

Abstract
Objective:
Autoantibodies targeting voltage-gated potassium channel (VGKC) complexes cause a spectrum of neuronal hyperexcitability disorders. We investigated pain as a manifestation of VGKC-complex autoimmunity.
Methods:
We reviewed the prevalence and characteristics of pain in VGKC-complex-immunoglobulin G (IgG)–seropositive patients in 25 months of comprehensive service testing for neural autoantibodies, subtyped positive sera for LGI1-IgG and CASPR2-IgG specificities, and reviewed pain prevalence in autoimmune control patients.
Results:
VGKC-complex-IgG was identified in 1,992 patients of 54,853 tested (4%). Of 316 evaluated neurologically at Mayo Clinic, 159 (50%) had pain, in isolation (28%) or with accompanying neurologic manifestations (72%), and not attributable to alternative cause. Pain was subacute in onset, chronic in course, neuropathic, nociceptive, regional, or diffuse and sometimes attributed to fibromyalgia (6%) or psychogenic cause (13%). Most patients had normal peripheral nervous system function, measured by neuropathy impairment scores and nerve conduction. Evidence of neuronal hyperexcitability (hyperhidrosis, quantitative heat-pain hyperalgesia, or electromyographic excitability) was 25-fold more common in pain patients. Pain management required multiple medications in 70% (narcotics, 30%); 13 of 16 patients reported pain relief with immunotherapy. Pain was significantly associated with CASPR2-IgG-positivity (16% positive with pain, 7% without pain; p = 0.014) but not with LGI1-IgG. Less than 10% of 167 patients with neural autoantibodies other than VGKC-complex-IgG reported pain.
Conclusions:
Chronic idiopathic pain is a syndromic manifestation of VGKC-complex autoimmunity. Hyperexcitability of nociceptive pathways is implicated. CASPR2-IgG significantly associates with pain, but in most patients the antigenic VGKC-complex molecule remains to be determined. VGKC-complex autoimmunity represents an important new direction for pain research and therapy.
The cause of most chronic pain disorders is unknown. Neuronal hyperexcitability conferred by DNA variants in the voltage-gated sodium channel 1.7 gene (Nav1.7) was recently identified in patients with idiopathic chronic pain with and without loss of small unmyelinated C fibers.1 An inward rectifying channel (potassium/sodium hyperpolarization-activated cyclic nucleotide-gated ion channel 2) also has been identified as an important regulator of nociceptive pain.2 Neuronal voltage-gated potassium channels (VGKC) act synergistically with these cation channels to maintain nociceptive afferent sensory neural thresholds.3,4
Autoantibodies targeting neuronal VGKC-complexes containing Kv1 channels were first reported in patients with motor nerve hyperexcitability and hyperhidrosis (Isaacs syndrome).5,6 Patient serum was reported to induce peripheral motor nerve hyperexcitability, attributed to suppression of voltage-gated outward K+ currents.7 Subsequent reports documented limbic encephalitis as another manifestation of VGKC autoimmunity (Morvan syndrome when accompanied by neuromyotonia, sleep disturbance, and autonomic dysfunction).8–10 The autoimmune neuronal VGKC hyperexcitability spectrum recognized today includes seizures, psychosis, and gut dysmotility.11–13 Pain has not been the focus of any reports.
Autoantibodies detected in diagnostic radioimmunoprecipitation assays infrequently bind to VGKC channel protein subunits, but rather to coprecipitating proteins in a multimolecular antigenic complex containing Kv1 channels.14,15 The leucine-rich glioma-inactivated 1 protein (LGI1, thought to be CNS-restricted) and contactin-associated protein-2 (CASPR2, expressed in both peripheral and central nervous systems) are 2 defined antigens. LGI1-binding antibodies are reportedly most common with limbic encephalitis,14,15 and CASPR2-binding antibodies with acquired neuromyotonia.15,16 Functions of LGI1 and CASPR2 are poorly understood, but their modulation of potassium channel function and location at synapses and nodal regions suggests a role in regulating neural excitation.17,18 Regardless of the molecular identity of the marker autoantibody's target, the neurologic manifestations of early-diagnosed VGKC-complex autoimmunity are generally responsive to immunotherapy.15,16 In this study we systematically investigated the prevalence and characteristics of pain in a large unselected cohort of patients in whom VGKC-complex-IgG was detected in serum by comprehensive evaluation for markers of neurologic autoimmunity.
