NPPA, ATP7B, GCH1 |
| GOTERM_BP_FAT | GO:0008015 ~ blood circulation | 7 | 10.45 | 2.41E-04 |
MTHFR, COL1A2, SERPING1, CFTR, ATP1A2, NPPA, GCH1 |
| GOTERM_BP_FAT | GO:0003013 ~ circulatory system process | 7 | 10.45 | 2.41E-04 |
MTHFR, COL1A2, SERPING1, CFTR, ATP1A2, NPPA, GCH1 |
| GOTERM_MF_FAT | GO:0050662 ~ coenzyme binding | 7 | 10.5 | 2.59E-04 |
DBT, GCDH, FMO2, ETFDH, PNPO, CRYM, GCH1 |
| SP_PIR_KEYWORDS | Blood coagulation | 4 | 5.97 | 4.62E-04 | FGB, F8, SERPING1, PROS1 |
| SP_PIR_KEYWORDS | Flavoprotein | 5 | 7.46 | 5.00E-04 | GCDH, MTHFR, FMO2, ETFDH, PNPO |
| GOTERM_CC_FAT | GO:0031093 ~ platelet alpha granule lumen | 4 | 5.97 | 6.78E-04 | FGB, F8, SERPING1, PROS1 |
| GOTERM_BP_FAT | GO:0006694 ~ steroid biosynthetic process | 5 | 7.46 | 6.92E-04 |
HSD3B2, DHCR7, CFTR, SRD5A2, NR0B1 |
| GOTERM_BP_FAT | GO:0042592 ~ homeostatic Process | 12 | 17.91 | 7.17E-04 |
PTHLH, SLC26A4, CTSK, CASR, OTC, IKBKG, SLC25A38, LIPG, ATP1A2, ATP6V0A4, RAD50, ATP7B |
| GOTERM_BP_FAT | GO:0055114 ~ oxidation Reduction | 11 | 16.42 | 7.76E-04 |
HSD3B2, GCDH, MTHFR, CYP2C19, DHCR7, FMO2, ETFDH, F8, HGD, PNPO, SRD5A2 |
| GOTERM_CC_FAT | GO:0060205 ~ cytoplasmic membrane-bounded vesicle Lumen | 4 | 5.97 | 8.35E-04 | FGB, F8, SERPING1, PROS1 |
| GOTERM_CC_FAT | GO:0031983 ~ vesicle lumen | 4 | 5.97 | 9.52E-04 | FGB, F8, SERPING1, PROS1 |
Table S3.
Major enriched (p < 0.001) categories for genes harbouring multiple mutations in stop codons
| Category | Term | Count | % | p value | Genes |
|---|---|---|---|---|---|
| SP_PIR_KEYWORDS | DNA-binding | 8 | 42.11 | 9.77E-04 |
SOX10, PHOX2B, MECP2, PAX6, HR, SHOX, ATM, FOXE3 |
| SP_PIR_KEYWORDS | Peters' anomaly | 2 | 10.53 | 0.0047 | PAX6, FOXE3 |
| SP_PIR_KEYWORDS | Transcription regulation | 7 | 36.84 | 0.0082 |
SOX10, PHOX2B, MECP2, PAX6, HR, SHOX, FOXE3 |
| GOTERM_MF_FAT | GO:0043565 ~ sequence-specific DNA binding | 5 | 26.32 | 0.0086 |
SOX10, PHOX2B, PAX6, SHOX, FOXE3 |
| GOTERM_MF_FAT | GO:0003700 ~ transcription factor activity | 6 | 31.58 | 0.0089 |
SOX10, PHOX2B, PAX6, HR, SHOX, FOXE3 |
| SP_PIR_KEYWORDS | Transcription | 7 | 36.84 | 0.0092 |
SOX10, PHOX2B, MECP2, PAX6, HR, SHOX, FOXE3 |
Mutability of the DNA sequence encompassing the mutated stop codons
The dinucleotide mutabilities within the pentanucleotides flanking the naturally mutated stop codons and the randomly mutated HGMD control stop codons were calculated in order to determine whether there was any bias in the mutability of the various dinucleotides that occur within the three types of stop codon, taking the flanking nucleotides into consideration. A strong positive correlation was noted between the distributions of mutation-harbouring dinucleotides and randomly simulated mutations within the stop codons of HGMD control sequences (Pearson's correlation r = 0.975; p = 8.04 × 10-8) with respect to the frequencies of 12 dinucleotides. No significant differences were found in dinucleotide-wise comparisons (Table 2), however, indicating that there is no evidence for a nearest nucleotide-directed bias in stop codon mutability.
