

Abstract
Understanding the nature of evolutionary relationships among persons and populations is important for the efficient application of genome science to biomedical research. We have analysed 8,525 autosomal single nucleotide polymorphisms (SNPs) in 84 individuals from four populations: African-American, European-American, Chinese and Japanese. Individual relationships were reconstructed using the allele sharing distance and the neighbour-joining tree making method. Trees show clear clustering according to population, with the root branching from the African-American clade. The African-American cluster is much less star-like than European-American and East Asian clusters, primarily because of admixture. Furthermore, on the East Asian branch, all ten Chinese individuals cluster together and all ten Japanese individuals cluster together. Using positional information, we demonstrate strong correlations between inter-marker distance and both locus-specific FST (the proportion of total variation due to differentiation) levels and branch lengths. Chromosomal maps of the distribution of locus-specific branch lengths were constructed by combining these data with other published SNP markers (total of 33,704 SNPs). These maps clearly illustrate a non-uniform distribution of human genetic substructure, an instructional and useful paradigm for education and research.
Keywords: population genomics, population genetics, microarray, genotyping, evolution, admixture
Introduction
The completion of the primary human genome sequence was announced in 2003 and millions of single nucleotide polymorphisms (SNPs) are already available in public databases [eg The SNP Consortium (TSC), dbSNP, HGVbase]. Paralleling these advances in our knowledge of the human genome have been remarkable breakthroughs in genotyping technologies, providing >1,000-fold increases in genotyping capacity. Thus, we are on the brink of an unprecedented understanding of human variation and the evolution of our species. A detailed understanding of the extent, pattern and meaning of human variation is fundamental to the effective application of genomics to studies of human biology. For example, understanding the amount of genetic structure present in human populations is relevant to epidemiological studies as, if uncontrolled for, it can produce false-positive results in association studies [1] and lower statistical power in linkage analyses [2]. Additionally, patterns of structure within and between human populations can be important in terms of epidemiological risks and the evaluation of drug response [3].
Previous studies have used relatively small numbers of genetic markers to explore the geographical patterns of human genetic variation, resulting in an incomplete picture of human diversity at the genomic level [4,5]. Only recently has it become possible to carry out studies with thousands of markers on a genome-wide scale under the new paradigm of population genomics, which models genetic variation at both a genomic and a locus-specific level [6,7]. We analysed the genetic variation of 8,525 autosomal markers in four population samples and two Centre d'Etude du Polymorphisme Humain (CEPH) family trios. The SNP multilocus genotype data were collected using a new method developed by Affymetrix, called 'whole genome amplification' (WGA) [8]. A sample of 78 unrelated individuals--38 European-Americans, 20 African-Americans and 20 East Asians (ten Chinese and ten Japanese)--was selected from the TSC core panel of individuals for analysis on the WGA microarrays. The two CEPH family trios (mother, father and child) are of European-American ancestry. We combined these new data with a recently available dataset consisting of 26,530 SNPs compiled from a public database [9]. Positional information was available for 33,704 of these 36,347 SNPs and was used to investigate human population substructure at a locus-specific level, whereby all genomic regions are not averaged together, but investigated as individual data elements.
Methods
Population samples
The DNA samples we analysed were from two publicly available sample sets curated at the Corriell Institute (Camden, NJ): TSC and CEPH. Two family trios (mother, father and child) were selected from the CEPH family mapping panels and were European-American. The four population samples were subsets of a commonly used set of samples assembled by TSC for the purposes of SNP verification and allele frequency estimation. From these TSC panels, we included 38 European-Americans, 20 African-Americans, ten Chinese and ten Japanese. The Chinese and Japanese subjects were ascertained in the USA, but were of Chinese and Japanese ancestry.
SNP genotyping
WGA technology was used to genotype individuals in this study. Details of this method have been published elsewhere,[8], but, briefly, fractions of the genome are obtained by restriction enzyme digestion of genomic DNA, ligated with adaptors and subsequently amplified with a universal primer. The amplified target is fragmented, labelled with terminal transferase and biotin-ddATP (dideoxy Adenosine Triposphate) and hybridised overnight to synthetic microarrays [10]. Genotypes are called by interpreting signals from allele-specific probes using a model-based algorithm. The accuracy of this method is >99.5 per cent. SNPs were chosen from the TSC database on the basis of their predicted location on 400-800 base pair fragments generated by in silico digestion of human genome sequences with various restriction enzymes.
Statistical analyses
Individual genetic distances were estimated using the allele sharing distance (ASD) [11]. The tree of individuals, based on the ASD distance, was constructed using the neighbour-joining method [12] with the Molecular Evolutionary Genetics Analysis software package (MEGA version 2.1) [13]. The tree branching pattern was evaluated by bootstrapping, and was based on 100 replicates. The principal coordinates analysis (PCA) was carried out with NTSYS software [14]. The computer program STRUCTURE 2.0 [15] was used to infer relative individual admixture levels in the sample. The analysis was carried out with an admixture model of K = 3 (three populations), the model previously determined to show the highest posterior probabilities for these data. A total of 25,000 simulation iterations were run for the burn-in period and 75,000 additional iterations were run to get parameter estimates. For estimations of individual admixture in the African-American sample, we included only the European-American and African-American subjects and set K = 2 with independent alphas. The average individual admixture in the African-American sample was 0.25.
Locus-specific branch lengths (LSBLs), x, y and z, were calculated using pairwise FST distances, dAB, dBC and dAC, where x = (dAB + dAC - dBC)/2, y = (dAB, + dBC - dAC)/2, z = (dAC + dBC - dAB)/2. A, B and C are the three populations under consideration. Figure 1 shows these calculations. LSBL correlations were estimated after transforming the data to more closely approach normality using the inverse transformation after adding 0.35 to make each measure positive. Computer simulations of the coalescent process were performed using Hudson's program, called ms[16]. Comparisons between the real data distributions of LSBL and the simulated results were conducted using the Kolmogorov-Smirnov (KS) test.
