

Abstract
The aldehyde dehydrogenase (ALDH) gene superfamily encodes enzymes that are critical for certain life processes and detoxification via the NAD(P)+-dependent oxidation of numerous endogenous and exogenous aldehyde substrates, including pharmaceuticals and environmental pollutants. Analysis of the ALDH gene superfamily in the latest databases showed that the human genome contains 19 putatively functional genes and three pseudogenes. A number of ALDH genes are upregulated as a part of the oxidative stress response and inexplicably overexpressed in various tumours, leading to problems during cancer chemotherapy. Mutations in ALDH genes cause inborn errors of metabolism -- such as the Sjögren - Larsson syndrome, type II hyperprolinaemia and γ-hydroxybutyric aciduria -- and are likely to contribute to several complex diseases, including cancer and Alzheimer's disease. The ALDH gene products appear to be multifunctional proteins, possessing both catalytic and non-catalytic properties.
Keywords: human genome, aldehyde dehydrogenase gene family, genetic polymorphism, evolution, crystallins
Introduction
Aldehyde dehydrogenases (ALDHs; EC1.2.1.3) represent a group of enzymes that oxidise a wide range of endogenous and exogenous aldehydes to their corresponding carboxylic acids [1]. Endogenous aldehydes are formed during the metabolism of amino acids, carbohydrates, lipids, biogenic amines, vitamins and steroids. Biotransformations of a large number of drugs and environmental chemicals also generate aldehydes. Aldehydes are highly reactive electrophilic compounds which interact with thiol and amino groups, the resulting effects vary from physiological and therapeutic to cytotoxic, mutagenic or carcinogenic. In this respect, ALDHs efficiently oxidise and, in most instances, detoxify a significant number of chemically diverse aldehydes which otherwise would be harmful to the organism. Strong evidence supporting this notion comes from the fact that mutations in ALDH genes cause inborn errors of metabolism associated with clinical phenotypes -- such as Sjögren - Larsson syndrome (SLS), type II hyperprolinaemia and γ-hydroxybutyric aciduria [2]. In addition, mutations in ALDH genes contribute to clinically relevant diseases such as cancer and Alzheimer's disease.
There are instances, however, in which ALDHs catalyse reactions yielding chemically reactive or bioactive metabolites that are essential to the organism. Several ALDH enzymes -- including ALDH1A1, ALDH1A2 and ALDH1A3 -- catalyse the irreversible oxidation of retinal to retinoic acid [3]. Whereas the light-absorbing properties of retinal are a necessary element for vision, the carboxylic acid isomers, all-trans-retinoic acid and/or 9-cis-retinoic acid, serve as ligands for the retinoic receptor (RAR) and the retinoid X receptor (RXR) that mediate gene expression for growth and development [4]. The importance of ALDH enzymes in retinoic acid formation became evident from the fact that homozygous disruption of the mouse Aldh1a2 gene results in an embryonic lethal phenotype due to defects in early heart morphogenesis [5,6], whereas Aldh1a3 null mice die shortly after birth, due to respiratory distress caused by choanal atresia [7].
Formation of retinoic acid and γ-aminobutyric acid (GABA) are among the most intriguing functions of ALDHs regarding bioactivation. GABA is implicated in the regulation of the GABAergic, dopaminergic and opioid systems. Even though the main pathway for GABA synthesis is the decarboxylation of L-glutamate, this neurotransmitter can also be formed from putrescine by direct oxidative deamination to give γ-aminobutyraldehyde, which is then converted into GABA by an ALDH [8]. All in all, the ALDH gene family represents a truly diverse group of proteins which are critical to metabolism.
Multiple function(s) of the ALDH enzymes
Although the major function of ALDH enzymes is the NAD(P)+-dependent aldehyde oxidation, it has become increasingly clear that some, if not most, ALDHs exhibit multiple functions (Figure 1). For example, ALDH1A1, ALDH2, ALDH3A1 and ALDH4A1 are known to catalyse ester hydrolysis, suggesting that the ALDHs may have more than one catalytic function [9]. Indeed, it has recently been suggested that ALDH2 also possesses nitrate reductase activity, which catalyses the formation of 1,2-glyceryl dinitrate and nitrite from nitroglycerin within mitochondria, leading to the production of cGMP and vasorelaxation [10].
Figure 1.
