

Abstract
Objective:
The aim of our study was to examine the relationship between corticostriatal Aβ-amyloid deposition and cognitive dysfunction in a cohort of patients with Parkinson disease (PD) at risk for dementia.
Methods:
This was a cross-sectional study of 40 patients with PD with mild cognitive impairment (MCI) or other known dementia risk factors. Subjects underwent dynamic Aβ-amyloid and vesicular monoamine transporter 2 PET imaging using [11C] Pittsburgh compound B (PiB) and [11C]dihydrotetrabenazine (DTBZ), respectively, and neuropsychological assessment. PiB and DTBZ PET data were analyzed using the Logan graphical method to determine cerebral PiB deposition relative to the cerebellar hemispheres and striatal DTBZ binding relative to occipital neocortex. Component z scores were calculated for individual cognitive domains (memory, visuospatial processing, working memory/attention, and executive function) and combined linearly for global estimation of cognition. Correlation of cognitive function and cortical PiB binding was investigated.
Results:
Elevated cerebral PiB binding at levels seen in patients with AD was infrequent (6 of 40 subjects). Mean cortical PiB binding in the entire cohort was 1.16 ± 0.16 (distribution volume ratio; range 0.96–1.78). A significant correlation was noted between cortical PiB binding and global composite cognitive function (r = −0.55, p < 0.005) as well as the Wechsler Adult Intelligence Scale score (r = −0.54, p = 0.0004).
Conclusion:
Elevated cerebral Aβ-amyloid deposition at levels seen in Alzheimer disease is uncommon in subjects with PD at risk for dementia. In our sample, the prevalence of markedly elevated PiB binding was significantly lower than that found in prior studies of cognitively normal elderly individuals. Neocortical PiB binding correlated robustly with measures of cognitive impairment in our cohort.
Dementia is frequent, especially in the advanced stages of Parkinson disease (PD), with a reported mean prevalence of 40%.1,2 Its underlying pathology is complex and incompletely understood. Some postmortem studies have suggested limbic and neocortical Lewy body deposition to be the main determinant of cognitive decline.3–5 Others suggest an important role for Aβ-amyloid plaques and neurofibrillary pathology,6,7 whereas a recent pathologic study supports a combination of pathologies as being the crucial determinant of PD dementia (PDD).8
11C-Pittsburgh compound B (PiB) is a PET Aβ-amyloid ligand; studies with 11C-PiB show robust neocortical binding in subjects with Alzheimer disease (AD).9 Previous in vivo imaging studies indicated lower levels of neocortical PiB binding in patients with PD or PDD compared with those with AD.10–14 In contrast, subjects with the closely related entity of dementia with Lewy bodies (DLB), may exhibit elevated neocortical PiB binding, an interesting observation, because the PDD-DLB distinction is currently based on the relative timing of onset of dementia and parkinsonism.13,14
The prior pathologic literature suggests a significant contribution of Aβ-amyloid deposition to cognitive impairment in PD. Whether this is a late phenomenon has not been addressed. Prior studies focused predominantly on assessment of differential Aβ-amyloid deposition between subjects with different types of dementia and control subjects.
The aim of our investigation was to quantify cortical and striatal Aβ-amyloid deposition in a cohort of 40 subjects with PD with mild cognitive impairment (MCI)15,16 or other risk factors for dementia. We examined the relationship between corticostriatal Aβ-amyloid deposition and cognitive function in this cohort.
METHODS
Subjects and clinical test batteries.
This cross-sectional study involved 40 subjects with PD (32 men and 8 women) and the presence of mild cognitive symptoms or known risk factors for PD-associated dementia, specifically older age, prominent gait and balance impairments, or long duration of PD.2,17 Subject recruitment was from a university-based movement disorders subspecialty clinic between 2008 and 2011. Subjects met the UK Parkinson's Disease Society Brain Bank Research Center Clinical diagnostic criteria for PD18 and did not have a diagnosis of dementia on routine clinical evaluation. (+)-[11C]dihydrotetrabenazine (DTBZ) PET confirmed the presence of nigrostriatal dopaminergic denervation, which is a feature of PD.
Standard protocol approvals, registrations, and patient consents.
All study procedures were approved by the local institutional review board. All subjects provided written informed consent for all study procedures before enrollment.
Subjects underwent structured evaluations of PD motor features and PET imaging in the morning after dopaminergic medications were withheld overnight (“off” state). Clinical evaluations included assessment of modified Hoehn and Yahr stage19 and rating with the Movement Disorder Society revised Unified Parkinson's Disease Rating Scale (UPDRS).
Neuropsychological examination.
