

Abstract
Since 1998, the bioinformatics, systems biology, genomics and medical communities have enjoyed a synergistic relationship with the GeneCards database of human genes (http://www.genecards.org). This human gene compendium was created to help to introduce order into the increasing chaos of information flow. As a consequence of viewing details and deep links related to specific genes, users have often requested enhanced capabilities, such that, over time, GeneCards has blossomed into a suite of tools (including GeneDecks, GeneALaCart, GeneLoc, GeneNote and GeneAnnot) for a variety of analyses of both single human genes and sets thereof. In this paper, we focus on inhouse and external research activities which have been enabled, enhanced, complemented and, in some cases, motivated by GeneCards. In turn, such interactions have often inspired and propelled improvements in GeneCards. We describe here the evolution and architecture of this project, including examples of synergistic applications in diverse areas such as synthetic lethality in cancer, the annotation of genetic variations in disease, omics integration in a systems biology approach to kidney disease, and bioinformatics tools.
Keywords: GeneCards, GeneDecks, Partner Hunter, Set Distiller, omics, genomics, human genes, database, synthetic lethality, genetic variations
GeneCards system evolution and architecture
From the very beginning, the core GeneCards features included two important components: the capability to view integrated details about a gene in 'card' format and a full text-based search engine. GeneCards has evolved by constantly adding new data sources and data types (eg protein expression and gene networks), revamping the search engine to improve results and performance, and expanding the original gene-centric dogma to encompass sets of genes.
Currently, GeneCards automatically mines over 90 sources in an offline process and constructs a consolidated gene list. First, the complete current snapshot of the HUGO Gene Nomenclature Committee (HGNC)-approved symbols[1] is used as the core gene list. Next, human Entrez Gene[2] entries that are different from the HGNC genes are added. Finally, human Ensembl[3] records are matched against the emerging gene list via GeneLoc's exon-based unification algorithm;[4] those that are not found to be equivalent to others in the set are included as novel Ensembl-based GeneCards gene entries. These primary sources provide annotations for aliases, descriptions, previous symbols, gene category, location, summaries, paralogues and non-coding RNA (ncRNA) details. Once the gene list is in place with these significant annotations, over 90 data sources--including those noted above and others[4-9]--are mined for thousands of additional descriptors.
The data for each gene are collected into a text file which is used to display the web-card. In addition to the legacy text file format, the complex data model of GeneCards version 3 is stored in relational databases [10]. One database ('by resource') stores the data largely in the originally mined architecture, and another database ('by function') supports the website and has over 130 tables and views, with an average volume of hundreds of thousands of records. The largest table has over 6.5 million rows. This compendium is modelled into 40 entities, with hundreds of hierarchical relationships. The introduction of the relational database enables the execution of complex queries in the advanced search mode and sophisticated functionalities for sets of genes. The 'by function' data model is strongly influenced by the organisation of information in sections on the web-card (eg first descriptions, then integrated locations, followed by all disorders and so on), an organisation based on integrated scientific logic, which also keeps track of originating sources of information.
The GeneCards search is made possible by Lucene-based Solr technology,[11,12] coupled with our original database crawler,[10] enabling new levels of meta-annotation for field-specific dissections. In GeneCards Version 3, the search also introduces new features, including stemming (using the grammatical root along with its inflections) and proximity relations for multi-word searches (using the distance between found instances of each searched word, for relevance). Users can home in on their most desired results by viewing 'minicards' and examining expanded annotations on their chosen GeneCards gene.
More specialised capabilities that exploit the wealth of the GeneCards data are available from the GeneCards Suite: GeneNote and GeneAnnot for transcriptome analyses, GeneLoc for genomic locations and markers, GeneALaCart for batch queries and GeneDecks for finding functional partners and for gene set distillations [4,7,13,14].