Table 2.
The proportion of mutations found within dinucleotides in the mutated stop codon-flanking pentanucleotides as compared with randomly generated HGMD controls
| Dinucleotide | Occurrence of nonstop mutations in mutated sequence dataset (%) | Occurrence of random mutations within HGMD control sequences (%) | p value (after correction for multiple testing) |
|---|---|---|---|
| AA | 25 (21.00) | 348 (20.57) | 0.907 |
| AC | 6 (5.04) | 71 (4.196) | 0.636 |
| AG | 18 (15.13) | 303 (17.91) | 0.534 |
| AT | 16 (13.44) | 238 (14.066) | 1.0 |
| CT | 23 (19.33) | 318 (18.79) | 0.903 |
| GG | 1 (0.84) | 35 (2.07) | NA* |
| GA | 32 (26.89) | 424 (25.06) | 0.663 |
| GC | 1 (0.84) | 25 (1.48) | NA* |
| GT | 21 (17.65) | 259 (15.31) | 0.511 |
| TT | 10 (8.4) | 155 (9.16) | 1.0 |
| TA | 36 (30.25) | 606 (35.82) | 0.235 |
| TG | 49 (41.18) | 602 (35.58) | 0.236 |
*Sample size of mutated sequences too small to generate p values. (Note that four dinucleotides (CC, CA, CG and TC) cannot occur in conjunction with any stop codon-spanning pentanucleotide and were therefore omitted from this analysis.)
Sequence context around stop codons that have been subject to nonstop mutations
In eukaryotic cells, the translational efficiency and readthrough potential of the three different stop codons have been reported to vary as a consequence of the influence of the surrounding nucleotide sequence [21-26]. With respect to human gene sequences, Ozawa et al. reported that the first three nucleotide positions after the stop codon are highly conserved, with G and A predominating at the +1 position, and C at the +4 position [24]. Again in the context of human genes, Liu reported a preponderance of C immediately upstream of the stop codon (at position -1) and G or T at position +1 [26]. Our HGMD control dataset exhibits similar sequence characteristics to those stop codon datasets reported by Ozawa et al [24]. and Liu [26]. This sequence bias flanking human stop codons represents, in effect, a consensus sequence for the translational termination signal that extends beyond the confines of the stop codon itself. With this in mind, we next examined the flanking sequences of the mutated stop codons in order to ascertain whether the local DNA sequence context could influence the likelihood that the associated nonstop mutations would come to clinical attention.
We first examined the frequencies of six nucleotides on either side of the stop codon in both 87 mutated and 1,692 control sequences. When considering the entire stop codon dataset (which includes sequences flanking the TAA, TAG and TGA stop codons on the 5' side at positions -1 to -6, and on the 3' side at positions +1 to +6), we observed a significant paucity in G at the -2 position (p = 0.0063) (Supplementary Table S4 (Table 7)). When considering the three types of stop codon separately, there was a significant excess (p = 0.0016) of G and a significant paucity of A (p = 0.0047) two nucleotides downstream of TAA stop codons (Table 3). Similarly, in the regions flanking TGA stop codons, we noted a significant excess of T at the +6 position (p = 0.0094) (Supplementary Table S5 (Table 8)). Although it is conceivable that TAA stop codons with a G at +2 and TGA stop codons with a T at +6 may be more prone to mutate than other sequences, we prefer the alternative explanation, that mutations occurring in TAA and TGA stop codons embedded within these sequence contexts are more likely, for whatever reason, to come to clinical attention. No significant difference was noted between the flanking regions of mutated and control TAG stop codons (data not shown).
Table S4.
Frequency of nucleotides present in regions flanking the 87 mutated stop codons.
| Base | -6 | -5 | -4 | -3 | -2 | -1 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 25 | 25 | 12 | 25 | 29 | 16 | 31 | 20 | 19 | 13 | 28 | 20 |
| C | 18 | 20 | 27 | 26 | 24 | 27 | 15 | 26 | 26 | 25 | 22 | 28 |
| G | 24 | 23 | 28 | 14 | 7 | 24 | 28 | 28 | 19 | 21 | 21 | 19 |
| T | 20 | 19 | 20 | 22 | 27 | 20 | 13 | 23 | 28 | 16 | 20 |
Position 0, corresponding to the stop codon, is not shown. Nucleotide frequencies that are significantly higher/lower (p < 0.01) in comparison to the HGMD control dataset are shown underlined
Table 3.