Subjects were examined while taking their usual dopaminergic medications to minimize bias in cognitive assessment by motor deficits. The Montreal Cognitive Assessment (MOCA) was used as a measure of global cognitive integrity. Tests included the California Verbal Learning Test as a measure of verbal memory.20 Executive functions were assessed with the Wechsler Adult Intelligence Scale–III (WAIS-III) Picture Arrangement test,21 Delis-Kaplan Executive Function System Sorting and Letter Verbal Fluency subtests,22 and Stroop Color Word Interference test.23 A switching version of the Stroop 3 test, in which subjects name ink color, unless the word is surrounded by a box, in which case they read the word itself (Stroop 4), was used to assess cognitive flexibility.24 Attention/psychomotor speed was assessed as absolute times on the Stroop 1 and 2 subsets.23 Visuospatial function was assessed with the Benton Judgment of Line Orientation test.25 Composite z scores were calculated for different cognitive domains (memory, executive, attention, and visuospatial functions) based on normative data. Global cognitive performance was calculated as the average z score for the 4 cognitive domains. The WAIS III Information subtest was used as a measure of semantic knowledge.21
Imaging techniques.
All subjects underwent brain MRI for anatomic coregistration, Aβ-amyloid PET imaging with Pittsburgh compound B (11C-PiB) and nigrostriatal terminal imaging with DTBZ PET.
MRI was performed on a 3-T Philips Achieva system (Philips, Best, the Netherlands) using an 8-channel head coil and the ISOVOX examination card protocol primarily designed to yield isotropic spatial resolution. A standard 3-dimensional T1-weighted series was performed in the sagittal plane using repetition time/echo time/inversion time = 9.8/4.6/1,041 msec; turbo factor = 200; single average; field of view = 240 × 200 × 160 mm; and acquired matrix = 240 × 200; 160 slices were reconstructed to 1-mm isotropic resolution.
PiB and DTBZ PET imaging was performed in 3-dimensional imaging mode using an ECAT HR+ tomograph (Siemens Molecular Imaging, Inc., Knoxville, TN), which acquires 63 transaxial slices (slice thickness = 2.4 mm; intrinsic in-plane resolution = 4.1 mm full-width at half-maximum over a 15.2-cm axial field of view). A NeuroShield (Scanwell Systems, Montreal, Canada) head-holder/shielding unit was used to reduce the contribution of detected photon events originating from the body outside the scanner field of view. Before PiB and DTBZ injections, a 5-minute transmission scan was acquired using rotating 68Ge rods for attenuation correction of emission data. All subjects were studied supine, with eyes and ears unoccluded, resting quietly in a dimly lit room.
11C-PiB was synthesized by published methods.9 11C-PiB PET scans were performed using a bolus/infusion protocol acquiring 17 frames over 80 minutes, with a priming bolus of 40% of the radioactive dose followed by continuous infusion of the remaining 60% over the 80-minute study using a dose of 666 MBq (18 mCi)26 Emission data were collected in 16 sequential emission scans: 4 × 30 second, 3 × 1 minute, 2 × 2.5 minutes, 2 × 5 minutes, and 6 × 10 minutes.
No carrier–added (+)-[11C]DTBZ (250–1,000 Ci/mmol at the time of injection) was prepared as reported previously.27 Dynamic PET scanning was performed for 60 minutes immediately after a bolus injection of 55% of 555 MBq (15 mCi) of (+)-[11C]DTBZ dose (containing less than 50 μg of cold DTBZ mass) over the first 15–30 seconds of the study, whereas the remaining 45% of the dose was continuously infused over the next 60 minutes.28 A series of 15 frame sequence of scans over 60 minutes was obtained as follows: 4 × 30 seconds, 3 × 1 minute, 2 × 2.5 minutes, 2 × 5 minutes, and 4 × 10 minutes.
Image postprocessing.