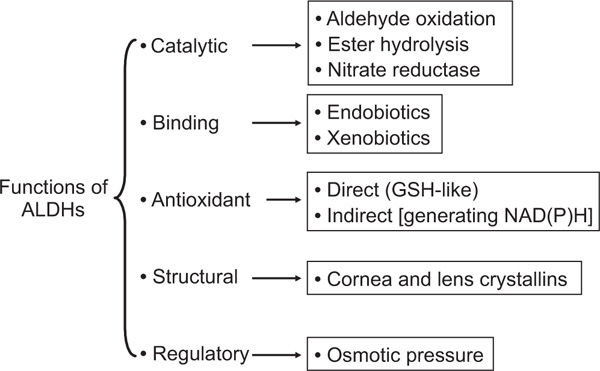
Multiple functions of aldehyde dehydrogenase (ALDH) enzymes. Endobiotics, endogenous compounds. Xenobiotics, foreign chemicals.
Aside from their catalytic properties, ALDH proteins are capable of non-catalytic interactions with chemically diverse endogenous compounds and chemotherapeutic agents. In this context, ALDH1A1 has been identified as an androgen-binding protein prominently expressed in human genital fibroblasts; as a cholesterol-binding protein in bovine lens epithelium; and as a cytosolic thyroid hormone-binding protein in Xenopus [11]. ALDH1A1 has also been identified as a flavopyridol-binding protein in non-small cell lung carcinomas and as a daunorubicin binding protein in rat liver [1]. Similar to ALDH1A1, ALDH2 also displays binding capabilities with exogenous compounds, which became evident from its identification as an acetaminophen binding protein [1].
In addition, it has been suggested that some ALDHs may play a critical role in cellular homeostasis by maintaining redox balance [12]. For example, it has been proposed that ALDH3A1 may scavenge hydroxyl radicals via the -SH groups of Cys and Met residues, and that both ALDH3A1 and ALDH1A1 may contribute to the antioxidant capacity of the cell by generating NADPH and/or NADH [13]. The enzymatic activity of ALDH3A1 generates NADPH, which is linked to the regeneration of reduced glutathione (GSH) from its oxidised form (GSSG) via the glutathione reductase/peroxidase system. NAD(P)H may also function as a direct antioxidant by reducing glutathiyl radicals (GSz) or tyrosyl radicals [14]. The expression of ALDH3A1 and ALDH1A1 at very high concentrations in the mammalian cornea and lens (crystallins) has led to additional hypotheses regarding the multifunctional properties of these proteins -- including a structural function contributing to transparency [15,16]. Finally, the ALDH7A1 gene product is similar to the green garden pea '26g protein' involved in the regulation of turgor pressure, suggesting that the ALDH7A1 protein might have osmoregulatory properties.
Evolution of the ALDH genes
ALDHs have a wide distribution in nature, ranging from bacteria and yeasts to plants and animals [17]. Sequence comparisons indicate extensive similarity between bacterial and human ALDHs and suggest that the superfamily has a common ancestral gene, dating back to ~3 billion years ago [18]. A systematic nomenclature scheme for the ALDH gene superfamily (in animals, plants, bacteria and yeasts) has been developed, based on evolutionary divergence [18], which has been implemented with biannual updates [19,20] and is available via the internet (http://www.aldh.org).
ALDH proteins are conveniently classified into families and subfamilies based on the percentage of amino acid identity. Proteins sharing ≥ 40 per cent identity are assigned to a particular family designated by an Arabic numeral, whereas those sharing ≥ 60 per cent identity are classified in the same subfamily designated by a letter. These cut-off values follow the original recommendations by Margaret Dayhoff and were first applied to the cytochrome P-450 superfamily [21]. At present, more than 130 additional gene superfamilies and large gene families follow this same format.
Endogenous functions of ALDHs
Antioxidants and oxidative stress increase the expression of certain ALDH genes, leading to increased protection of the cell against insult by environmental chemicals and drugs [22]. Increased expression of certain ALDHs in tumour cells, however, leads to decreased cellular sensitivity to cyclophosphamide and other oxazaphosphorines and, thus, to clinical problems in the treatment of cancer patients [23]. The reason for certain ALDHs -- and other non-P450 members of the [Ah] battery -- to be upregulated in some tumours [24] remains an enigma.