The GeneCards project's instantiation of data management planning, implementation, releases and versioning, with examples of its sources, technologies, data models, presentation needs, de novo insights, algorithms,[14-16] quality assurance, user interfaces and data dumps, is described in detail by Mayer et al.[17] Over the years the life cycle has included project planning phases followed by implementation, development and semi-automated quality assurance, and deployment approximately three times a year, cycling back into new planning phases for subsequent revisions. Technologies used include Eclipse, Apache, Perl, XML, PHP, Propel, Java, R and MySQL. This platform enables user capacities that allow targeted searches, including search 'by section'. Importantly, because GeneCards mines from so many sources, each specific search amounts to obtaining knowledge from judiciously selected excerpts from many of these sources.
GeneCards utilisation examples
Several past projects used GeneCards as a major information source for their bioinformatic analyses. In one example, the Kestler group from the University of Ulm (Germany) built a software tool, IdeogramBrowser, which provides karyotypic visualisation of multiple DNA copy number aberrations that are often found in different types of cancer [18]. They employ the available characterisation of such structural variation events by high-density single nucleotide polymorphism (SNP) microarrays with high resolution (500,000 SNPs per genome). Their novel open-source software tool covers multiple aberration profiles, which are then directly deep linked to GeneCards so as to provide information on the relevant genes. Visualisation of consensus regions together with gene representation allows the explorative assessment of the data. Another project is the Extensible MicroArray Analysis System (EMAAS) application created by Butcher and colleagues from Imperial College (UK) and the National Cancer Institute at Frederick (MD, USA) to provide simple, robust access to updated resources for microarray analysis [19]. When looking at specific gene information, their program generates an interactive expression profile plot and concomitantly brings forth the respective GeneCards information, thereby allowing further scrutiny of experimental data. Finally, Ferrari and colleagues at the University of Modena (Italy),[20] utilised the GeneAnnot member of the GeneCards Suite[7] to help form a reliable reconstruction of expression levels in transcriptome analyses and to overcome the problem posed by the existence of more than one probe set per gene. The latter often leads to inconsistent expression signals for a given transcript when focusing on a gene's differential tissue expression. Ferrari et al. developed a novel set of custom chip definition files (CDFs) and the corresponding bioconductor libraries for Affymetrix human GeneChips, based on the information contained in the GeneAnnot database and utilising only probes matching a single gene. Such GeneAnnot-based CDFs are freely distributed to users, along with supplementary information (CDF libraries, installation guidelines and R code, CDF statistics and analysis results).
Synthetic lethality in cancer
Synthetic lethality is a situation where a mutation in one gene does not affect cell viability, but a mutation in one or more additional genes causes the cell's demise. Those two genes are considered to be in synthetic lethality interaction. This phenomenon is interpreted as genetic buffering in an organism where two or more genes are effectively functional paralogues. Synthetic lethality is suspected to have consequences in several applications, in particular in the field of cancer chemoresistance [21].
Most methods for identifying functional paralogues rely on sequence similarity. Such methods are incomplete, however, since sequence-based homology is not always synonymous with functional similarity. The Partner Hunter mode of GeneDecks (http://www.genecards.org/index.php?path=/GeneDecks) is designed to create a similarity metric based on a broader set of shared annotations between genes [14]. This helps to emphasise the functional similarity between two genes which might not be easily identified using sequence similarity alone. When comparing a given query gene with all remaining candidate genes in the GeneCards database, Partner Hunter calculates a score reflecting the degree of annotation sharing for ten attributes, including phenotypes, domains, tissue expression pattern and disorders. This overlap of descriptors between query and potential functional paralogues also takes into account the descriptor's frequency in the database, generating a statistical significance assessment. Tissue expression pattern and bone fide sequence paralogy are given special treatment by calculating the Pearson correlation for the expression profile and giving an 'exact match' score for the paralogy attribute. Each attribute is multiplied by the user-assigned weight, and the overall sum gives the total similarity score. Annotation-based partners are sorted thereafter.