All dynamic PET imaging frames were spatially coregistered in subjects with a rigid body transformation to reduce the effects of subject motion during the imaging session. These motion-corrected PET frames were spatially coregistered to the MRI scan using SPM8 software (Wellcome Trust Centre for Neuroimaging, London, UK). IDL image analysis software (Research Systems, Inc., Boulder, CO) was used to manually trace volumes of interest (VOIs) on the MRI scan. Traced VOIs included the striatum (caudate and putamen), cerebellum, and thalamus. Cortical VOI definition used semiautomated thresholding delineation of the cortical gray matter signal on the MRI scans. General cortical area was determined by VOI placement and then was thresholded (based on signal intensity) to filter out any white matter. The entire neocortex was included in the VOI. Time-activity curves for each VOI were generated from the spatially aligned PET frames. PiB and DTBZ distribution volume ratio (DVR) was then estimated by using the Logan plot graphical analysis method with the cerebellar hemisphere gray matter and the occipital neocortex as reference tissue for PiB and DTBZ, respectively.29,30 In addition, voxel-wise PiB DVR maps were generated using the same calculation as described for the VOIs. These maps were sampled with Neurostat (University of Washington, Seattle), resulting in surface renderings of cortical PiB DVR. PiB scans were analyzed qualitatively in isolation, both in transaxial and as surface presentations, by an investigator experienced with PiB PET evaluation and blinded to all clinical data (K.A.F.). Transaxial assessments were classified as abnormal when frontal cortical DVR exceeded that of subjacent white matter, a threshold previously used in our laboratory to identify significant amyloid deposition in AD and DLB.31 PiB surface projection maps were classified as abnormal when frontal, temporal, and posterior cingulate cortical DVRs were conspicuously elevated against the cerebellum and the remainder of the cerebral hemispheres.
Statistical analyses.
The potential relationship between global cognitive ability and PiB accumulation in the cerebral cortex was assessed as our a priori primary hypothesis via Pearson correlation coefficient estimation. Exploratory relationships among the clinical, neuropsychological, and imaging measures were assessed with principal component analysis (PCA). Specifically, each variable was normalized within the study cohort, followed by PCA using correlation matrices and Varimax rotation to optimize distinction of variable loadings. PCA included the representative variables: cortical Aβ-amyloid deposition (cortical PiB DVR); nigrostriatal projection integrity (striatal DTBZ DVR); clinical PD measures (Hoehn and Yahr score, duration of disease, and UPDRS motor score); and cognitive indices (MOCA score, composite average z score for global cognitive function). Relationships among high-loading variables within individual principal components were further assessed using Pearson correlation analyses. Analyses were performed using SYSTAT 12.0 (SYSTAT Software Inc., San Jose, CA).
RESULTS
Thirty subjects had evidence of mild cognitive impairment based on domain-specific z scores; 5 subjects had normal range cognitive abilities and 5 subjects met the neuropsychological criteria for mild dementia (table 1).
Table 1.
Summary of clinical and imaging characteristics
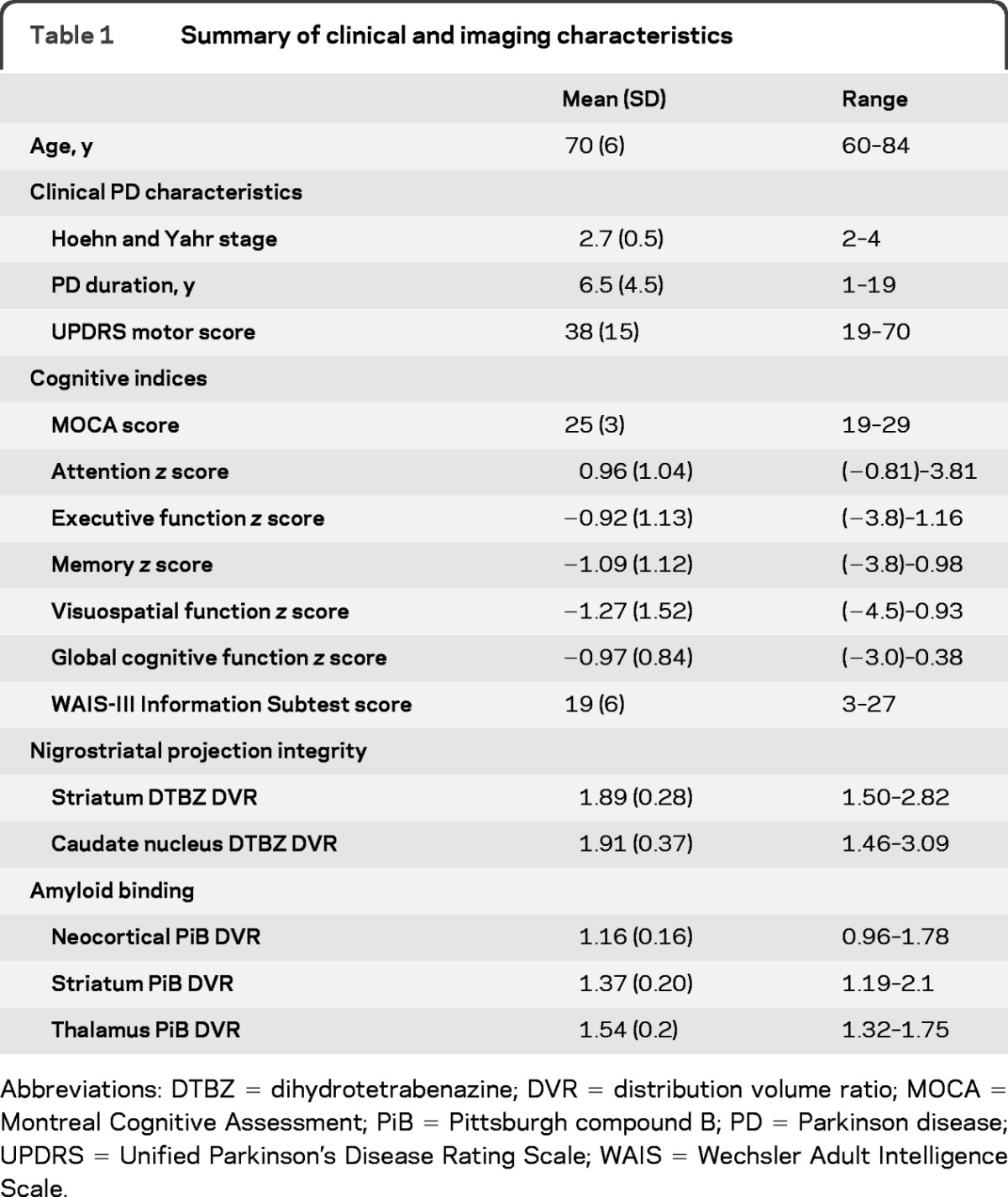