METHODS
Participants and clinical evaluation.
The Mayo Clinic Institutional Review Board approved the study. We reviewed medical records of VGKC-complex-IgG-positive patients evaluated at Mayo Clinic June 1, 2008, to June 30, 2010. As disease control population, we selected 167 patients in whom PCA-1-IgG, ANNA-1-IgG, or amphiphysin-IgG was detected during the same period. At least 2 study neurologists (C.J.K., P.A., a.m., S.J.P.) reviewed demographic, clinical, and laboratory data. Abstracted pain descriptors (terminology of the International Association for the Study of Pain [IASP])19 included onset (rapid [maximal within 2 weeks], insidious [progressively worsening more than 3 months], chronic [present more than 6 months]), distribution (head, face, abdominal viscera, total body, extremities), neuropathic quality (burning, tingling, lancinating, allodynic, pruritic), and nociceptive qualifiers (visceral, deep, superficial). “Visceral” descriptors included diffuse and difficult-to-localize pain; “deep” was localized and dull; “superficial” was sharp and well-defined on the body surface. Patients with incidental pain from a clearly definable cause (e.g., trauma, migraine, or arthritis) were classified in the nonpain cohort. The Neuropathy Impairment Score (NIS; based on sum of motor, sensory, and reflex abnormalities) was calculated for all study patients (0 = no impairment; 288 = areflexia, extremity, and bulbar paralysis and extremity insensitivity).20 We reviewed neurologic test results (EMG, quantitative sensory21 and autonomic functions) for evidence of neuron loss or hyperexcitability,22,23 and clinical characteristics and outcomes of patients who received immunotherapy.
Serologic evaluation.
Radioimmunoprecipitation screening assay for VGKC-complex autoantibodies.
Multimolecular complexes were solubilized in digitonin from porcine cerebral cortical membranes and ligated with 125I-α-dendrotoxin added in slight excess. After incubating duplicate aliquots 16 hours at 4°C with 5 μL of patient serum, we added goat IgG reactive with human IgG, and counted γ-emission from washed immune precipitates. After correction for precipitation of 125I-α-dendrotoxin alone,24 results are expressed as nanomoles of VGKC-complex bound per liter of serum (normal range, 0.00–0.02 nmol/L).
Assays for LGI1-IgG and CASPR2-IgG.
All sera yielding positive VGKC-complex-IgG results were absorbed at 1:10 dilution with bovine liver powder (to remove nonorgan-specific autoantibodies) and analyzed by immunofluorescence for IgG reactive with LGI1 or CASPR2 (in-house-validated kit assay [EUROIMMUN, Lübeck, Germany] incorporating as substrate fixed HEK 293 cells, nontransfected and transfected with plasmid encoding human LGI1 or CASPR2 proteins). FITC-conjugated goat IgG (human IgG-specific) detected bound IgG. Controls included sera from 126 healthy subjects.
Assays for other neural autoantibodies.
A standardized immunofluorescence assay was used to detect IgG neural autoantibodies (anti-neuronal nuclear [ANNA-1, 2, 3], amphiphysin, Purkinje cell cytoplasmic [PCA]-1, 2, Tr]) and non-neuronal antibodies.24 Radioimmunoprecipitation assays detected antibodies to neuronal voltage-gated calcium channels (P/Q-type and N-type), muscle and neuronal ganglionic (α3) nicotinic acetylcholine receptors, and glutamic acid decarboxylase 65-isoform. Recombinant Western blot detected collapsin response-mediator protein–5-IgG. ELISA detected striational antibodies and latex agglutination assays detected thyroglobulin and thyroid microsomal (thyroperoxidase) antibodies.24
Statistical analyses.
To determine whether variables other than VGKC-complex-IgG might contribute to the pain presentation, we performed multivariate logistic regression analysis for significant association with neurologic presentations, coexisting autoantibodies, sex, or age. We compared VGKC-complex-IgG values for groups with and without pain using 2-tailed Fisher exact test. Two-sided p < 0.05 was considered significant. We also compared neurologic manifestations and demographic features in the 2 groups. To determine whether serum VGKC-complex-IgG values differed significantly between seropositive patients with and without pain, we compared median values of each group; for subgroup analysis we divided the patients into 3 groups based on serum VGKC-complex-IgG values: low (0.03–0.09 nmol/L), medium (0.10–0.99 nmol/L), and high (>1.00 nmol/L).