Synthetic lethality was the subject of the European Union (EU)-funded consortium, SYNLET (http://synlet.izbi.uni-leipzig.de/), which investigated the resistance of neuroblastoma cells to vincristine, an example of the well-known phenomenon of acquisition of chemotherapeutic drug resistance by cancer cells [22]. In a comparative molecular analysis performed by the consortium on vincristine resistance attainment utilising cell lines, the consortium was able to identify the significant involvement of actin-associated features with vincristine resistance. Using a computational screening procedure, the consortium identified synthetic lethal hub proteins involved in actin-related processes having synthetic lethal interactions with downregulated features individually found in all chemoresistant cell lines tested, therefore promising an improved therapeutic window [22]. The computational screening procedure used, among other routines, the advanced search of the GeneCards database to select for all actin-related genes and GeneCards' gene-orthologue mapping in conjunction with synthetic lethality information obtained in yeast whole-genome analysis [23].
Annotations for genetic variations in human disease
The development of next generation sequencing, coupled with massively parallel DNA-enrichment technologies such as sub-genome capture and sample indexing, has allowed the sequencing of targeted regions of the human genome, including genes of interest and linkage regions for many samples at once. This provides a powerful approach to identifying new candidate genes for monogenic diseases, and may thus contribute substantially to the genetic aetiology of many disorders for which the disease-causing mutation has not yet been found [24]. For example, a significant portion of known genes for X-linked mental retardation (XLMR) reside on chromosome X [25,26]. In this realm, GeneCards became highly instrumental in research within a consortium for mutation discovery involving one of the present authors (D.L.), as well as D. Goldstein from Duke University (Durham NC, USA) and E. Pras from the Sheba Medical Center (Tel Hashomer, Israel). A directed capture-based exome sequencing of expanded territories related to X chromosome genes allowed the discovery of new mutations for XLMR, which will shed new light on the mechanism of the disease. The GeneLoc suite member, which presents an integrated chromosome map,[4] was used to collect the coordinates for the exons and introns in addition to regulatory and conserved regions for chromosome X. This is crucial for designing a custom-made capture chip. Similarly, GeneCards aided in the discovery of mutations underlying two other significant monogenic diseases, microcephaly and cerebellar ataxia. In the process of sifting the numerous candidate variations, GeneCards has aided in the understanding of the function of the relevant genes and proteins, by highlighting their involvement in the molecular pathways and tissue expression sections. The final result of this mode of utilisation was the narrowing down of a long list of candidate genes, based on integrated annotations in GeneCards, which helped to decide the most likely gene candidates and eventually led to successful mutation discovery for the diseases.
In this context, a fundamental tool is the GeneCards Suite member GeneALaCart, a gene-set-orientated batch query engine [10]. Here, a set of candidate genes is entered along with a list of requested fields. A convenient tabular output helps to identify and sort the candidate genes and their variations. In a neuro-informatics project, headed by M. Kimpel at the Indiana University School of Medicine (Indianapolis, IN, USA), various annotations, such as pathways and summaries, were retrieved using GeneALaCart and used to prune a dataset containing hundreds of genes to those most relevant for alcohol addiction (personal communication). Other examples include a study of human gene expression in the brain and the blood[27] and another seeking candidate genes related to the involvement of omega-3 fatty acids in mental disorders [28]. GeneCards and its associated suite member tools are also used by professional practitioners for the counselling of subjects with genetic diseases. GeneLoc has aided J. Kitchen from the Samaritan Center (Detroit, MI, USA) with finding useful genome-wide polymorphic markers that are closely linked to causative genes crucial for the genetic counselling of future parents. An illustrative example of user interactions which promote an improvement in GeneCards is the collaboration with S. Horowitz--also a genetic counsellor--from the Center for Clinical Genetics at the Hadassah University Medical Center (Jerusalem, Israel), whereby gene annotation summaries were added to the GeneALaCart repertoire upon her specific request. These, along with other GeneALaCart fields, such as genomic location and disease relationships, are used for genetic counselling.