Numerous polymorphisms exist in the human ALDH genes, some of which cause inborn errors of metabolism and contribute to clinically relevant diseases [2]. Polymorphism in the ALDH2 gene is associated with altered acetaldehyde metabolism, alcohol-induced 'flushing' syndrome, decreased risk for alcoholism and increased risk of ethanol-induced cancers. The genetic ALDH2 deficiency has also been reported as a risk factor in late-onset Alzheimer's disease [25]. Epidemiological studies have revealed conflicting evidence about the association between the ALDH2 polymorphism and ethanol-induced hypertension [11]. Polymorphisms in the ALDH3A2, ALDH4A1, ALDH5A1 and ALDH6A1 genes are associated with metabolic diseases, which, in most cases, are characterised by neurological complications. Mutations in ALDH3A2 are the molecular basis for SLS, an autosomal recessive disorder characterised by congenital ichthyosis, mental retardation, spasticity, ocular abnormalities and pruritus [26,27]. Premature birth has also been observed in 73 per cent of children with SLS [28]. Loss of ALDH4A1 function causes type II hyperprolinaemia, an autosomal recessive disorder characterised by plasma accumulation of proline and Δ1-pyrroline-5-carboxylate, as well as neurological manifestations such as seizures and mental retardation [29]. Loss of ALDH5A1 function leads to γ-hydroxybutyric aciduria, a rare autosomal recessive disorder in GABA metabolism associated with accumulation of both GABA and γ-hydroxybutyric acid in blood serum and cerebrospinal fluid [30]. ALDH6A1 (methylmalonic semialdehyde dehydrogenase) deficiency is an inborn metabolic disorder that results in developmental delay [31].
Latest genes in the ALDH database
A search of the Human Gene Nomenclature Committee (HGNC) database using 'ALDH' produced 20 'hits': 19 ALDH genes plus AGPS (encoding alkylglycerone phosphate synthase). This latter entry appears in the database because one of its aliases is 'ALDHPSY'. A search of the HGNC database using 'aldehyde dehydrogenase' produced 21 hits -- the 19 putatively functional ALDH genes plus two others, AASDHPPT and ADH5 (Table 1). After various analyses, it was concluded that these latter two, evolutionarily, do not belong to the ALDH gene superfamily. AASDHPPT was found to belong to the 4'-phosphopantetheinyl transferase superfamily (pfam01648; ACPS) and ADH5 belongs to the alcohol dehydrogenase family (pfam00107: ADH_zinc_N). These two genes appear in the HGNC database in response to the cue 'aldehyde dehydrogenase', because these two words are included within their names: aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl transferase and form aldehyde dehydrogenase. Interestingly, the Enzyme Commission (EC) database gives L-aminoadipate-semialdehyde dehydrogenase the number EC 1.2.1 [31]. -- meaning that it is closely related functionally to the other ALDH activities (EC 1.2.1.3).
Table 1.
Human ALDH genes listed in the Human Gene Nomenclature Committee database, plus three pseudogenes
| Approved gene symbol | Approved gene name | Chromosomal location |
|---|---|---|
| AASDHPPT | Aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl transferase | 11q22 |
| ADH5 | Alcohol dehydrogenase 5 (class III), chi polypeptide | 4q21-q25 |
| ALDH1A1 | Aldehyde dehydrogenase 1 family, member A1 | 9q21 |
| ALDH1A2 | Aldehyde dehydrogenase 1 family, member A2 | 15q21.2 |
| ALDH1A3 | Aldehyde dehydrogenase 1 family, member A3 | 15q26 |
| ALDH1B1 | Aldehyde dehydrogenase 1 family, member B1 | 9p13 |
| ALDH1L1 | Aldehyde dehydrogenase 1 family, member L1 (formyltetrahydrofolate dehydrogenase) |
3q21.3 |
| ALDH1L2 | Aldehyde dehydrogenase 1 family, member L2 | 12q23.3 |
| ALDH2 | Aldehyde dehydrogenase 2 family (mitochondrial) | 12q24.2 |
| ALDH3A1 | Aldehyde dehydrogenase 3 family, member A1 | 17p11.2 |