Frequency of nucleotides present in regions flanking the mutated TAA stop codon (N = 40).
| Base | -6 | -5 | -4 | -3 | -2 | -1 | 1 | 2 | 3 | 4 | 5 | 6 |
| A | 14 | 13 | 7 | 10 | 10 | 5 | 17 | 6 | 11 | 7 | 18 | 11 |
| C | 7 | 9 | 15 | 10 | 13 | 13 | 9 | 10 | 12 | 13 | 9 | 14 |
| G | 8 | 10 | 11 | 5 | 12 | 12 | 11 | 15 | 9 | 9 | 7 | 8 |
| T | 11 | 8 | 7 | 15 | 10 | 10 | 3 | 9 | 8 | 11 | 6 | 7 |
Position 0, corresponding to the stop codon, is not shown. Nucleotide frequencies that are significantly higher/lower (p < 0.01) in comparison with the HGMD control dataset are shown underlined
Table S5.
Frequency of nucleotides present in regions flanking the mutated TGA stop codon (N = 35).
| Base | -6 | -5 | -4 | -3 | -2 | -1 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 9 | 9 | 4 | 12 | 12 | 9 | 12 | 9 | 8 | 6 | 10 | 6 |
| C | 7 | 8 | 10 | 13 | 10 | 11 | 4 | 12 | 8 | 9 | 8 | 9 |
| G | 12 | 10 | 11 | 7 | 5 | 9 | 13 | 10 | 9 | 9 | 8 | 8 |
| T | 7 | 8 | 10 | 3 | 8 | 6 | 6 | 4 | 10 | 11 | 9 | 12 |
Position 0 corresponding to the stop codon is not shown. Nucleotide frequencies that are significantly higher/lower (p < 0.01) in comparison to the HGMD control dataset are shown in bold
The nucleotide frequencies of the flanking regions of the stop codons that harboured single and multiple mutations were also analysed separately, and compared both with the HGMD control dataset and with each other. Supplementary Table S6 (Table 9) presents the comparison of sequences containing only single mutations with sequences in the HGMD control dataset. These sequences exhibit a significant paucity of G at the -2 (p = 0.0078) and -3 (p = 0.0096) positions relative to the controls. However, no significant difference was apparent between those sequences harbouring multiple mutations and controls (data not shown).
Table S6.
Frequency of nucleotides occurring within regions flanking mutated stop codons harbouring single nonstop mutations.
| Base | -6 | -5 | -4 | -3 | -2 | -1 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 21 | 19 | 11 | 21 | 21 | 14 | 26 | 15 | 16 | 11 | 23 | 16 |
| C | 14 | 17 | 19 | 19 | 19 | 19 | 11 | 19 | 22 | 21 | 18 | 23 |
| G | 19 | 18 | 22 | 9 | 5 | 17 | 21 | 23 | 13 | 14 | 14 | 14 |
| T | 14 | 14 | 16 | 19 | 23 | 18 | 10 | 11 | 17 | 22 | 13 | 15 |
Position 0 corresponding to the stop codon is not shown. Frequencies which are significantly higher/lower (p < 0.01) in comparison with corresponding HGMD controls are shown underlined
Sequence context around the next in-frame stop codon downstream of the stop codons that have been subject to nonstop mutations
The DNA sequences around the next downstream in-frame stop codon were analysed using the same method as described above. The regions flanking the next in-frame stop codons located downstream of the mutated stop codons were compared with their counterparts in the HGMD control sequences. This analysis was performed for each of the three codon types (TAA, TAG and TGA) separately and for all the mutated stop codons combined. When analysing all downstream in-frame stop codons together, a significant excess of T was observed at the +6 position (p = 0.0051; Supplementary Table S7 (Table 10)). When the three types of stop codon were examined separately, the only significant difference noted was in the sequences surrounding the next in-frame TGA stop codons, where an excess of C was found at the +6 position (p = 0.0019; Supplementary Table S8 (Table 11)), as compared with the TGA codons in the control dataset. Taken together, these findings suggest that, in general, there is no obvious difference between the sequences surrounding the next downstream in-frame stop codons and their counterparts in the HGMD control sequences. However, it is possible that the nucleotide occurring at position +6 relative to the downstream alternative in-frame stop codon could influence the likelihood that a given nonstop mutation might come to clinical attention.