Figure 1.
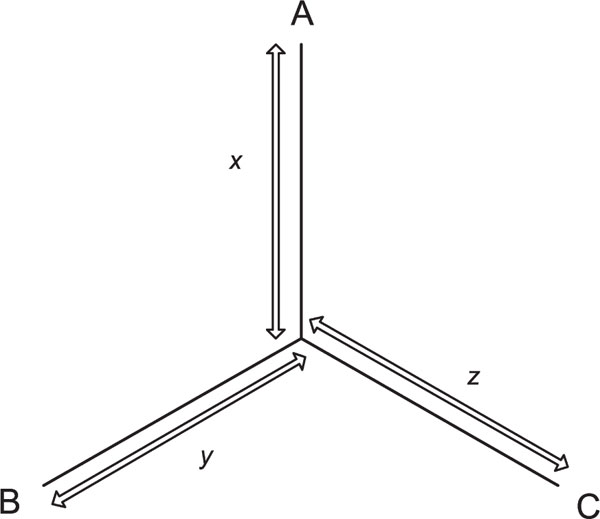
Diagram demonstrating how branch lengths are estimated for a network with three populations. Locus-specific branch lengths x, y and z, are calculated using pairwise FST distances, dAB, dBC and dAC, wher x = (dAB + dAC - dAB)/2, y = (dAB +dBC - dAC)/2, z = (dAC + dBC - dAB)/2 and A, B and C are the three populations under consideration.
Results
Individual-based analyses
The relative proportion of genetic variance due to differences between populations was estimated using Weir's FST [17] Figure 2 shows a histogram of the FST distribution for this sample. The average level of FST for autosomal SNPs, 0.132, is very similar to FST values previously described for major continental groups,[9], confirming the well-known fact that most variability in human populations is observed within populations and that a minor fraction of genetic variation (5-15 per cent) is due to differences between major continental groups [4,5,18-21]. With such a large number of loci, the distribution of FST provides important details regarding variation among loci at the level of genetic differentiation. Most SNPs show low FST values (66 per cent have FST < 0.15 and only a small fraction show high FST (4 per cent have FST > 0. 40).
Figure 2.
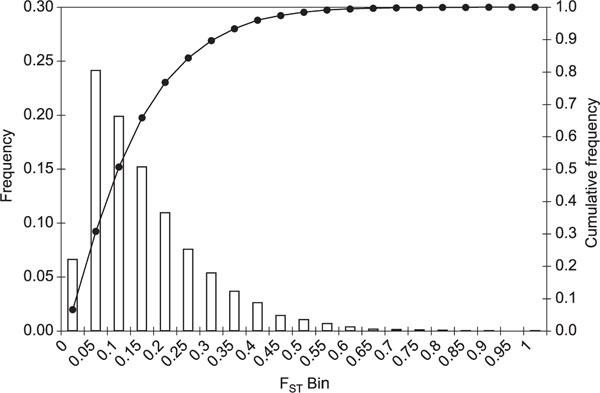
Distribution of FST. FST was calculated using Weir and Cockerham's unbiased estimator (1984) [17]. The histogram shows bin distribution, as indicated on the x-axis, and the cumulative distribution, represented by a line.
Phylogenetic and clustering relationships were studied among the 84 persons using the ASD method [11] to estimate the average distance between all pairwise combinations of individuals. Matrices of ASD measures were used to reconstruct individual trees with the neighbour-joining method [12] and prepare a two-dimensional plot based on a PCA. Additionally, we used subsets of the total marker panels based on FST level (both high and low FST markers) to explore the effects of allele frequency difference on the results. Finally, we used pairwise FST measures to calculate LSBLs for each of the populations. Markers were grouped by the distance between them and tested for the level of correlation in branch length values.
Figure 3A shows a neighbour-joining tree of individuals, constructed with the ASD measure matrix using 8,525 autosomal SNPs. There are three clear clusters on the tree showing high bootstrap values. These clusters coincide with individual geographical origins: all European-Americans cluster together (98 per cent bootstrap), all African-Americans cluster together (100 per cent bootstrap) and all East Asians cluster together (100 per cent bootstrap). The East Asian and European-American samples form tighter, more star-like, branching patterns radiating from focused points than the African-American sample. This difference in branching pattern may be due either to variation in individual admixture levels [22,23] or to higher levels of genetic diversity in sub-Saharan Africa. Notable is that, within the East Asian branch, there is a bifurcation between clusters of Japanese and Chinese individuals. Although these clusters are monophyletic, the bootstrap interval is only 57 per cent, indicating that there is not significant internal consistency supporting this split and there are likely to be a limited number of loci differentiating these two closely related populations. When comparing the Japanese and Chinese samples, the pairwise FST is 0.045 (449 SNPs with FST >0.20). Interestingly, one European-American subject, Eu5, stands apart from the European-American cluster and branches closer to the internode than to other European-Americans in the sample. The members of the two families form two clusters on this tree and, as expected, the child-parent distance is approximately half the distance between unrelated persons (data not shown).
Figure 3A.