Omics integration
GeneCards strives to consolidate a complete human gene compendium and to create an annotation network for connecting genes. One could traverse this web to integrate various omics data via its gene-centric framework in order to understand underlying complex patterns. This is exemplified by work in the context of the EU consortium, SysKid (http://www.syskid.eu/), which has 25 participating groups from 16 countries. The strategic aim of the consortium is the use of systems biology to enable novel chronic kidney disease (CKD) diagnosis and treatment. GeneCards is being used as a consortium tool in ways that far transcend its local utilisation by the Lancet group. Different types of CKD-related omics data have been collected, such as transcriptome (including microRNA expression), proteome, metabolome and SNP associations with genes. GeneCards assists in finding genes and pathways related to such data, so as to implicate them in the disease and help to develop new methods of diagnosis and treatments. A crucial component in this process is Set Distiller, part of the GeneDecks suite member of GeneCards. Set Distiller is an analysis tool that ranks descriptors by their degree of sharing within a given gene set [14]. In a pilot study, six metabolites suggested by consortium members as strong candidate CKD biomarkers were analysed. This resulted in the finding of shared descriptors between the genes for each metabolite, thus ranking the relevance of the metabolites for the kidney disease [29]. This capacity is now being augmented by a weighting algorithm to prioritise the metabolite-related gene sets.
The consortium has established a GeneKid database, moulded after the GeneCards design, to hold the omics information as it arrives from consortium members. The GeneKid database consists of 18 tables that hold omics data as the main entities, together with the study and samples from which they originated. An essential aspect of creating an integrated omics network is linking each of the GeneKid's omics data entries to a human gene, thereby 'symbolising' (ie finding the correct official HUGO nomenclature committee symbols) for all annotations through one shared entity. This is often a non-trivial task due to the heterogeneity and non-uniqueness of the gene identifiers provided by the experimental laboratories. An especially challenging relevant task is associating genes with cellular metabolites, an important aspect of the SysKid effort. There is scant gene-association information for many metabolites, therefore, a requirement arose to enhance GeneCards' capacities in this respect. This is an example of the two-way interaction often occurring between users and GeneCards developers. As a result, two new compound-gene association sources have just been added to GeneCards (Version 3.06) in the drugs and compounds section (Figure 1). These are The Human Metabolome Database (HMDB)[30] and DrugBank,[31] bioinformatics and cheminformatics resources that combine information about drugs and their targets.
Figure 1.
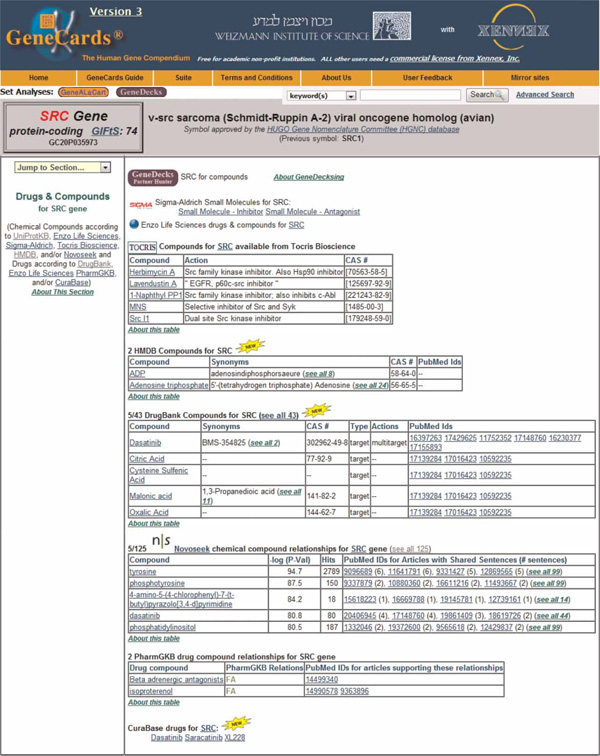
GeneCards Drugs and Compounds section, containing data from nine sources, including two new ones which were incorporated to further enable metabolomics analyses for the SysKid project.