Abbreviations: DTBZ = dihydrotetrabenazine; DVR = distribution volume ratio; MOCA = Montreal Cognitive Assessment; PiB = Pittsburgh compound B; PD = Parkinson disease; UPDRS = Unified Parkinson's Disease Rating Scale; WAIS = Wechsler Adult Intelligence Scale.
The pattern of PiB binding in 34 subjects was below the pathologic range identified in patients with AD and in elderly individuals without dementia with increased risk of conversion to AD.32,33 Four of the 6 subjects with pathologically elevated neocortical PiB DVRs had dementia based on neuropsychological assessments, despite prior clinical assessment as not having dementia. Two subjects with markedly elevated cortical PiB binding had MCI by neuropsychological criteria. On qualitative blinded interpreter evaluation of PiB images, there was complete agreement between the transaxial frontal cortical/white matter PiB ratio assessments and the surface mapped cortical PiB DVR classifications. Subjects with elevated PiB binding had a similar pattern of cortical DVR, with prominent involvement of frontal lobes and posterior cingulate cortices, substantial but lower involvement of temporal neocortices, and relative sparing of primary somatomotor and occipital cortices (figure 1). This pattern is similar to that observed in AD and DLB.34
Figure 1. Representative surface maps of Pittsburgh compound B (PiB) distribution volume ratio (DVR) in Parkinson disease (PD).

Shown are surface projection representations of 11C-PiB DVRs in representative subjects with PD (rows). Images are in pseudocolor according to the key at the bottom right, ranging from a minimum DVR of 0 to a maximum DVR of 3. The top 2 rows depict subjects classified as having mild dementia (DEM) after neuropsychometric evaluation. The next 2 rows depict subjects classified as having mild cognitive impairment (MCI). The bottom row depicts a subject with normal-range neuropsychological abilities (NML). Subjects in the top 3 rows were classified as PiB-positive by qualitative assessment of these projection images as well as in transaxial assessments of frontal cortical/subcortical white matter PiB DVR. Note a similar pattern of regional cortical PiB DVRs, with the most conspicuous elevations above cerebellar background in the frontal and temporal lobes and in the posterior cingulate cortex. This pattern may also be present, albeit at low intensity, even in the subjects classified as PiB-negative in the bottom 2 rows.
There was generally strong intercorrelation among telencephalic regional PiB DVR values although thalamic PiB binding correlated weakly with neocortical PiB binding. Mean neocortical PiB binding was selected for further exploratory analysis. There was a strong intercorrelation between DTBZ binding in the caudate nucleus and total striatal binding. Striatal DTBZ binding was selected for further exploration. Among cognitive measures, all domain-specific measures were strongly correlated with the aggregate measure (global cognitive z score); thus, the global score was used for further exploratory analyses. PCA revealed 2 components accounting collectively for more than 63% of the total variance (table 2). Component 1 (eigenvalue 2.63) accounted for 36% of the variance and included 3 variables with loading values >0.5. These are cortical PiB binding, global cognitive function, and MOCA scores. Component 2 (eigenvalue 1.17) accounted for 28% of the variance and included 3 factors with loading values >0.5: DTBZ binding in the striatum, UPDRS motor score, and PD duration. Association of striatal DTBZ binding with disease duration is consistent with our prior results.35
Table 2.