RESULTS
Demographics and neurologic accompaniments.
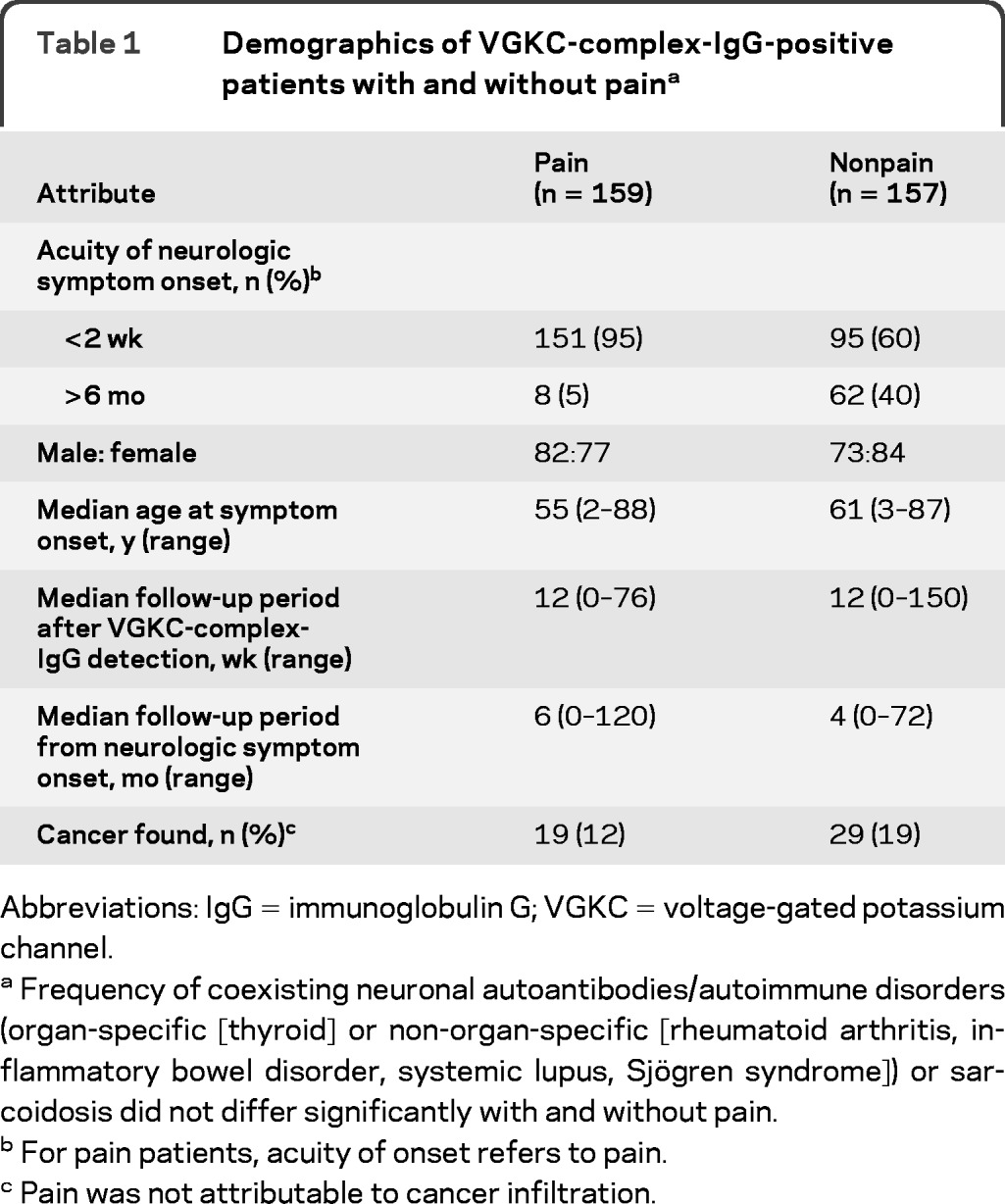
From June 1, 2008, through June 30, 2010, serum samples from 54,853 patients with diverse neurologic presentations were submitted to the Mayo Clinic Neuroimmunology Laboratory for comprehensive service evaluation of neural autoantibody profiles. VGKC-complex-IgG was detected in 1,992 patients; 316 were evaluated neurologically at this institution (table 1). Pain was an initial symptom in 159 patients (50%), 82 male and 77 female. Pain symptoms in 151 patients (95%) were subacute in onset (maximum severity by 2 weeks). In 80 patients followed beyond 6 months, pain became chronic; 70% required multiple pain medications (30% required narcotics). Patients who received narcotics were evaluated by a pain specialist, as were 12% of the remaining pain cohort.