A literature mining of papers of 17 omics studies related to CKD[29] assisted in benchmarking the GeneKid pipeline (Figure 2). Additional benchmarking was performed based on a list of 26 initial biomarkers (22 proteins, two peptides, one autoantibody and one nucleotide), prioritised by the extent of relevant gene annotations, using the GeneCards database for obtaining diseases, compounds and pathway relationships. When the experimental identifier could not easily be associated with a gene, an exhaustive effort was made using any available identifier, such as probeset, protein or SNP identifier, again highlighting the power of GeneCards' integration. A consortium user interface was constructed, enabling basic services such as browsing the GeneKid database by study, sample and experiment information to allow the 25 collaborating groups to obtain access to interim results. This capacity is strongly dependent on GeneCards' concepts and architecture. One of the key features assisting the consortium is the information within GeneCards about products such as antibodies and silencing RNA kits affiliated with specific genes of interest. These help to expedite the execution of relevant SysKid experiments, and in the development of proprietary diagnostic tools. This applies particularly to a shortlist of seven candidate CKD genes which are now being tested. Such use exemplifies the power of the products feature within GeneCards. Notably, ~15 per cent of all users who browse GeneCards use one or more of these links.
Figure 2.
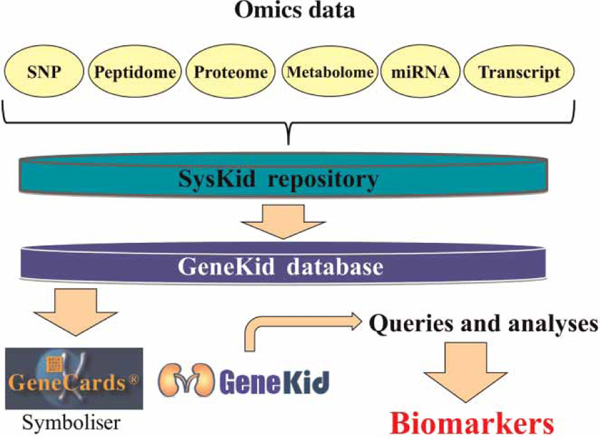
SysKid data pipeline. Starting with various omics experiments, data are fed into the SysKid repository and subsequently into the GeneKid database where each data entry is linked to a human gene. Further queries and analyses will lead to the isolation of potential kidney disease markers. Abbreviations: SNP, single nucleotide polymorphism; miRNA, microRNA.
Ongoing GeneCards expansions
Animal models
The afore-mentioned SYNLETexample of transferring experimental knowledge from one organism, namely yeast, to another (human) has emphasised the need for additional annotations derived from various model organisms to our human-centric database. This importantly includes enrichment with orthologues from species not yet covered, by adding to the current sources (eg HomoloGene[32] and others) additional orthologues from Ensembl,[33] thus increasing gene orthologue mapping. One model organism for which integration work has begun is zebrafish (Danio rerio), because of its importance as a model for human disease and drug discovery [34]. A major aim is to obtain additional information about phenotypes that can be incorporated in GeneCards' function section. This will be followed by other animal models, such as Caenorhabditis elegans and Drosophila melanogaster. Some product links to rat animal models have recently been added, with more species and products planned.
Tissue proteomics profiling
Several studies have found a moderate-to-weak correlation between the expression levels of protein and mRNA for a given tissue [35-37]. These may be attributed to experimental imprecision or biological origin, such as post-transcriptional regulation [36]. For years, GeneCards has displayed mRNA expression levels for different normal and cancerous human tissues, obtained from both inhouse and external microarray experiments [9]. Due to the above considerations, we have now decided to complement such data with a pilot quantitative tissue proteomics display in GeneCards' protein section. This was done via a collaboration with E. Kolker and colleagues at Seattle Children's Hospital (Seattle, WA, USA), who have created a database for protein expression for a total of nine normal tissues, as well as cancerous cell lines and body fluids, based on published mass spectrometry experiments. The total number of genes covered by this dataset is about 8,000, but most of them have coverage for a relatively small fraction of the nine tissue-related sample types (Figure 3). We intend collaboratively to broaden these data by seeking additional sample types for which similar information is available, as well as to integrate more than one source of certain tissues. This addition will allow users to compare transcriptome and proteome expression patterns for numerous genes.
Figure 3.