Component loading factors for principal components
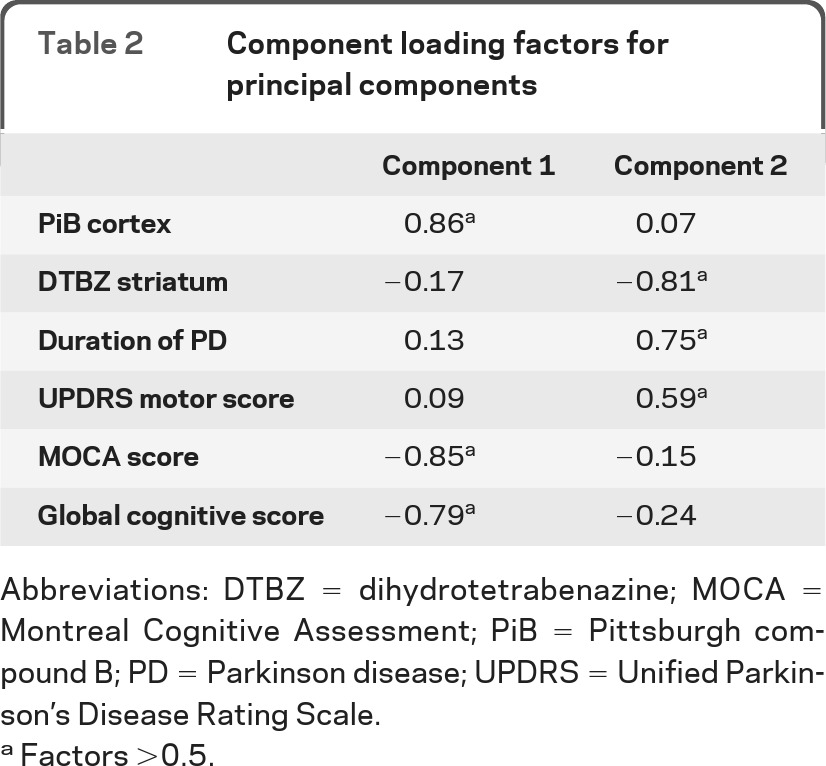
Abbreviations: DTBZ = dihydrotetrabenazine; MOCA = Montreal Cognitive Assessment; PiB = Pittsburgh compound B; PD = Parkinson disease; UPDRS = Unified Parkinson's Disease Rating Scale.
Factors >0.5.
There was significant correlation between cortical PiB binding and global composite cognitive z scores (r = −0.55, p = 0.0006) (figure 2). There was also significant correlation between cortical PiB binding and the WAIS score (r = −0.54, p = 0.0004).
Figure 2. Relationship of global cognition to neocortical Pittsburgh compound B (PiB) binding (A) and relationship of Wechsler Adult Intelligence Scale (WAIS) score to neocortical PiB binding (B).

(A) r = −0.55; p = 0.0006. (B) r = −0.54; p = 0.0004. DVR = distribution volume ratio.
DISCUSSION
Dementia is nearly universal in advanced PD, and cognitive impairment is common in mild to moderate PD. Cognitive impairment in PD probably results from a combination of pathologies including α-synuclein and Aβ-amyloid deposition, as well as cholinergic and dopaminergic denervation. The relative roles of these different pathologies have not been intensively investigated in the early stages of cognitive impairment in PD.
Our findings in patients with PD with dementia or at high risk for the development of dementia suggest infrequent AD-range levels of cortical Aβ-amyloid and significant correlation between neocortical Aβ-amyloid deposition and cognitive function. Specifically, there were 6 subjects (15%) from our cohort with AD-range neocortical PiB binding. Four of these 6 subjects were found to have dementia on detailed neuropsychological testing. Only 2 of 30 subjects with MCI (7%) exhibited AD-range neocortical PiB binding. None of the 5 cognitively intact subjects with PD showed AD-range PiB binding. In comparison, previously published large cohorts showed AD-range cortical PiB binding in 68% of subjects with MCI33 and 25%−33% of elderly healthy subjects.33,32
Our findings of relatively low neocortical PiB binding are in agreement with prior in vivo imaging studies in patients with PD and PDD. One study did not find evidence of abnormal PiB retention in early-stage PD, although these subjects were not at high risk of dementia in the short term.36 A different group reported abnormal neocortical PiB binding in only 2 of 10 subjects with PDD.12 A study comparing subjects with DLB and PD reported high global neocortical Aβ-amyloid burden in DLB, with subjects with PD manifesting low neocortical amyloid burden.13 Increased striatal Aβ-amyloid deposition was noted also in 2 of 10 subjects with PDD without elevated neocortical Aβ-amyloid. One postmortem study described a greater frequency of striatal Aβ-amyloid deposition in PDD than in PD.37 A study comparing cortical PiB retention in subjects with PD, PDD, DLB, and AD and healthy control subjects reported higher cortical Aβ-amyloid in the DLB group than in the PDD group, with PiB binding in the DLB group being comparable to PiB binding in the AD group.14 Aβ-amyloid deposition in the PDD group was low and comparable to that in the PD and healthy control groups. A more recent PiB study10 found no major differences in the degree of cortical or caudate nucleus PiB binding among patients with PD (with or without MCI), PDD, and DLB, with the majority of patients in each group showing PiB binding below the range typical of AD. Although this study did not report significant differences in mean values of PiB binding among the subject groups, the MCI and frank dementia groups showed greater variability in PiB binding values.