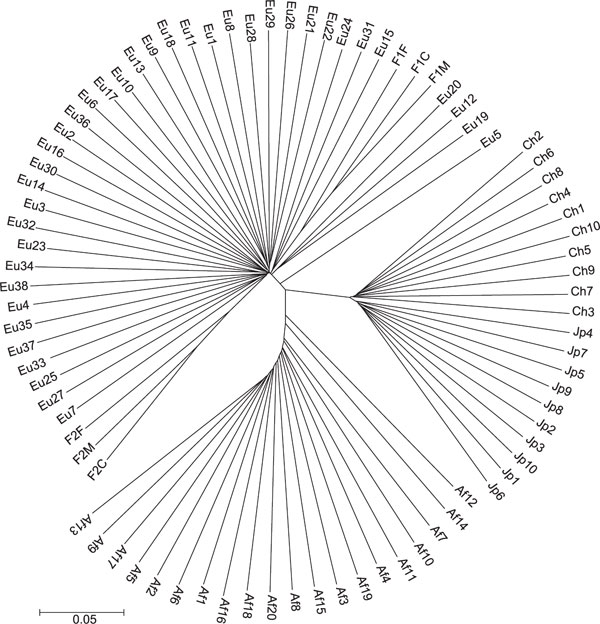
Relationships among individuals were reconstructed using the neighbour-joining method on a matrix of allele sharing distances (ASDs). Unrelated individuals from four populations are shown: African-American (labelled AfX; n = 20); European-American (labelled EuX; n = 38; Japanese (labelled JpX; n = 10); and Chinese (labelled ChX; n = 10), along with two nuclear family trios (mother, father and child, labelled FXM, FXF and FXC, respectively). This figure shows phylogeny constructed using all 8,525 autosomal single nucleotide polymorphisms. Figure 3B shows phylogeny constructed using the 1,000 highest FST markers. Figure 3C shows the tree of individuals constructed using the 1,000 lowest FST markers. It is notable that the family relationships are still evident in the two trios when using markers like these, which clearly contain no information regarding population differences.
The 8,525 markers were ranked by FST level, calculated using the three primary groupings (African-American, European-American and East Asian). We selected the 1,000 loci with the highest levels of FST (FST > 0.281) and the 1,000 loci with the lowest levels of FST (FST < 0.0143) and constructed neighbour-joining trees using the ASD. These two trees are shown in Figures 3B and 3C. There are several reasons for separating and displaying these data in this fashion. First, we can investigate the structure of the tree using markers that are ancestry informative with respect to how FST has been defined (Figure 3B). Some of the SNPs in the high- FST category may have been subject to, or tightly linked to, markers under recent directional selection. Because of the stochastic nature of genetic drift, however, some high FST markers can result from a completely neutral evolutionary history [20]. As expected, this tree exhibits much longer internal branches separating population groups and shorter terminal branches. Additionally, the previously-noted feathered clustering of African-American individuals using the complete data (Figure 3A) is accentuated in this tree. Secondly, we can display and investigate the topology of the tree drawn from the 1,000 lowest-FST SNPs (Figure 3C). Although these SNPs, or nearby variations, may have been subject to balancing or overdominant selection, the bulk of the genome is low-FST (Figure 2). As such, these markers might be a better representation of the evolutionary history of 'average' regions of the genome [6]. This tree is strikingly star-like, with very little internal structure and all individuals radiating from the centre.
Figure 3B.

Tree of individuals constructed using the 1,000 highest FST markers.
Figure 3C.
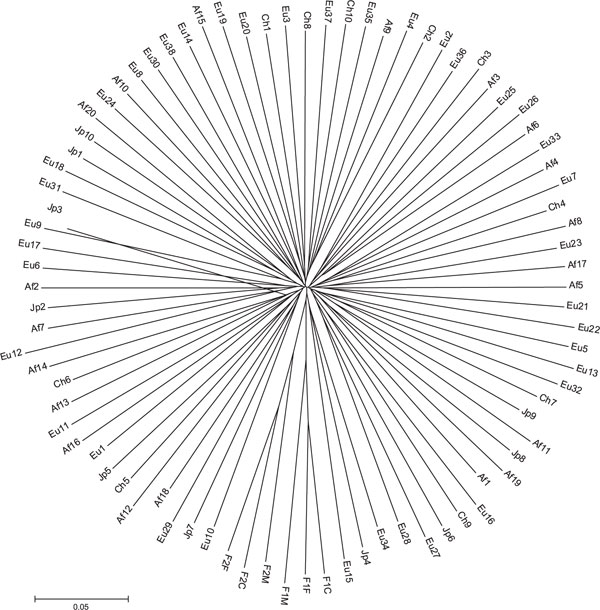
Tree of individuals constructed using the 1,000 latest FST markers.
Although trees provide a useful means of representing evolutionary relationships among populations and individuals, there is another important group of methods independent of certain assumptions inherent in phylogenetic analyses. For this reason, we performed a PCA using ASD. Some 25.4 per cent of the total variation in these distance measures is explained by the first two principal coordinates, which are plotted in Figure 4. Like the tree, the PCA plots show substantial clustering of individuals by population. The East Asian individuals form the tightest cluster, followed by the European-Americans. The African-Americans cluster in a linear fashion approaching the European-American cluster, suggesting that European gene flow is an important aspect of the diversity among African-American individuals and a likely explanation for why African-American branches are more widely spaced than the other two populations in the trees (Figure 3A and 3B). Additionally, the European-American individual noted as an outlier in the tree of individuals (Eu5) is the European-American outlier on the PCA plot moving away from the European cluster.
Figure 4.