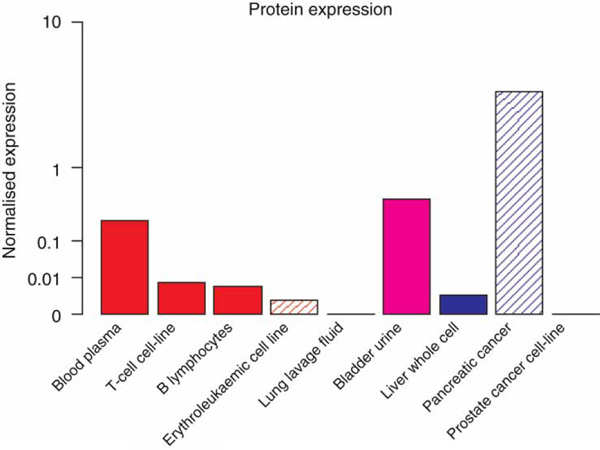
Protein expression profile of the KRT1 gene for normal human tissues, cell lines and body fluids. Expression levels of three plasma samples were merged using the geometric mean, and the expression values for each tissue were normalised using the total amount of protein extracted. The same principle employed for displaying mRNA expression levels, whereby both high and low levels are emphasised using a unique y-axis,[15] was also implemented for protein tissue production. Solidly filled boxes denote normal tissues, whereas striped boxes denote cancerous ones.
RNA genes
A major challenge of the post-genome era is to obtain a truly comprehensive list of all human genes. This is hard to achieve for obvious reasons, including ambiguities in gene identification within genomic sequences. One of the most important expansion targets is ncRNA genes. GeneCards currently mines a total of 14,315 such genes (Version 3.06) and their annotations from Ensembl (including the ncRNA subsection), HGNC,[38] the National Center for Biotechnology Information (NCBI)'s Entrez Gene and miRBase [39]. An immediate goal is to begin mining and integration of several of the numerous RNA gene databases, each providing partial information about the RNA gene universe. One target is to include new RNA gene types such as lncRNA, piRNA and snoRNA [38]. Another is to introduce some of the following new sources: fRNADB,[40] NONCODE,[41] RNAdb[42] and/or RFAM [43].
Gene and protein identifier mapping
Many interesting biological and bioinformatics applications require the integration of data from various sources, and have taken advantage of the rich annotation within GeneCards to facilitate the translation of identifiers (including symbols, aliases and database-specific identifications) and annotations (eg location on the chromosome via the GeneLoc algorithm[4]), from one system to another. Examples include combining microarray data with pathway (as done in the SYNLET project), and/or disease databases, matching names and descriptions used in the literature with official gene symbols; developing GeneAnnot-based custom CDFs;[20] and associating gene symbols with vendor products. We intend strongly to enhance this central GeneCards' capacity, with clear examples of a need for symbol management and integration for RNA genes, and for gene-to-protein identifier mapping in an upcoming effort to add proteome expression summaries for human tissues, in collaboration with E. Kolker.
Online analytical processing (OLAP)
OLAP is a designated tool for sifting through data and quickly locating trends that are worthy of further scrutiny [44]. This functionality is currently used most widely for decision support in financial management, but also can be of great benefit for biological and pharmaceutical researchers. The classical OLAP model of multi-dimensional data separates facts (records) into dimensions and measures, where the measure is the value obtained in the coordinates determined by the dimensions, and queries are made only on the latter. Applying the OLAP model to biological annotation data is not trivial, since the queries are made on both the measure (eg how many genes participate in the cell cycle pathway) and the dimensions (eg how many pathways are related to genes on chromosome 11), but this hurdle may be overcome, as reported in OLAP models for geographical data [45,46]. Another aspect involved in OLAP development is devising biological visualisation methods that will make querying and analysing results an intuitive process. We intend to employ one such OLAP technology,[47] namely the Mondrian system (http://mondrian.pentaho.com/), to enable traversals over annotations and navigations through the vast amounts of data from omics experiments.