Table S7.
Frequencies of nucleotides flanking the next downstream in-frame stop codon in mutated sequences.
| Base | -6 | -5 | -4 | -3 | -2 | -1 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 9 | 10 | 14 | 16 | 8 | 9 | 13 | 10 | 17 | 14 | 16 | 15 |
| C | 13 | 11 | 7 | 7 | 12 | 17 | 12 | 17 | 11 | 16 | 9 | 15 |
| G | 8 | 15 | 11 | 9 | 8 | 10 | 12 | 12 | 9 | 9 | 10 | 12 |
| T | 16 | 10 | 14 | 15 | 19 | 11 | 11 | 9 | 10 | 8 | 12 | 5 |
Position 0, corresponding to the stop codon, is not shown. Frequencies which are significantly higher/lower (p < 0.01) in comparison with the corresponding HGMD controls are shown underlined
Table S8.
Frequencies of nucleotides flanking the next downstream in-frame TGA stop codon.
| Base | -6 | -5 | -4 | -3 | -2 | -1 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 4 | 6 | 9 | 9 | 3 | 1 | 5 | 6 | 7 | 6 | 9 | 4 |
| C | 7 | 8 | 4 | 3 | 6 | 11 | 7 | 10 | 8 | 10 | 5 | 12 |
| G | 6 | 8 | 6 | 4 | 5 | 7 | 8 | 4 | 4 | 4 | 4 | 6 |
| T | 8 | 3 | 6 | 9 | 11 | 6 | 6 | 6 | 6 | 5 | 7 | 3 |
Position 0, corresponding to the stop codon, is not shown. Frequencies which are significantly higher/lower (p < 0.01) in comparison with the corresponding HGMD controls are shown in bold
The distance to the next stop codon is a key determinant of whether a given nonstop mutation will come to clinical attention
We next explored the possibility that the distance from the mutated stop codon to the next in-frame stop codon downstream might influence the likelihood that a given nonstop mutation would come to clinical attention. We reasoned that the greater the distance between the mutated stop codon and the next viable alternative downstream stop codon, the more likely it would be that the mRNA/ protein would be unstable/degraded and hence that the nonstop mutation would give rise to a deleterious and clinically observable phenotype. Conversely, the presence of an alternative in-frame stop codon in the immediate vicinity of the mutated natural stop codon could yield a near-normal or at least ameliorated clinical phenotype. Since such phenotypes would be less likely to come to clinical attention, we might therefore expect there to be a paucity of alternative in-frame stop codons in the immediate vicinity of the mutated stop codons as compared with their counterparts derived from the HGMD control sequences. This was, indeed, what was found when mutated and control sequences were compared. Although a relatively strong correlation was noted between the distributions of the distances (Pearson's correlation 0.75; p = 0.008), the number of alternative in-frame stop codons was found to be significantly lower among the mutated sequences than in the controls, but only in the range 0-49 nucleotides downstream of the mutated stop codon (p = 7.81 × 10-4). This implies that at least some stop codon mutations with alternative stop codons 0-49 nucleotides downstream of the mutated stop codon will not have come to clinical attention, possibly because they will have given rise to stable mRNAs that were (i) not subject to nonstop mRNA decay and (ii) consequently translated into proteins of near-normal length and biological function.
Although the number of in-frame stop codons in the HGMD control dataset approximates to a Zipfian distribution, and steadily decreases with increasing distance from the original stop codon (Figure 1), we noted a significant excess (by comparison with the controls) of downstream in-frame stop codons within 150-199 nucleotides of the mutated stop codon (p = 8.551 × 10-4). A signifi-cant (p = 6.558 × 10-6) excess of in-frame stop codons within 100-299 nucleotides was also noted as compared with the HGMD controls. One possible explanation could be that the recruitment of these alternative stop codons at an intermediate distance from the mutated stop codon may serve to trigger nonstop mRNA decay, thereby dramatically decreasing the amount of protein product produced and giving rise to a clinical phenotype that is more likely to come to clinical attention. Confirmation or otherwise of this postulate must await the emergence of a clearer understanding of the mechanism of nonstop mRNA decay in mammalian cells.