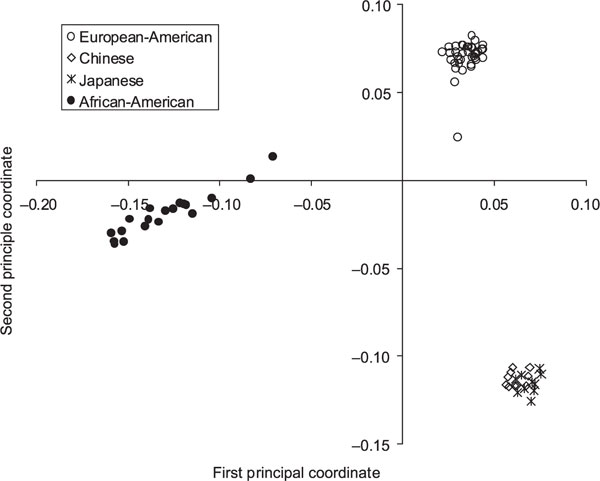
A principal coordinates analysis representation of the allele sharing distance. Populations included are indicated by the symbols listed in the key. Note that subject Eu5 is the open circle spaced away from the primary cluster of European-Americans.
Locus-specific analyse
Tree-based and PCA approaches quantify the average evolutionary relationships among individuals and populations, and FST calculated from many loci quantifies the average amount of genetic variation due to differences among groups or geographical regions. As illustrated in Figures 2, 3B and 3C, not all loci across the genome have experienced the same amount of evolutionary change. Rather, most loci have undergone only marginal changes in allele frequency, while a smaller number of loci have undergone very large changes in frequency. Although FST can be used to quantify the degree of evolution at a particular locus, and this approach has proven successful in several studies, it is not without some drawbacks.
One particular drawback is that FST is sensitive to changes in any of the populations included in the analysis. Any one (or more) of the populations could have different allele frequencies from the others, leading to a higher FST. With this in mind, we extended this approach to quantify the degree of evolution at a particular locus by calculating LSBLs (see Figure 1 and Methods section). This approach geometrically isolates allele frequency change, allowing specification of not only the amount of evolution that has occurred, but also the population(s) that underwent changes at particular loci.
A test of the appropriateness of LSBL as a measure of evolution is to examine the extent of the relationship between genomic proximity and branch length level. Figure 5 shows the relationship between correlation for pairs of syntenic SNPs in terms of branch length for each population and full FST and gap size (distance between SNP pairs). X-linked and Y-linked SNPs were excluded from this analysis because the different effective population size, the potentially stronger effects of natural selection and the lower recombination rate for markers on these chromosomes might artificially exaggerate correlations. Clearly, closely spaced SNPs show more similarity in branch length, and this correlation decays as a function of gap size. Additionally, there is a notable transition in the shape of the curve between 30 kilobases (kb) and 50 kb, where a rapid decrease in correlation becomes more gradual.
Figure 5.
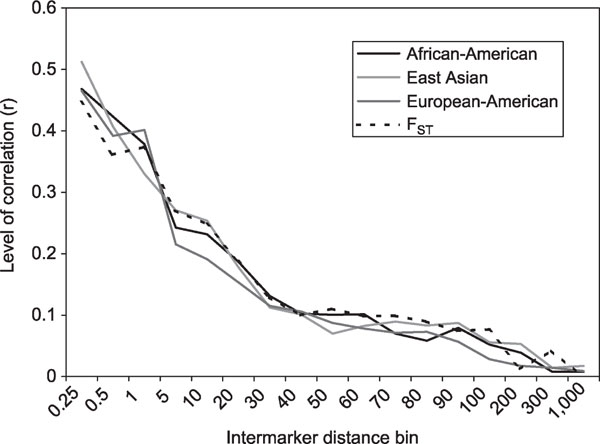
Levels of correlation between autosomal single nucleotide polymorphism loci as a function of inter-marker distance. Inter-marker distance bin, labelled by upper bin cutoff in kilobases, is shown on the x-axis. Markers, ranked by chromosomal position and then individually compared with markers further down on the same chromosome, were placed in the appropriate bin based on inter-marker distance. All markers in each bin were used to calculate the correlation between the FST level for adjacent markers.
Figure 6.
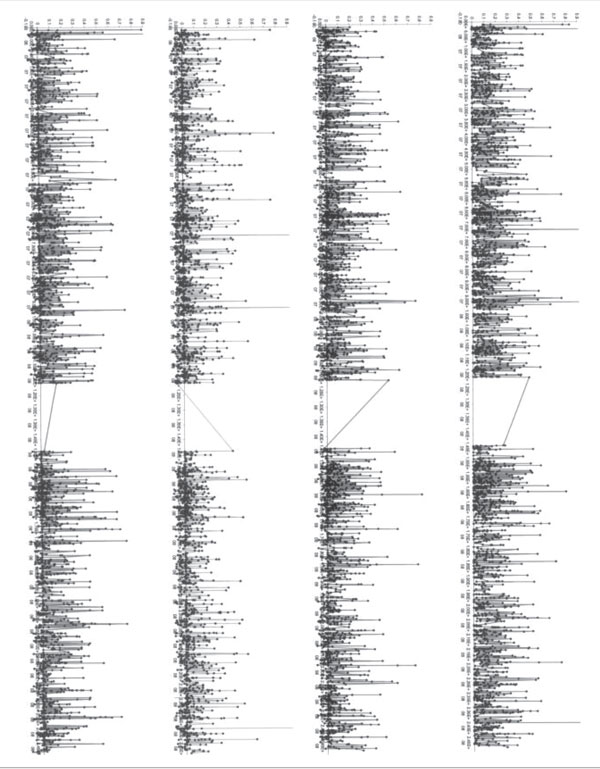
(see page 281). Chromosomal distribution of locus-specific branch length and FST for 3,258 markers localised to chromo-some 1. The top panel (A) shows the full FST, African-American locus specific branch length (LSBL) in the second panel (B), European-American LSBL in the third panel (C) and East Asian LSBL in the fourth panel (D). The value for each SNP is indicated with a black point and lines are drawn connecting adjacent points. Note that higher resolution colour plots for each chromosome are included in the Online Supplementary material (www.anthro.psu.edu/biolab).