Conclusion
The human genome project is currently at a stage where huge amounts of inter-individual comparative data are becoming available. An example is the new capacity, afforded by next-generation DNA sequencing, for performing whole-exome or whole-genome analyses of hundreds of human individuals. This data avalanche is at present partly addressed by the GeneCards variation section. The synergy between GeneCards integrative architecture and multi-source mining, and user base feedback mechanisms, enhances the probability of GeneCards' continuously being an informative genome annotation and research tool.
References
- HGNC. http://www.genenames.org/
- Entrez gene. http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene
- Ensembl. http://www.ensembl.org/index.html
- Rosen N, Chalifa-Caspi V, Shmueli O, Adato A. et al. GeneLoc: Exon-based integration of human genome maps. Bioinformatics. 2003;19(Suppl 1):i222–i224. doi: 10.1093/bioinformatics/btg1030. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D. et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult CJ, Eppig JT, Kadin JA, Richardson JE. et al. The Mouse Genome Database (MGD): Mouse biology and model systems. Nucleic Acids Res. 2008;36:D724–D728. doi: 10.1093/nar/gkm961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifa-Caspi V, Yanai I, Ophir R, Rosen N. et al. GeneAnnot: Comprehensive two-way linking between oligonucleotide array probesets and GeneCards genes. Bioinformatics. 2004;20:1457–1458. doi: 10.1093/bioinformatics/bth081. [DOI] [PubMed] [Google Scholar]
- Consortium TU. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2008;36:D190–D195. doi: 10.1093/nar/gkn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H. et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safran M, Dalah I, Alexander J, Rosen N. et al. GeneCards Version 3: The human gene integrator. Database (Oxford) 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solr. http://lucene.apache.org/solr/
- Lucene. http://lucene.apache.org/
- Shmueli O, Horn-Saban S, Chalifa-Caspi V, Schmoish M. et al. GeneNote: Whole genome expression profiles in normal human tissues. C R Biol. 2003;326:1067–1072. doi: 10.1016/j.crvi.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Stelzer G, Inger A, Olender T, Iny-Stein T. et al. GeneDecks: Paralog hunting and gene-set distillation with GeneCards annotation. OMICS. 2009;13:477–487. doi: 10.1089/omi.2009.0069. [DOI] [PubMed] [Google Scholar]
- Safran M, Chalifa-Caspi V, Shmueli O, Olender T. et al. Human gene-centric databases at the Weizmann Institute of Science: GeneCards, UDB, CroW 21 and HORDE. Nucleic Acids Res. 2003;31:142–146. doi: 10.1093/nar/gkg050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel A, Inger A, Stelzer G, Strichman-Almashanu L. et al. GIFtS: Annotation landscape analysis with GeneCards. BMC Bioinformatics. 2009;10:348. doi: 10.1186/1471-2105-10-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer B, Harel A, Dalah S, Pretrokovski S, In: Bioinformatics for Omics Data. Meyer B, editor. Humana Press, Totowa, NJ; 2011. Omics data management and annotation; pp. 71–96. [Google Scholar]
- Muller A, Holzmann K, Kestler HA. Visualization of genomic aberrations using Affymetrix SNP arrays. Bioinformatics. 2007;23:496–497. doi: 10.1093/bioinformatics/btl608. [DOI] [PubMed] [Google Scholar]
- Barton G, Abbott J, Chiba N, Huang DW. et al. EMAAS: An extensible grid-based rich internet application for microarray data analysis and management. BMC Bioinformatics. 2008;9:493. doi: 10.1186/1471-2105-9-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Bortoluzzi S, Coppe A, Sirota A. et al. Novel definition files for human GeneChips based on GeneAnnot. BMC Bioinformatics. 2007;8:446. doi: 10.1186/1471-2105-8-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Fechete R, Barth S, Olender T, Munteanu A. et al. Synthetic lethal hubs associated with vincristine resistant neuroblastoma. Mol Biosyst. 2010;7:200–214. doi: 10.1039/c0mb00082e. [DOI] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Dixon S, Vizeacoumar FJ. et al. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Methods Enzymol. 