Table 1.
Demographics of VGKC-complex-IgG-positive patients with and without paina
Abbreviations: IgG = immunoglobulin G; VGKC = voltage-gated potassium channel.
Frequency of coexisting neuronal autoantibodies/autoimmune disorders (organ-specific [thyroid] or non-organ-specific [rheumatoid arthritis, inflammatory bowel disorder, systemic lupus, Sjögren syndrome]) or sarcoidosis did not differ significantly with and without pain.
For pain patients, acuity of onset refers to pain.
Pain was not attributable to cancer infiltration.
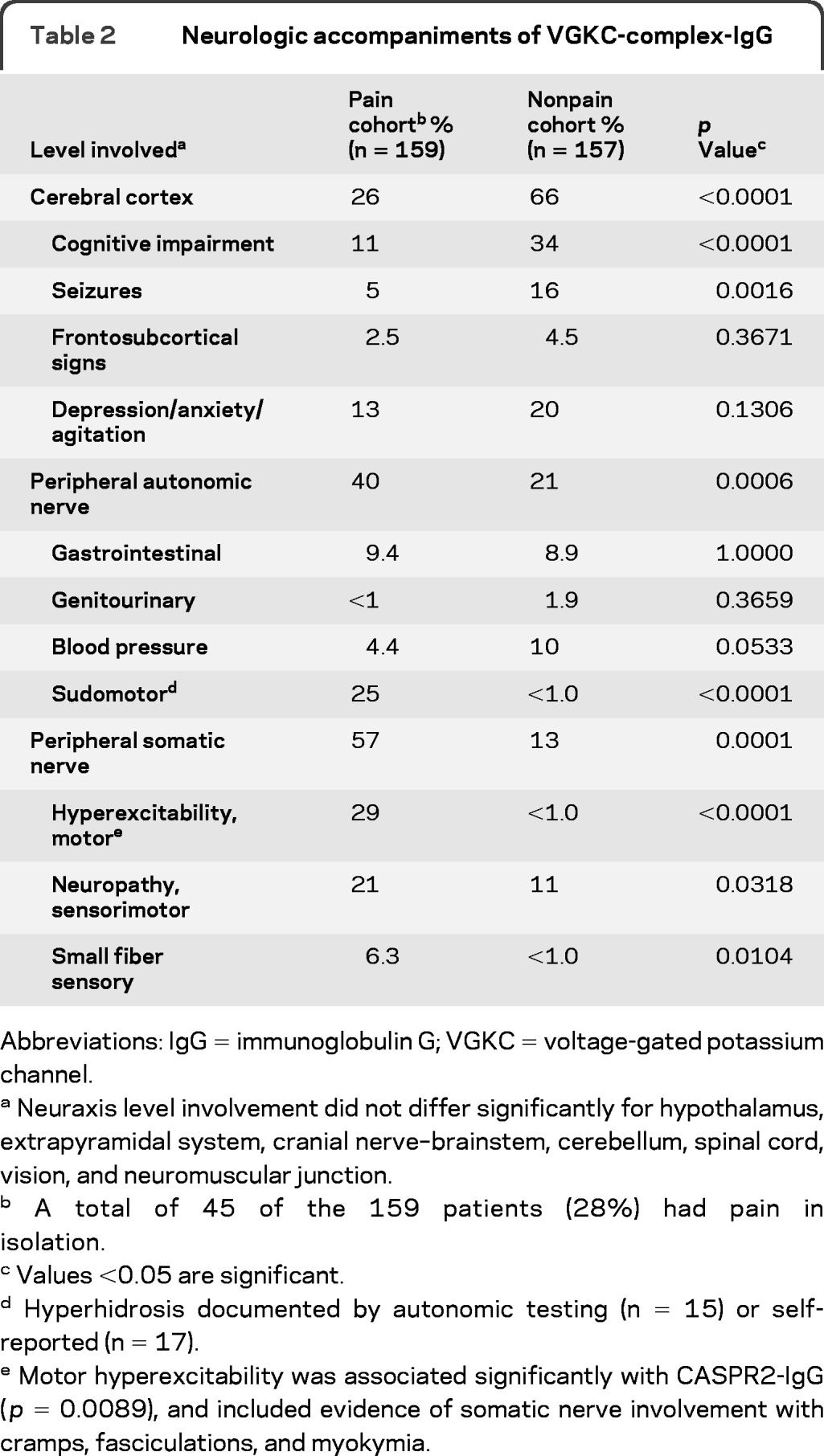
Pain was the sole symptom in 45 patients (28%). Table 2 summarizes coexisting neurologic manifestations in the remaining 72%. Somatic and small fiber neuropathies were diagnosed more commonly in the pain cohort (27%) than in the nonpain cohort (12%; p = 0.03 and p < 0.0001, respectively), and 11% had episodic global hyperhidrosis (none in the nonpain cohort). Twenty-nine percent of the pain cohort, but less than 1% of the nonpain cohort, complained of cramps, myokymia, or fasciculations (p < 0.0001). Cognitive impairment, seizures, frontosubcortical manifestations, depression, or anxiety occurred in 26% of the pain cohort and in 66% of the nonpain cohort (p < 0.0001).