An interesting issue raised by our findings and those of prior investigations of PD with PiB imaging is the low frequency of AD-range elevated neocortical PiB binding in our study cohort compared with results reported in unaffected elderly subject cohorts. Only 15% (6 of 40) of our subjects exhibited AD-range neocortical PiB binding, a contrast with the 25%−30% reported in large studies of PiB binding in unaffected elderly subjects. Could PD prevent the pathologic deposition of fibrillar Aβ-amyloid? This is unlikely. Our results indicate elevated PiB binding in our most cognitively impaired subjects with PD. Subject selection bias is the likely explanation for the relatively low frequency of AD-range PiB neocortical PiB binding in our study cohort. We suggest that the majority of previously asymptomatic subjects with high neocortical Aβ-amyloid burdens and significant nigrostriatal degeneration will present with dementia and will be classified as having DLB (or AD if parkinsonism is not yet clinically evident). By convention, these subjects would not be recruited to studies such as ours, focusing on PD and excluding those with overt dementia within the first year of symptoms. This explanation is consistent also with previous reports of higher neocortical Aβ-amyloid burden and PiB binding in DLB as opposed to PDD.
Although average cortical PiB binding values in our PD MCI cohort are not in the AD range, PiB binding correlated significantly with measures of cognitive impairment. Our study findings support more indirect evidence from other Aβ-amyloid imaging studies for a possible role of amyloidopathy in the development of cognitive impairment in PD. These prior imaging studies predominantly focused on subjects with established PDD and for the most part did not include subjects at earlier stages of cognitive impairment. Only one published study includes a small PD MCI cohort of 9 subjects. In this study, subjects with PDD and DLB with higher vs lower PiB binding were compared. Higher PiB binding was associated with worse global cognitive impairment but not with any other clinical or neuropsychological features, including earlier onset or faster rate of progression of cognitive impairment. Of note, the authors reported that 3 of the PiB positive subjects were “too impaired to allow neuropsychological testing.”10 These authors did not report regression analysis based on continuous PiB levels and individual cognitive scores. A different study reported that Aβ-amyloid deposition in the parietal and posterior cingulate cortices in the PD and PDD groups was related to visuospatial impairment.14 They found no significant correlations between Aβ-amyloid retention and executive or memory functions. A study comparing PiB-positive with PiB-negative subjects with PDD and DLB found that PiB-positive subjects with dementia had lower Mini-Mental Status Examination scores than PIB-negative subjects with dementia.12 PiB-positive subjects in this study had a higher age at onset of parkinsonism and onset of dementia. Cumulative data suggest that the concurrent presence of amyloidopathy in PD may contribute to cognitive impairment.
Reported findings accord with prior neuropathology studies suggesting a role for Aβ-amyloid plaques, possibly in conjunction with Lewy body deposition, in the development of cognitive impairment in PD.7,8,38 Our results are supported also by a study suggesting an AD-like CSF biomarker profile in PDD and PD with MCI.39 Another study showed correlations between CSF Aβ42 and cognitive parameters in a set of patients with PD without dementia.40
Our findings suggest that subjects with PD at risk for dementia do not show AD-range neocortical Aβ-amyloid deposition. This observation is in contrast with previously reported findings of up to 33% of healthy elderly subjects, as well as subjects with DLB, showing AD-range Aβ-amyloid deposition. Differences in amyloid deposition may provide insight into the differential pathogenesis of PDD and DLB. Despite their non-AD range levels, neocortical Aβ-amyloid deposition in subjects with PD and MCI is probably of pathophysiologic significance, with significant correlations with cognitive function. Further longitudinal imaging studies will be particularly helpful in delineating the role of Aβ-amyloid in cognitive decline in PD.