Summary statistics describing the distributions of hetero-zygosity, LSBLs and FST are provided in Table 1. We computed the unbiased heterozygosity and LSBL for the autosomal SNPs and the X-linked SNPs separately, to make comparisons between these two classes of markers. Interestingly, significant differences in heterozygosity are seen for two of the three populations. European-Americans and East Asians both exhibit lower heterozygosity levels for X-linked markers compared with autosomal markers, while the African-American sample shows no significant difference (p = 0.18). Conversely, the West African-American sample shows a substantially higher average LSBL for X-chromosomal markers compared with autosomal markers (0.113 and 0.069, respectively) relative to European-Americans and East Asians.
Table 1.
Summary statistics for heterozygosity, branch lengths and FST.
| Measure | Marker type | African-American | European-American | East Asian | Full F ST |
|---|---|---|---|---|---|
| Heterozygosity1 | Autosomal | 0.321 (0.027) | 0.317 (0.031) | 0.290 (0.035) | - |
| X-linked | 0.317 (0.028) | 0.280 (0.038) | 0.245 (0.041) | - | |
| p-value | 0.180 | 1.6 × 10 -12 | 1.4 × 10 -15 | - | |
| LSBL raw dat 2 | Autosomal | 0.069 (0.014) | 0.039 (0.010) | 0.066 (0.015) | 0.130 |
| X-linked | 0.113 (0.031) | 0.054 (0.017) | 0.080 (0.023) | 0.194 | |
| p-value | 5.5 × 10-33 | 9.0 × 10-7 | 0.0001 | 3.6 × 10-63 | |
| LSBL adjusted3 | Autosomal | 0.114 | 0.046 | 0.055 | 0.163 |
| X-linked | 0.170 | 0.063 | 0.068 | 0.242 |
1Average (variance) for the unbiased heterozygosity calculated for n = 32,527 autosomal and n = 1,175 X-linked single nucleotide polymorphism (SNP) loci. Autosomal SNPs were compared with X-linked SNPs using a t-test and p-values are shown;
2Average (variance) for the locus-specific branch length and FST shown for n = 32,527 autosomal and n = 1,175 X-linked SNP loci. LSBL = locus-specific branch length;
3Admixture-adjusted average. African-American allele frequencies were adjusted to account for an average admixture level of 25 per cent European using the formula, pAf = (pAA - (m)pEA)/(1 - m); where m is the admixture rate (25 per cent European), pAA is the African-American allele frequency, pEA is the European-American allele frequency and pAf is the predicted African allele frequency.
In addition to examining the branch lengths using measured allele frequencies, we adjusted for, and analysed, the effects of admixture. The European admixture rate in this sample was measured with STRUCTURE 2.0 [15] to be 25 per cent -- a reasonable level, given what has been observed in other African-American populations [22,23]. The effect of gene flow is to decrease genetic distance, in this case between the African-American and European-American samples. This results in shorter African-American and European-American branches, and relatively longer East Asian branches. Indeed, when controlling for gene flow, average branch lengths change substantially: African autosomal branch length increases to 0.114 from a raw level of 0.069, European-American branch length increases to 0.046 from 0.039 and East Asian branch length decreases to 0.055 from 0.066.
It is important to know if the admixture adjustment affected all markers similarly. Therefore, we calculated the correlation between branch lengths for the unadjusted and the admixture adjusted branch lengths and found a high correlation (R2 levels of 0.944, 0.977 and 0.963 for the African-American, European-American and East Asian branch lengths, respectively).
Regions where multiple SNPs showing high LSBL are in close genomic proximity indicate locations that have recently undergone dramatic changes in allele frequency because of either random genetic drift or natural selection. Therefore, it may be instructive to plot the LSBL estimates relative to their chromosomal positions. Genomic positions were obtained for a total of 33,704 SNPs from a combined set of markers (9,817 total markers from the WGA chip and 26,530 from using a recent version of the dbSN [9] (January, 2003). The results have been plotted for each of the 23 chromosomes and are presented in the online supplementary material (http://www.anthro.psu.edu/biolab). Figure 1 presents chromosome 1 as an example of these plots. Patterns of high and low FST levels are clarified and decomposed by branch length values. It is usually the case that high branch lengths for linked SNPs in particular populations result in clusters of high FST levels. This is reasonable because branch lengths are calculated from the three pairwise FST values. As such, the impression is that the full FST plot is noisier, having more spikes (single high values) than the branch length plots and a higher level of extreme values for FST compared with branch lengths.
Finally, we conducted coalescent simulations to investigate the selectively neutral expectations for the LSBL statistic. The simulations were performed using an island model, where the levels of migration between demes were adjusted so that the average LSBL levels of the observed and simulated data matched. These simulations are summarised relative to the distributions of the real data in Figure 7. Although the distributions of the simulations are different from the real data, using the KS test (KS = 0.145, p < 0.0001 for African-Americans; KS = 0. 133, p < 0.0001 for East Asians; KS = 0.167, p < 0.0001 for European-Americans), there are some similarities. First, the basic shape of the distributions is the same, with a highly skewed distribution where the bulk of the SNPs have low LSBL, with decreasing numbers showing higher LSBL levels. Secondly, the same relative differences among the three populations are found in the simulations. Namely, the European-American group shows more loci having shorter LSBL levels than the other two populations. It is likely that more realistic simulations, using, for example, a stepping-stone model of population divergence or expansion, would provide results that show a better fit to the observed distribution, allowing us to contrast observed distributions and particular loci or groups of loci with a neutral model of evolution.
Figure 7.