Table 2.
Neurologic accompaniments of VGKC-complex-IgG
Abbreviations: IgG = immunoglobulin G; VGKC = voltage-gated potassium channel.
Neuraxis level involvement did not differ significantly for hypothalamus, extrapyramidal system, cranial nerve–brainstem, cerebellum, spinal cord, vision, and neuromuscular junction.
A total of 45 of the 159 patients (28%) had pain in isolation.
Values <0.05 are significant.
Hyperhidrosis documented by autonomic testing (n = 15) or self-reported (n = 17).
Motor hyperexcitability was associated significantly with CASPR2-IgG (p = 0.0089), and included evidence of somatic nerve involvement with cramps, fasciculations, and myokymia.
The frequency of cancer detection did not differ significantly (12% pain cohort vs 19% nonpain cohort, p = 0.1197; table 1); 53% of the pain cohort, and 57% of the nonpain cohort, were evaluated by CT body, PET imaging, colonoscopy, or mammography.
Pain characteristics.
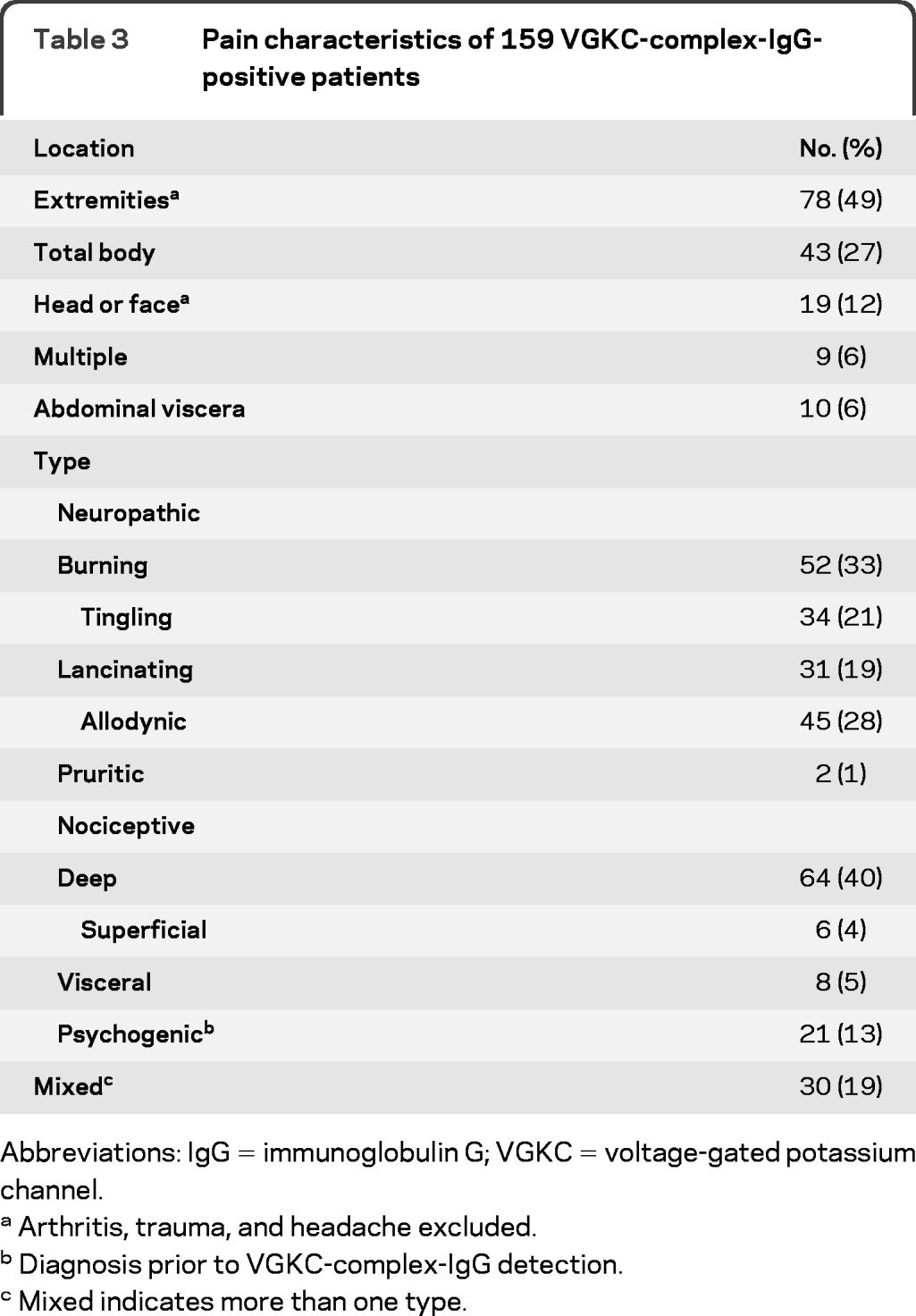
Table 3 summarizes the pain location and qualifiers. Extremities were affected most commonly (49%). Twenty-seven percent presented with total body pain; before VGKC-complex-IgG detection, 13% were diagnosed as psychogenic and 6.2% as fibromyalgia (multiple painful trigger points). Atypical head and face pain was documented in 12% of patients. In the entire group, pain was descriptively neuropathic (58%) or nociceptive (47%). Descriptors included burning (33%), allodynic (28%), tingling (21%), lancinating (19%), and uncommonly pruritic (1%).
Table 3.
Pain characteristics of 159 VGKC-complex-IgG-positive patients
Abbreviations: IgG = immunoglobulin G; VGKC = voltage-gated potassium channel.
Arthritis, trauma, and headache excluded.
Diagnosis prior to VGKC-complex-IgG detection.
Mixed indicates more than one type.
Neurologic examination and investigation.