| ALDH3A2 | Aldehyde dehydrogenase 3 family, member A2 | 17p11.2 |
| ALDH3B1 | Aldehyde dehydrogenase 3 family, member B1 | 11q13 |
| ALDH3B2 | Aldehyde dehydrogenase 3 family, member B2 | 11q13 |
| ALDH4A1 | Aldehyde dehydrogenase 4 family, member A1 | 1p36 |
| ALDH5A1 | Aldehyde dehydrogenase 5 family, member A1 (succinate-semialdehyde dehydrogenase) |
6p22 |
| ALDH6A1 | Aldehyde dehydrogenase 6 family, member A1 | 14q24 |
| ALDH7A1 | Aldehyde dehydrogenase 7 family, member A1 | 5q31 |
| ALDH7A1P1 | Aldehyde dehydrogenase 7 family, pseudogene 1 | 5q14 |
| ALDH7A1P2 | Aldehyde dehydrogenase 7 family, pseudogene 2 | 7q36 |
| ALDH7A1P3 | Aldehyde dehydrogenase 7 family, pseudogene 3 | 10q21 |
| ALDH8A1 | Aldehyde dehydrogenase 8 family, member A1 | 6q24.1-q25.1 |
| ALDH9A1 | Aldehyde dehydrogenase 9 family, member A1 | 1q22-q23 |
| ALDH16A1 | Aldehyde dehydrogenase 16 family, member A1 | 19q13.33 |
| ALDH18A1 | Aldehyde dehydrogenase 18 family, member A1 | 10q24.3-q24.6 |
The two most recently discovered ALDH genes are ALDH1L2 and ALDH16A1. The ALDH1L2 protein is very similar to ALDH1L1, which is better known as 10-formyltetrahydrofolate dehydrogenase (TFDH), a bifunctional enzyme formed from the fusion of two unrelated genes; TFDH is highly expressed in human liver, kidney and pancreas [32]. The deduced amino acid sequence of ALDH1L1 contains three domains -- including the amino terminal (residues 1 - 203), which is approximately 30 per cent identical to phosphoribosylglycinamide formyltransferase (EC 2.1.2.2), and the carboxyl terminal (residues 417 - 902), which belongs to the ALDH superfamily [33]. The intermediate domain (residues 204 - 416) does not appear to have any known catalytic function, although it shows significant homology with the structural domain of a calmodulin-like protein [34]. This intermediate domain is apparently an essential structural element that aligns the two functional domains together for 10-formyl-tetrahydrofolate (10-FTHF) dehydrogenase activity [35], -- the primary function of this enzyme [31]. This multidomain enzyme catalyses: (a) the NADP+-dependent oxidation of 10-FTHF to tetrahydrofolate (THF); (b) the NADP+-dependent oxidation of 2-propanal and acetaldehyde and; (c) the NADP+-independent hydrolysis of 10-FTHF to formate and THF [34]. ALDH1L1 is involved in formate metabolism, as well as the regulation of 10-FTHF and THF, which are principal sources of folate in the cell.
The ALDH1L2 gene encodes a protein that is 72.3 per cent identical with the ALDH1L1 and is also a fusion gene, comprising three domains: (a) the formyl-trans-N-formyl transferase (pfam00551) at the amino terminal (residues 23 - 202); (b) the formyltransferase carboxyl terminal domain (pfam02911) in the middle (residues 226 - 327); and (c) the aldehyde dehydrogenase domain at the carboxyl terminal (residues 451 - 910). No functional data have yet been reported for the ALDH1L2 protein. It is worth mentioning that the BLAST scores for the ALDH domain in both the ALDH1L1 and ALDH1L2 genes are much higher than those for the other two domains, which is strong evidence to support the notion that these two genes should be listed as ALDH genes.
The ALDH16A1 gene, located at 19q13.33, was identified recently by the National Institutes of Health Mammalian Gene Collection (MGC) Program, which represents a multi-institutional effort to identify and sequence a cDNA clone containing a complete open reading frame for each human and mouse gene [36]. The ALDH16A1 gene encodes a protein of 802 amino acids (listed in databases as 'hypothetical protein MGC10204'), which is ~35 per cent identical to putatively membrane-anchored ALDHs found in bacterial species such as Sinorhizobium meliloti 102. Putative orthologues of ALDH16A1 are found in mouse, rat and chimpanzee, and exhibit around 72 - 74 per cent amino acid identity with the human ALDH16A1.