Figure 1.
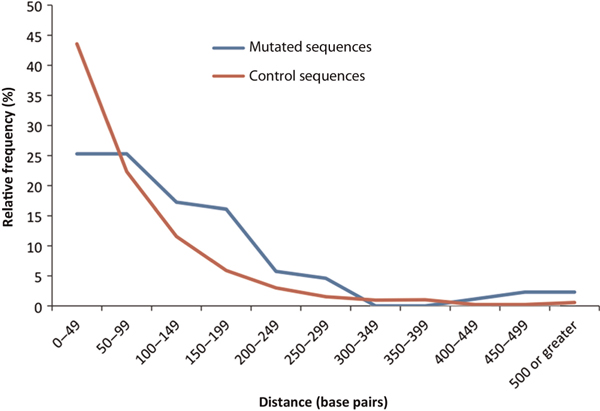
Distribution of distances (in nucleotides) to the next in-frame stop codon in mutated and HGMD control DNA sequences.
Figure 2 depicts a comparison of the single (N = 69 in 69 genes) and multiple (N = 18 in 18 genes) nonstop mutations with respect to the distribution of distances to the next downstream in-frame stop codon in each sequence. If those nonstop mutations which occurred within sequences lacking alternative in-frame stop codons in the range 0-49 nucleotides from the mutated codon did indeed display an increased likelihood of coming to clinical attention, then we might reasonably expect those sequences harbouring multiple nonstop mutations to exhibit an even greater paucity of alternative downstream in-frame stop codons in this size range relative to those sequences harbouring only one nonstop mutation. Although only 18 sequences harboured multiple nonstop mutations (yielding very small sample sizes in each distance category and precluding formal statistical assessment), only one (corresponding to 5.5 per cent of the total number of multiple nonstop mutations) of these sequences bearing multiple nonstop mutations was characterised by an alternative in-frame stop codon within 50 nucleotides downstream of the mutated stop codon, as opposed to 21 sequences with single mutations (30.9 per cent of the total number of single nonstop mutations) (Figure 2). This finding is therefore wholly compatible with our postulate that nonstop mutations occurring within DNA sequences lacking alternative in-frame stop codons in the immediate vicinity of the mutated stop codon display an increased likelihood of coming to clinical attention, possibly because the resulting extended mRNAs are more likely to be subject to nonstop mRNA decay.
Figure 2.
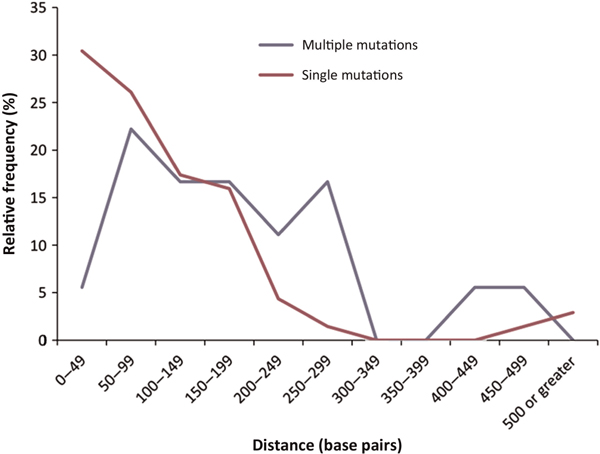
Distribution of distances to the next in-frame stop codon in DNA sequences harbouring single (N = 69) and multiple (N = 18) mutations.