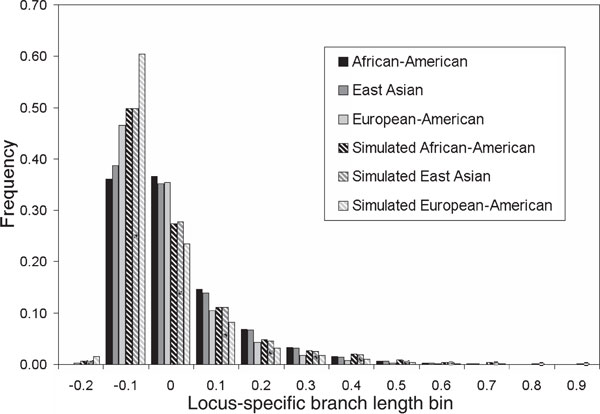
Distribution of locus-specific branch length (LSBL) levels for real data and coalescent simulations. Shown are the histogram distributions of the LSBL levels for the three main population groups. East Asian (Japanese and Chinese) are shown in dark grey, European-American in light grey and African-American in black. Coalescent simulations conditioned on the average LSBL are shown as striped bars of the same shades.
Discussion
The large number of markers used in these analyses provides an unprecedented level of resolution facilitating the study of human history at the genomic level. Our investigation of multilocus genotype data on 8,525 autosomal SNPs reinforces two observations consistently reported in the literature. First, the root of the tree branches from within the African-American clade, as has been previously observed [24,25]. This is consistent with palaeontological evidence indicating an African origin of our species [26]. Secondly, most human genetic variation is found within populations, while a minor proportion of the total variance is due to differences between continental population groups. The average FST in this study (13.2 per cent) is similar to values reported in numerous previous studies,[4,5,9,18,12,22,23] beginning with the classic paper by Richard Lewontin in 1972 [21]. The average FST obtained with this study's markers is not significantly different from the average FST obtained in a recent independent study based on SNPs typed in these same three populations (13.2 per cent versus 12.3 per cent; t-test p > 0.05) [9]. Additionally, the average FST level for X-chromosome SNPs is greater than that for autosomal SNPs (Table 1: 19.4 per cent vs 13.0 per cent; t-test p < 3.6 × 10-63 Likewise, the LSBL was observed to be significantly higher for X-chromosomal markers compared with autosomal markers in all three populations. A faster rate of evolution for X-chromosome markers has been noted previously [27,28] and the potential causes discussed in detail [29]. A higher average level of X-chromosomal differentiation is consistent with the action of higher selection pressure, especially in the African-American sample, where the heterozygosity is not decreased; however, additional population samples without admixture are needed, particularly West African samples, before conclusions to this effect are drawn.
Geographic distribution
Although genetic variation between major continental groups represents a minor fraction of the total variation observed in humans, it is misleading to describe variation between world populations as negligible. There is a wide dispersion of FST values at the genomic level. Most of the 8,525 markers analysed in this study show small allele frequency differences between populations; however, there is a subset of markers showing very high FST values and, as illustrated in Figures 3A, 3B and 4, these loci can have major effects on the observed clustering of populations and individuals according to geographical origin. At the continental level, bootstrap support is high for individuals belonging to major branches of this tree. Japanese and Chinese individuals cluster in two separate groups within the East Asian branch, but show a lower bootstrap level. Demographic factors, restricted gene flow and natural selection driving adaptation to different environments have resulted in genetic divergence between major human continental groups that can be captured at both the population and the individual level using a large number of markers. These results confirm and extend earlier work by Cavalli-Sforza and colleagues [4,30]. As demonstrated by Mountain and Cavalli-Sforza, clustering relationships are expected to change as additional populations are analysed and the number of subjects is increased [30]. To some unknown extent, the large degree of separation observed in the PCA plot and on the trees is a result of having data representing geographically extreme populations to the exclusion of intermediate groups.
It is also important to note that admixture can have a profound impact on the genetic clustering of individuals [31,32]. By contrast with the patterns observed for European-Americans and East Asians on both the PCA plot and the trees, African-American individuals do not cluster tightly or in a similarly globular fashion. We estimated relative individual ancestry levels in the African-American sample using STRUCTURE 2.0 [15] and compared these with the branching pattern of the tree of individuals. There is remarkable correspondence between the individual admixture estimates obtained with STRUCTURE and both tree branching order (ρ = 0.983, p < 0.0001) and the PCA results (ρ = 0.988, p < 0.0001). While the role of admixture in the origins of African-American populations is widely appreciated,[22,23], the extent to which non-European ancestry is present in European-Americans has received much less attention [32]. One individual (Eu5--Coriell# NA17205) among the 44 European-Americans stands out from the others on both the trees and the PCA graph, suggesting a significant proportion of non-European ancestry. Indeed, this person clusters with South Asians (from India) in separate analyses (data not shown). These results emphasise that in some contemporary populations, quantitative descriptions of the genetic clustering of individuals [28] may be more appropriate than dichotomous classifications [1,11,33,34].
In addition to considering its influence on genetic clustering, it is important to recognise how admixture can affect the magnitude of LSBLs. As shown in Table 1, the effect of European ancestry in the African-American sample is both to shorten African-American and European-American branches and to lengthen East Asian branches. Since more individuals of European ancestry were used to identify and validate the TSC SNPs, ascertainment bias may be affecting the overall magnitude of branch length [35]. Although admixture has a dramatic effect on average branch length, however, there is a high correlation between LSBLs calculated with raw allele frequencies and those calculated with ancestry-adjusted allele frequencies.