Finally, there are three pseudogenes -- ALDH7A1P1, ALDH7A1P2 and ALDH7A1P3 -- located at Chr 5q14, 7q36 and 10q21, respectively, of which only ALDH7A1P1 meets the HGNC criteria for a pseudogene (at least 50 per cent amino acid identity across 50 per cent of the open reading frame); however, the names ALDH7A1P2 and ALDH7A1P3 are proposed because sequence homology with the ALDH7A1 gene is significantly higher than that with any other ALDH gene. An alternative nomenclature system for naming four various types of pseudogenes has recently been proposed [37]. As is commonly seen with pseudogenes in the mammalian genome, 37 all three of these pseudogenes are found at chromosomal locations that differ from that of the ALDH7A1 functional gene from which the pseudogenes clearly originated.
Conclusions
The authors' current analysis of the ALDH genes within the human genome is now probably complete, and it can be concluded that the human ALDH gene superfamily comprises 19 genes in 11 families and four subfamilies (Figure 2). The ALDH1 family contains six functional genes: the cytosolic ALDH1A1 and the mitochondrial ALDH1B1 may be involved in acetaldehyde metabolism; ALDH1A1 also participates in retinal oxidation and the detoxification of cyclophosphamide; the ALDH1A2 and ALDH1A3 proteins are integral to the oxidation of retinal to retinoic acid; the ALDH1L1 gene codes for 10-FTHF dehydrogenase; the ALDH1L2 gene product is very similar to that of ALDH1L1, but no functional data are available yet. The ALDH2 family has a single member, encoding the mitochondrial ALDH that exhibits the highest affinity for acetaldehyde and is critical in ethanol metabolism. Although ALDH2 officially qualifies as a seventh member of the ALDH1 family, its longstanding name of "ALDH2" associated with ethanol emtabolism has been grandfathered into the more recent nomenclature system based on evolutionary divergence [18]. The ALDH3A subfamily contains the dioxin-inducible ALDH3A1 and ALDH3A2, which are primarily involved in the oxidation of medium- and long-chain aliphatic and aromatic aldehydes. The ALDH3B subfamily consists of two structurally related genes, ALDH3B1 and ALDH3B2; as yet, there are no functional data for either gene product. ALDH5A1 encodes the succinic semialdehyde dehydrogenase. ALDH6A1 encodes the acetyl CoAdependent methylmalonate semialdehyde dehydrogenase. The ALDH7A1 gene product, also known as 'antiquitin', is similar to the green garden pea 26 g protein involved in the regulation of turgor pressure. ALDH8A1 appears to metabolise retinal. ALDH9A1 codes for an enzyme that participates in the metabolism of γ-aminobutyraldehyde and aminoaldehydes derived from polyamines. The ALDH16A1 gene encodes an 802-amino acid protein with as-yet unknown function. Finally, the ALDH18A1 gene encodes Δ1-pyrroline-5-carboxylate synthetase, which qualifies for classification in the ALDH superfamily based on the sequence homology of one of the protein domains (residues 361 - 772).
Figure 2.
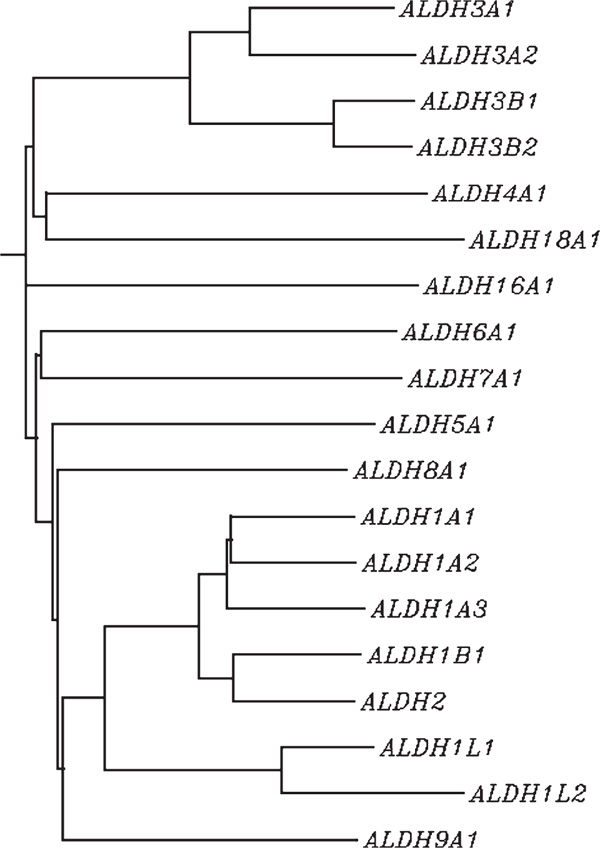
Dendrogram of the 19 human aldehyde dehydrogenase (ALDH) genes that are bona fide members of the ALDH superfamily. To avoid additional clutter, alternative splice variants of ALDH genes have not been included in the construction of this tree or the three pseudogenes listed in Table 1. This neighbour-joining method gives various branches of different lengths, reflecting that evolutionary divergence is not the same between different branches of the gene tree.