ACKNOWLEDGMENT
The authors thank Christine Minderovic, V.A. Rogers, the UMHS PET technologists, cyclotron operators, and chemists for their assistance.
GLOSSARY
- AD
Alzheimer disease
- DLB
dementia with Lewy bodies
- DTBZ
dihydrotetrabenazine
- DVR
distribution volume ratio
- MCI
mild cognitive impairment
- MOCA
Montreal Cognitive Assessment
- PCA
principal component analysis
- PiB
Pittsburgh compound B
- PD
Parkinson disease
- PDD
Parkinson disease dementia
- UPDRS
Unified Parkinson's Disease Rating Scale
- VOI
volume of interest
- WAIS
Wechsler Adult Intelligence Scale
AUTHOR CONTRIBUTIONS
M.P. researched data, contributed to discussion, wrote the manuscript, and reviewed and edited the manuscript. N.I.B, R.A.K, and K.A.F. researched data, contributed to discussion, and reviewed and edited the manuscript. M.L.T.M.M and R.L.A. contributed to discussion and reviewed and edited the manuscript. Statistical analysis was performed by M.P, K.A.F., and N.I.B.
DISCLOSURE
M. Petrou receives research support from the Radiological Society of North America, the NIH, and the Department of Veterans Affairs. N. Bohnen receives research support from the NIH and the Department of Veterans Affairs. M. Müller reports no disclosures. R. Koeppe serves on the Board of the International Society of Cerebral Blood Flow and Metabolism; receives research support from NIH (NINDS, NIA); and is a consultant for Johnson & Johnson and Merck. R. Albin has received compensation for expert witness testimony in litigation regarding dopamine agonist-induced impulse control disorders. Dr. Albin serves on the editorial boards of Neurology®, Experimental Neurology, and Neurobiology of Disease; receives grant support from the NIH and the Department of Veterans Affairs; and has served on the Data Safety and Monitoring Boards for the QE3 and HORIZON trials. K. Frey receives research support from the NIH, GE Healthcare, and AVID pharmaceuticals (Eli Lilly subsidiary); serves as a consultant to AVID Pharmaceuticals, MIMVista, Inc., Bayer-Schering, and GE Healthcare; and holds equity (common stock) in GE, Bristol-Myers, Merck, and Novo-Nordisk. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord 2007;22:1689− 1707; quiz 1837 [DOI] [PubMed] [Google Scholar]
- 2. Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sorensen P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60:387–392 [DOI] [PubMed] [Google Scholar]
- 3. Aarsland D, Perry R, Brown A, Larsen JP, Ballard C. Neuropathology of dementia in Parkinson's disease: a prospective, community-based study. Ann Neurol 2005;58:773–776 [DOI] [PubMed] [Google Scholar]
- 4. Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology 2000;54:1916–1921 [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 2005;64:1404–1410 [DOI] [PubMed] [Google Scholar]
- 6. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 2006;67:1931–1934 [DOI] [PubMed] [Google Scholar]
- 7. Sabbagh MN, Adler CH, Lahti TJ, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord 2009;23:295–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Compta Y, Parkkinen L, O'Sullivan SS, et al. Lewy- and Alzheimer-type pathologies in Parkinson's disease dementia: which is more important? Brain 2011;134:1493–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klunk WE, Engler E, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease using the novel positron emission tomography tracer, Pittsburgh compound-B. Ann Neurol 2004;55:306–319 [DOI] [PubMed] [Google Scholar]
- 10. Foster ER, Campbell MC, Burack MA, et al. Amyloid imaging of Lewy body-associated disorders. Mov Disord 2010;25:2516–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Villemagne VL, Ong K, Mulligan RS, et al. Amyloid imaging with 18F-florbetaben in Alzheimer disease and other dementias. J Nucl Med 2011;52:1210–1217 [DOI] [PubMed] [Google Scholar]
- 12. Maetzler W, Reimold M, Liepelt I, et al. [11C]PIB binding in Parkinson's disease dementia. Neuroimage 2008;39:1027–1033 [DOI] [PubMed] [Google Scholar]
- 13. Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–1338 [DOI] [PubMed] [Google Scholar]
- 14. Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71:903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janvin CC, Larsen JP, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson's disease: progression to dementia. Mov Disord 2006;21:1343–1349 [DOI] [PubMed] [Google Scholar]