Genomic distribution
Although potentially useful and descriptive, qualitative and quantitative assessments of individual and population affiliations and phylogenies are heuristic rather than definitive statements regarding genetic variation. Average values may describe important historical and demographic aspects of both individuals and populations under consideration; however, the nature of human genomic variation is such that there is no one history [6]. Independent assortment, recombination, natural selection and genetic drift have resulted in tens of thousands of genomic regions, each with a unique history. The identification and subsequent exclusion of loci responding to selective pressures allows for a more realistic assessment of population demographics. This approach, first recognised by Lewontin and Krakauer,[36] has since been expanded upon in other analyses interrogating the genome for markers affected by natural selection [9,37-39]. Much of this focus has been concentrated on FST, which summarises the proportion of total variation due to group differences. We have used pairwise FST measures to calculate the three LSBLs, thus effectively decomposing the full FST into component parts. In this way, we can isolate and evaluate the population-specific changes in allele frequency.
To test whether LSBL captures evolutionary history, we compared branch length levels for pairs of markers (see Figure 5). Levels of correlation, which are high for closely spaced SNPs, decrease as a function of inter-marker spacing. On a genomic scale, nearby regions share more in terms of common evolutionary histories than do more widely spaced regions. A relationship between the correlation of FST for marker pairs and inter-marker distance was first shown by Akey et al [9]. using a subset of the data analysed here. These researchers found that the correlation observed between FST and inter-marker distance was stronger than a simulated coalescent distribution assuming selective neutrality. They interpreted this higher correlation as the footprint of adaptive hitchhiking. While adaptive selection has unquestionably occurred at particular genomic locations, these correlations represent summaries of data on markers from across the genome. Since demographic events will also affect the levels of linkage disequilibrium and haplotype block characteristics, they are expected to affect relationships between levels of evolution and inter-marker distance. Thus, more generally, it can be concluded that these correlations in branch length and FST levels are functions of the shared evolutionary histories of closely linked markers and reflect a non-uniform distribution of human genetic substructure across the genome.
The multiform distribution of genetic substructure has significant implications for research, not only in evolutionary, but also in biomedical contexts. Using LSBL to isolate allele frequency change allows for the identification and subsequent investigation of genomic regions that are candidates for having experienced recent directional selection by virtue of containing clusters of outlying SNPs. Additional work is required to develop statistics which combine positional and branch length information so that regions least likely to have been the result of genetic drift alone can be identified. The human genome has a multivariate history; consequently, efforts to control for population structure (eg genomic control,[40] structured association [41] and combined methods [42]) can and should be improved through marker selection efforts. It will ultimately be possible to produce data on the scale that we demonstrate here on a routine basis in disease association studies. Until that time, however, smaller sets of informative markers can be selected from large surveys because they are informative for particular axes across which population substructure is found. Such selected sets of markers can be used to efficiently detect and adjust for even very high levels of population substructure [32,42]. In sum, these analyses, which reveal a non-uniform distribution of human genetic substructure, suggest a paradigm relevant to the further explorations of genotype/phenotype relationships both within and among populations.
Acknowledgements
This work was supported in part by a grant: NIH/NHGRI (HG02154) to MDS. We would like to acknowledge helpful discussions with Rick Kittles, Nik Schork, Kateryna Makova and Bruce Lindsey.
References
- Risch N, Burchard E, Ziv E. et al. 'Categorization of humans in biomedical research: Genes, race and disease'. Genome Biol. 2002;3:1–12. doi: 10.1186/gb-2002-3-7-comment2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork N, Fallin D, Tiwari HK, In: Handbook of Statistical Genetics. Balding, D., Bishop, M. and Cannings, C, editor. John Wiley and Sons, Hoboken, NJ; 2001. 'Pharmacogenetics'; pp. 741–764. [Google Scholar]
- Burroughs VJ, Maxey RW, Levy RA. 'Racial and ethnic differences in response to medicines: Towards individualized pharmaceutical treatment'. J Natl Med Assoc. 2002;94:1–26. [PMC free article] [PubMed] [Google Scholar]
- Bowcock AM, Ruiz-Linares A, Tomfohrde J. et al. 'High resolution of human evolutionary trees with polymorphic microsatellites'. Natur. 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA, Pritchard JK, Weber JL. et al. 'Genetic structure of human populations'. Science. 2002;298:2381–2385. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- Cavali-Sforza LL. 'Population structure and human evolution'. Proc R Soc Lond B Biol Sci. 1966;164:362–379. doi: 10.1098/rspb.1966.0038. [DOI] [PubMed] [Google Scholar]
- Black WC IV, Baer CF, Antolin MF. et al. 'Population genomics: Genome-wide sampling of insect populations'. Annu Rev Entomol. 2001;46:441–469. doi: 10.1146/annurev.ento.46.1.441. [DOI] [PubMed] [Google Scholar]
- Kennedy GC, Matsuzaki H, Dong S. et al. 'Large-scale genotyping of complex DNA'. Nat Biotechnol. 2003;21:1233–1237. doi: 10.1038/nbt869. [DOI] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K. et al. 'Interrogating a high-density SNP map for signatures of natural selection'. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee M, Yang R, Hubbell E. et al. 'Accessing genetic information with high-density DNA arrays'. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Jin L. In: A unified approach to study hypervariable polymorphisms: Statistical considerations of determining relatedness and population distances DNA Fingerprinting: Current State of the Science. Pena, S.D.J., Jefferys, A.J., Epplen, J. and Chakraborty, R, editor. Vol. 67. Birkhauser, Basel, Switzerland; 1993. pp. 153–175. Vol. EXS. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. 'The neighbor-joining method: A new method for reconstructing phylogenetic trees'. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Jakobesen IB. et al. 'MEGA2: Molecular evolutionary genetics analysis software'. Bioinformatics. 2001;17:1244–1245. doi: 10.1093/bioinformatics/17.12.1244. [DOI] [PubMed] [Google Scholar]
- Rohlf FJ. 1992. NTSYS-pc version 1.70.