Acknowledgements
We thank Dr Elspeth Bruford and Tia Estey for valuable discussions and a critical reading of this manuscript. The writing of this paper was funded, in part, by NIH grants R01 EY11490 (VV) and P30 ES06096 (DWN).
References
- Vasiliou V, Pappa A, Petersen DR. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem Biol Interact. 2000;129:1–19. doi: 10.1016/S0009-2797(00)00211-8. [DOI] [PubMed] [Google Scholar]
- Vasiliou V, Pappa A. Polymorphisms of human aldehyde dehydrogenases. Consequences for drug metabolism and disease. Pharmacology. 2000;61:192–198. doi: 10.1159/000028400. [DOI] [PubMed] [Google Scholar]
- Duester G. Families of retinoid dehydrogenases regulating vitamin A function: Production of visual pigment and retinoic acid. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Fraulob V, Garnier JM. et al. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech Dev. 2002;110:165–171. doi: 10.1016/S0925-4773(01)00561-5. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Vermot J, Schuhbaur B. et al. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development. 2000;127:75–85. doi: 10.1242/dev.127.1.75. [DOI] [PubMed] [Google Scholar]
- Niederreither K, Subbarayan V, Dolle P. et al. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999;21:444–448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- Dupe V, Matt N, Garnier JM. et al. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc Natl Acad Sci USA. 2003;100:14036–14041. doi: 10.1073/pnas.2336223100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler N, Eichentopf B. 4-aminobutyrate in mammalian putrescine catabolism. Biochem J. 1975;152:201–210. doi: 10.1042/bj1520201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek NE. Human aldehyde dehydrogenases: Potential pathological, pharmacological, and toxicological impact. J Biochem Mol Toxicol. 2003;17:7–23. doi: 10.1002/jbt.10057. [DOI] [PubMed] [Google Scholar]
- Sydow K, Daiber A, Oelze M. et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V, Pappa A, Estey T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metab Rev. 2004;36:279–299. doi: 10.1081/DMR-120034001. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ. et al. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/S0006-2952(99)00310-X. [DOI] [PubMed] [Google Scholar]
- Uma L, Hariharan J, Sharma Y. et al. Corneal aldehyde dehydrogenase displays antioxidant properties. Exp Eye Res. 1996;63:117–120. doi: 10.1006/exer.1996.0098. [DOI] [PubMed] [Google Scholar]
- Kirsch M, De GH. NAD(P)H, a directly operating antioxidant? FASEB J. 2001;15:1569–1574. doi: 10.1096/fj.00-0823hyp. [DOI] [PubMed] [Google Scholar]
- Piatigorsky J. Enigma of the abundant water-soluble cytoplasmic proteins of the cornea: The "refracton hypothesis". Cornea. 2001;20:853–858. doi: 10.1097/00003226-200111000-00015. [DOI] [PubMed] [Google Scholar]
- Jester JV, Moller-Pedersen T, Huang J. et al. The cellular basis of corneal transparency: Evidence for "corneal crystallins". J Cell Sci. 1999;112:613–622. doi: 10.1242/jcs.112.5.613. [DOI] [PubMed] [Google Scholar]
- Vasiliou V. Aldehyde dehydrogenase genes. Adv Exp Med Biol. 1997;414:595–600. [PubMed] [Google Scholar]
- Vasiliou V, Bairoch A, Tipton KF. et al. Eukaryotic aldehyde dehydrogenase (ALDH) genes: Human polymorphisms, and recommended nomenclature based on divergent evolution and chromosomal mapping. Pharmacogenetics. 1999;9:421–434. [PubMed] [Google Scholar]
- Sophos NA, Vasiliou V. Aldehyde dehydrogenase gene superfamily: The 2002 update. Chem Biol Interact. 2003;143-144:5–22. doi: 10.1016/s0009-2797(02)00163-1. [DOI] [PubMed] [Google Scholar]