References
- Stenson PD, Mort M, Ball EV, Howells K. et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A, Angelini M, Garelli E, Tchernia G. et al. Nonsense-mediated and nonstop decay of ribosomal protein S19 mRNA in Diamond-Blackfan anemia. Hum Mutat. 2004;24:526–533. doi: 10.1002/humu.20117. [DOI] [PubMed] [Google Scholar]
- Ameri A, Machiah DK, Tran TT, Channell C. et al. A nonstop mutation in the factor (F)X gene of a severely haemorrhagic patient with complete absence of coagulation FX. Thromb Haemost. 2007;98:1165–1169. doi: 10.1160/th07-02-0125. [DOI] [PubMed] [Google Scholar]
- Doucette L, Green J, Fernandez B, Johnson GJ. et al. A novel, non-stop mutation in FOXE3 causes an autosomal dominant form of variable anterior segment dysgenesis including Peters anomaly. Eur J Hum Genet. 2011;9:293–299. doi: 10.1038/ejhg.2010.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang S, Wang W, Rich B, David R. et al. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene causing, respectively, nonclassic and classic 3beta-HSD deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87:2556–2563. doi: 10.1210/jc.87.6.2556. [DOI] [PubMed] [Google Scholar]
- Torres-Torronteras J, Rodriguez-Palmero A, Pinós T, Accarino A. et al. A novel nonstop mutation in TYMP does not induce nonstop decay in a MNGIE patient with severe neuropathy. Hum Mutat. 2011;32:E2061–E2068. doi: 10.1002/humu.21447. [DOI] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O'Donnell K, Guerrerio AL. et al. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- Schaeffer D, van Hoof A. Different nuclease requirements for exosome-mediated degradation of normal and nonstop mRNAs. Proc Natl Acad Sci USA. 2011;108:2366–2371. doi: 10.1073/pnas.1013180108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T, Aiba H. Translation of aberrant mRNAs lacking a termination codon or with a shortened 3'-UTR is repressed after initiation in yeast. EMBO J. 2005;24:1584–1595. doi: 10.1038/sj.emboj.7600636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Meaux S, van Hoof A. A genomic screen in yeast reveals novel aspects of nonstop mRNA metabolism. Genetics. 2007;177:773–784. doi: 10.1534/genetics.107.073205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimitsu N, Tanaka J, Pelletier J. Translation of nonSTOP mRNA is repressed post-initiation in mammalian cells. EMBO J. 2007;26:2327–2338. doi: 10.1038/sj.emboj.7601679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isken O, Maquat LE. Quality control of eukaryotic mRNA: Safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- Akimitsu N. Messenger RNA surveillance systems monitoring proper translation termination. J Biochem. 2008;143:1–8. doi: 10.1093/jb/mvm204. [DOI] [PubMed] [Google Scholar]
- Danckwardt S, Hentze MW, Kulozik AE. 3' end mRNA processing: Molecular mechanisms and implications for health and disease. EMBO J. 2008;27:482–498. doi: 10.1038/sj.emboj.7601932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs GH, Chen A, Stevens SG, Stockwell PA. et al. Transterm: A database to aid the analysis of regulatory sequences in mRNAs. Nucleic Acids Res. 2008;37:D72-D76. doi: 10.1093/nar/gkn763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao M, Barrero RA, Mukai Y, Motono C. et al. Large-scale analysis of human alternative protein isoforms: Pattern classi-fication and correlation with subcellular localization signals. Nucleic Acids Res. 2005;33:2355–2363. doi: 10.1093/nar/gki520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddle KJ, Goodship JA, Keavney B, Santibanez-Koref MF. Bases adjacent to mononucleotide repeats show an increased single nucleotide polymorphism frequency in the human genome. Bioinformatics. 2011;27:895–898. doi: 10.1093/bioinformatics/btr067. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Dennis G Jr, Sherman BT, Hosack DA, Yang J. et al. DAVID: Database for Annotation Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- McCaughan KK, Brown CM, Dalphin ME, Berry MJ. et al. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc Natl Acad Sci USA. 1995;92:5431–5435. doi: 10.1073/pnas.92.12.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassan M, Rousset JP. UAG readthrough in mammalian cells: Effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol Biol. 2001;2:3. doi: 10.1186/1471-2199-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Hatin I, Rousset JP. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001;2:787–793. doi: 10.1093/embo-reports/kve176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa Y, Hanaoka S, Saito R, Washio T. et al. Comprehensive sequence analysis of translation termination sites in various eukaryotes. Gene. 2002;300:79–87. doi: 10.1016/S0378-1119(02)01042-9. [DOI] [PubMed] [Google Scholar]
- Cridge AG, Major LL, Mahagaonkar AA, Poole ES. et al. Comparison of characteristics and function of translation termination signals between and within prokaryotic and eukaryotic organisms. Nucleic Acids Res. 2006;34:1959–1973. doi: 10.1093/nar/gkl074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. Comparative analysis of base biases around the stop codons in six eukaryotes. Bio Systems. 2005;81:281–299. doi: 10.1016/j.biosystems.2005.05.005. [DOI] [PubMed] [Google Scholar]