- Pritchard JK, Stephens M, Donelly P. 'Inference of population structure from multilocus genotype data'. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. 'Generating samples under a Wright-Fisher neutral model'. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. 'Estimating F-statistics for the analysis of population substructure'. Evolution. 1984;38:1358–1370. doi: 10.2307/2408641. [DOI] [PubMed] [Google Scholar]
- Romualdi C, Balding D, Nasidze IS. et al. 'Patterns of human diversity, within and among continents, inferred from biallelic DNA polymorphisms'. Genome Res. 2002;12:602–612. doi: 10.1101/gr.214902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde LB, Watkins WS, Bamshad MJ. et al. 'The distribution of human genetic diversity: A comparison of mitochondrial, auto-somal, and Y-chromosomal data'. Am J Hum Genet. 2000;66:979–988. doi: 10.1086/302825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton University Press, Princeton, NJ; 1994. [Google Scholar]
- Lewontin R. 'The apportionment of human diversity'. Evol Biol. 1972;6:381–398. [Google Scholar]
- Pfaff CL, Parra EJ, Bonilla C. et al. 'Population structure in admixed populations: Effects of admixture dynamics on the pattern of linkage disequilibrium'. Am J Hum Genet. 2001;68:198–207. doi: 10.1086/316935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra EJ, Marcini A, Akey J. et al. 'Estimating African American admixture proportions by use of population specific alleles'. Am J Hum Genet. 1998;63:1839–1851. doi: 10.1086/302148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins WS, Ricker CE, Bamshad MJ. et al. 'Patterns of ancestral human diversity: An analysis of Alu insertion and restriction site polymorphisms'. Am J Hum Genet. 2001;68:738–752. doi: 10.1086/318793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Takezaki N. 'The root of the phylogenetic tree of human populations'. Mol Biol Evol. 1996;13:170–177. doi: 10.1093/oxfordjournals.molbev.a025553. [DOI] [PubMed] [Google Scholar]
- Stringer C. 'Modern human origins: Progress and prospects'. Phil Trans R Soc Lond. 2002;357:563–579. doi: 10.1098/rstb.2001.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH. 'The relative rates of evolution of sex chromosomes and autosomes'. Am Nat. 1987;130:113–149. doi: 10.1086/284701. [DOI] [Google Scholar]
- Payseur BA, Cutter AJ, Nachman MW. 'Searching for evidence of natural selection in the genome using microsatellite variability'. Mol Biol Evol. 2002;19:1143–1153. doi: 10.1093/oxfordjournals.molbev.a004172. [DOI] [PubMed] [Google Scholar]
- Kayser M, Brauer S, Stoneking M. 'A genome scan to detect candidate regions influenced by Local Natural Selection in Human Populations'. Mol Biol Evol. 2003;20:893–900. doi: 10.1093/molbev/msg092. [DOI] [PubMed] [Google Scholar]
- Mountain J, Cavalli-Sforza LL. 'Multilocus genotypes, a tree of individuals and human evolutionary history'. Am J Hum Genet. 1997;61:705–718. doi: 10.1086/515510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue PM, Carpenter J, Parra EJ. et al. 'Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach using Markov chain simulation: Application to African-American populations'. Ann Hum Genet. 2000;64:171–186. doi: 10.1046/j.1469-1809.2000.6420171.x. [DOI] [PubMed] [Google Scholar]
- Shriver MD, Parra EJ, Dios S. et al. 'Skin pigmentation, biogeographical ancestry and admixture mapping'. Hum Genet. 2003;112:387–399. doi: 10.1007/s00439-002-0896-y. [DOI] [PubMed] [Google Scholar]
- Wilson JF, Weale ME, Smith AC. et al. 'Population genetic structure of variable drug response'. Nat Genet. 2001;29:265–269. doi: 10.1038/ng761. [DOI] [PubMed] [Google Scholar]
- Bamshad MJ, Wooding S, Watkins WS. et al. 'Human population genetic structure and inference of group membership'. Am J Hum Genet. 2003;72:578–589. doi: 10.1086/368061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountain J, Cavalli-Sforza LL. 'Inference of human evolution through cladistic analysis of nuclear DNA restriction polymorphisms'. Proc Natl Acad Sci USA. 1994;91:6515–6519. doi: 10.1073/pnas.91.14.6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC, Krakauer J. 'Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms'. Genetics. 1973;74:175–195. doi: 10.1093/genetics/74.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowcock AM, Kidd JR, Mountain JL. et al. 'Drift, admixture, and selection in human evolution: A study with DNA polymorphisms'. Proc Natl Acad Sci USA. 1991;88:839–843. doi: 10.1073/pnas.88.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA. 'Evaluating loci for use in the genetic analysis of population structure'. Proc Biol Soc. 1996;263:1619–1626. doi: 10.1098/rspb.1996.0237. [DOI] [Google Scholar]
- Vitalis R, Dawson K, Boursot P. 'Interpretation of variation across marker loci as evidence of selection'. Genetics. 2001;158:1811–1823. doi: 10.1093/genetics/158.4.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Roeder K. 'Genomic control for association studies'. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341X.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Donnelly P. 'Case-control studies of association in structured or admixed populations'. Theor Popul Biol. 2001;60:227–237. doi: 10.1006/tpbi.2001.1543. [DOI] [PubMed] [Google Scholar]
- Hoggart CJ, Parra EJ, Shriver MD. et al. 'Control of confounding of genetic associations in stratified populations'. Am J Hum Genet. 2003;72:1492–1504. doi: 10.1086/375613. [DOI] [PMC free article] [PubMed] [Google Scholar]