- Sophos NA, Pappa A, Ziegler TL. et al. Aldehyde dehydrogenase gene superfamily: The 2000 update. Chem Biol Interact. 2001;130-132:323–337. doi: 10.1016/s0009-2797(00)00275-1. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Wain HM. Update on human genome completion and annotations: Gene nomenclature. Hum Genomics. 2003;1:66–71. doi: 10.1186/1479-7364-1-1-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappa A, Chen C, Koutalos Y. et al. Aldh3a1 protects human corneal epithelial cells from ultraviolet- and 4-hydroxy-2-nonenalinduced oxidative damage. Free Radic Biol Med. 2003;34:1178–1189. doi: 10.1016/S0891-5849(03)00070-4. [DOI] [PubMed] [Google Scholar]
- Sladek NE. Aldehyde dehydrogenase-mediated cellular relative insensitivity to the oxazaphosphorines. Curr Pharm Des. 1999;5:607–625. [PubMed] [Google Scholar]
- Nebert DW, Gonzalez FJ. P450 genes: Structure, evolution and regulation. Annu Rev Biochem. 1987;56:945–993. doi: 10.1146/annurev.bi.56.070187.004501. [DOI] [PubMed] [Google Scholar]
- Kamino K, Nagasaka K, Imagawa M. et al. Deficiency in mitochondrial aldehyde dehydrogenase increases the risk for late-onset Alzheimer's disease in the Japanese population. Biochem Biophys Res Commun. 2000;273:192–196. doi: 10.1006/bbrc.2000.2923. [DOI] [PubMed] [Google Scholar]
- De Laurenzi LV, Rogers GR, Hamrock DJ. et al. Sjögren-Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet. 1996;12:52–57. doi: 10.1038/ng0196-52. [DOI] [PubMed] [Google Scholar]
- Willemsen MA, IJlst L, Steijlen PM. et al. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124:1426–1437. doi: 10.1093/brain/124.7.1426. [DOI] [PubMed] [Google Scholar]
- Willemsen MA, Rotteveel JJ, van Domburg PH. et al. Preterm birth in Sjögren-Larsson syndrome. Neuropediatrics. 1999;30:325–327. doi: 10.1055/s-2007-973513. [DOI] [PubMed] [Google Scholar]
- Valle D, Goodman SI, Applegarth DA. et al. Type II hyperprolinemia. Delta1-pyrroline-5-carboxylic acid dehydrogenase deficiency in cultured skin fibroblasts and circulating lymphocytes. J Clin Invest. 1976;58:598–603. doi: 10.1172/JCI108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl PL, Gibson KM, Acosta MT. et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology. 2003;60:1413–1417. doi: 10.1212/01.WNL.0000059549.70717.80. [DOI] [PubMed] [Google Scholar]
- Roe CR, Struys E, Kok RM. et al. Methylmalonic semialdehyde dehydrogenase deficiency: Psychomotor delay and methylmalonic aciduria without metabolic decompensation. Mol Genet Metab. 1998;65:35–43. doi: 10.1006/mgme.1998.2737. [DOI] [PubMed] [Google Scholar]
- Cook RJ, Lloyd RS, Wagner C. Isolation and characterization of cDNA clones for rat liver 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 1991;266:4965–4973. [PubMed] [Google Scholar]
- Krupenko SA, Wagner C, Cook RJ. Domain structure of rat 10-formyltetrahydrofolate dehydrogenase. Resolution of the aminoterminal domain as 10-formyltetrahydrofolate hydrolase. J Biol Chem. 1997;272:10273–10278. doi: 10.1074/jbc.272.15.10273. [DOI] [PubMed] [Google Scholar]
- Xiao C, Xin H, Dong A. et al. A novel calmodulin-like protein gene in rice which has an unusual prolonged C-terminal sequence carrying a putative prenylation site. DNA Res. 1999;6:179–181. doi: 10.1093/dnares/6.3.179. [DOI] [PubMed] [Google Scholar]
- Reuland SN, Vlasov AP, Krupenko SA. Disruption of a calmodulin central helix-like region of 10-formyltetrahydrofolate dehydrogenase impairs its dehydrogenase activity by uncoupling the functional domains. J Biol Chem. 2003;278:22894–22900. doi: 10.1074/jbc.M302948200. [DOI] [PubMed] [Google Scholar]
- Strausberg RL, Feingold EA, Grouse LH. et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci USA. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR, Zeldin D, Hoffman S. et al. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes including nomenclature recommendations for genes, pseudogenes, and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]