- 16. Caviness JN, Driver-Dunckley E, Connor DJ, et al. Defining mild cognitive impairment in Parkinson's disease. Mov Disord 2007;22:1272–1277 [DOI] [PubMed] [Google Scholar]
- 17. Hughes TA, Ross HF, Musa S, et al. A 10-year study of the incidence of and factors predicting dementia in Parkinson's disease. Neurology 2000;54:1596–1602 [DOI] [PubMed] [Google Scholar]
- 18. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442 [DOI] [PubMed] [Google Scholar]
- 20. Delis DC, Kramer JH, Kaplan E, Ober BA. California Verbal Learning Test Manual, Adult Version, 2nd ed. San Antonio, TX: The Psychological Corporation; 2000 [Google Scholar]
- 21. Wechsler D. WAIS III Technical Manual. San Antonio, TX: The Psychological Corporation; 1997 [Google Scholar]
- 22. Delis DC, Kaplan E, Kramer JH. eds. Delis-Kaplan Executive Function System (D-KEFS): Examiner's Manual. San Antonio, TX: The Psychological Corporation; 2001 [Google Scholar]
- 23. Stroop JR. Studies of interference in serial verbal reactions. J Exp Psychol 1935;18:643–662 [Google Scholar]
- 24. Bohnen N, Jolles J, Twijnstra A. Modification of the Stroop Color Word Test improves differentiation between patients with mild head injury and matched controls. Clin Neuropsychol 1992;6:178–184 [DOI] [PubMed] [Google Scholar]
- 25. Benton AL, Varney NR, Hamsher K. Judgment of Line Orientation, Form V. Iowa City: University of Iowa Hospitals; 1975 [Google Scholar]
- 26. Koeppe RA, Frey KA. Equilibrium analysis of [11C]PIB studies. Neuroimage 2008;41:T30 [Google Scholar]
- 27. Jewett DM, Kilbourn MR, Lee LC. A simple synthesis of [11C]dihydrotetrabenazine (DTBZ). Nucl Med Biol 1997;24:197–199 [DOI] [PubMed] [Google Scholar]
- 28. Koeppe RA, Frey KA, Kume A, Albin R, Kilbourn MR, Kuhl DE. Equilibrium versus compartmental analysis for assessment of the vesicular monoamine transporter using (+)-α-[11C]dihydrotetrabenazine (DTBZ) and positron emission tomography. J Cereb Blood Flow Metab 1997;17:919–931 [DOI] [PubMed] [Google Scholar]
- 29. Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 1996;16:834–840 [DOI] [PubMed] [Google Scholar]
- 30. Koeppe RA, Frey KA, Kuhl DE, Kilbourn MR. Assessment of extrastriatal vesicular monoamine transporter binding site density using stereoisomers of [11C]dihydrotetrabenazine. J Cereb Blood Flow Metab 1999;19:1376–1384 [DOI] [PubMed] [Google Scholar]
- 31. Burke JF, Albin RL, Koeppe RA, et al. Assessment of mild dementia with amyloid and dopamine terminal positron emission tomography. Brain 2011;134:1647–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology 2006;67:446–452 [DOI] [PubMed] [Google Scholar]
- 33. Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–1283 [DOI] [PubMed] [Google Scholar]
- 34. Frey KA, Koeppe RA. Surface projection maps of cortical amyloid binding reveal canonical patterns in neurodegenerative dementias. J Nucl Med 2012. ( suppl 1):144 Abstract [Google Scholar]
- 35. Bohnen NI, Albin RL, Koeppe RA, et al. Positron emission tomography of monoaminergic vesicular binding in aging and Parkinson disease. J Cereb Blood Flow Metab 2006;26:1198–1212 [DOI] [PubMed] [Google Scholar]
- 36. Johansson A, Savitcheva I, Forsberg A, et al. [11C]-PIB imaging in patients with Parkinson's disease: preliminary results. Parkinsonism Relat Disord 2008;14:345–347 [DOI] [PubMed] [Google Scholar]
- 37. Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Striatal beta-amyloid deposition in Parkinson disease with dementia. J Neuropathol Exp Neurol 2008;67:155–161 [DOI] [PubMed] [Google Scholar]
- 38. Kalaitzakis ME, Pearce RK, Gentleman SM. Clinical correlates of pathology in the claustrum in Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 2009;461:12–15 [DOI] [PubMed] [Google Scholar]
- 39. Montine TJ, Shi M, Quinn JF, et al. CSF Aβ42 and tau in Parkinson's disease with cognitive impairment. Mov Disord 2010;25:2682–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leverenz JB, Watson GS, Shofer J, Zabetian CP, Zhang J, Montine TJ. Cerebrospinal fluid biomarkers and cognitive performance in non-demented patients with Parkinson's disease. Parkinsonism Relat Disord 2011;17:61–64 [DOI] [PMC free article] [PubMed] [Google Scholar]