2010;470:145–179. doi: 10.1016/S0076-6879(10)70007-0. [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Deriziotis P, Lee C, Vives L. et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Smith R, Pleasance E, Whibley A. et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Bell RL, Ringham HN, Sullivan EV. et al. Ethanol-induced changes in the expression of proteins related to neurotransmission and metabolism in different regions of the rat brain. Pharmacol Biochem Behav. 2011;99:428–436. doi: 10.1016/j.pbb.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le-Niculescu H, Case NJ, Hulvershorn L, Patel SD. et al. Convergent functional genomic studies of omega-3 fatty acids in stress reactivity, bipolar disorder and alcoholism. Transl Psychiatry. 2011;1:e4. doi: 10.1038/tp.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechete R, Heinzel A, Perco P, Monks K. et al. Mapping of molecular pathways, biomarkers and drug targets for diabetic nephropathy. Proteomics Clin Appl. 2011;5:354–366. doi: 10.1002/prca.201000136. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Eisner R, HMDB: A knowledgebase for the human metabolome. 2009. pp. D603–D610. [DOI] [PMC free article] [PubMed]
- Knox C, Law V, Jewison T, Liu P. et al. DrugBank 3.0: A comprehensive resource for 'omics' research on drugs. Nucleic Acids Res. 2011;39:D1035–D1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;39(Suppl 1):D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensembl Pan Taxonomic Compara. http://fungi.ensembl.org/info/docs/compara/index.html
- Kari G, Rodeck U, Dicker AP. Zebrafish: An emerging model system for human disease and drug discovery. Clin Pharmacol Ther. 2007;82:70–80. doi: 10.1038/sj.clpt.6100223. [DOI] [PubMed] [Google Scholar]
- Fu N, Drinnenberg I, Kelso J, Wu J-R. et al. Comparison of protein and mRNA expression evolution in humans and chimpanzees. PLoS ONE. 2007;2:e216. doi: 10.1371/journal.pone.0000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox B, Kislinger T, Emili A. Integrating gene and protein expression data: Pattern analysis and profile mining. Methods. 2005;35:303–314. doi: 10.1016/j.ymeth.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Tian Q, Stepaniants SM, Mao M, Weng L. et al. Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol Cell Proteomics. 2004;3:960–969. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- Wright MW, Bruford EA. Naming 'junk': Human non-protein coding RNA (ncRNA) gene nomenclature. Hum Genomics. 2011;5:90–98. doi: 10.1186/1479-7364-5-2-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkn221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kin T, Yamada K, Terai G, Oxida H. et al. fRNAdb: A platform for mining/annotating functional RNA candidates from non-coding RNA sequences. Nucleic Acids Res. 2007;35:D145–D148. doi: 10.1093/nar/gkl837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Bai B, Skogerbo G, Cai L. et al. NONCODE: An integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005;33:D112–D115. doi: 10.1093/nar/gni113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KC, Stephen S, Engstrom PG, Tajal-Arifin K. et al. RNAdb -- A comprehensive mammalian noncoding RNA database. Nucleic Acids Res. 2005;33:D125–D130. doi: 10.1093/nar/gni117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate JG, Nawrocki EP. et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codd EF, Codd SB, Salley CT. Providing OLAP (On-Line Analytical Processing) to User-Analysis: An IT Mandate. Technical report, E.F. Codd and Associates. 1993.
- Bédard Y, Merrett T, Han J. In: Geographic Data Mining and Knowledge Discovery. Miller HJ, Han J, editor. Taylor and Francis, London; 2001. Fundamentals of spatial data warehousing for geographic knowledge discovery; pp. 53–73. [Google Scholar]
- Rivest S, Bédard Y, Marchand P. Toward better support for spatial decision making: Defining the characteristics of spatial on-line analytical processing (SOLAP) Geomatica. 2001;55:539–555. [Google Scholar]
- Alkharouf NW, Jamison DC, Matthews BF. Online analytical processing (OLAP): A fast and effective data mining tool for gene expression databases. J Biomed Biotechnol. 2005;2005:181–188. doi: 10.1155/JBB.2005.181. [DOI] [PMC free article] [PubMed] [Google Scholar]