Structured peripheral neurologic examination commonly revealed no abnormality in the pain cohort (median NIS, 0 [normal]; range, 0–84). Furthermore, the patient subset assigned a diagnosis of neuropathy had minimal abnormalities documented (median NIS, 6); most common was mild subjective loss of small fiber sensation (temperature and pain) in toes with preserved reflexes and strength. No patient had the local skin or trophic changes encountered in complex regional pain disorder. Family histories did not suggest inherited peripheral neuropathies.
Evaluations were nonrevealing for diabetes, thyroid, kidney, or connective tissue disease, infectious hepatitis, or plasma cell dyscrasia. Median fasting blood glucose values were normal (available for 130 of 159 pain patients and for 122 of 157 nonpain patients). The median hemoglobin A1c value (available for 124 of 159 pain patients and for 123 of 157 nonpain patients) did not differ significantly with and without pain (5.6%; 5.8%). Glucose tolerance test results (3 patients) were normal.
The 45 patients whose sole complaint was pain (28%) were commonly evaluated for occult neuropathy or CNS disease. No abnormalities were detected by EMG (64%), thermoregulatory sweat test (20%), autonomic reflex testing (27%), sural nerve biopsy (2%), or brain and spinal cord MRI (42%).
In evaluation for peripheral neuropathy, nerve conduction studies were performed in 94 (59%) of the entire pain cohort. Results for motor nerves (peroneal, tibial, ulnar) and sensory nerves (sural and median) were normal in 62%. The abnormality detected most commonly was a minor reduction of sural sensory nerve action potential amplitude. EMG revealed motor hyperexcitability (neuromyotonia, fasciculations, or cramp discharges) in 9 of 94 patients tested (10%), compared with 0 of 44 patients in the nonpain cohort (p < 0.001, table 2). After-discharges on F-wave stimulation were documented in 2 patients. Results for cramp-fasciculation-specific testing revealed no after-discharges (2, 5, 10 Hz, recording at abductor hallucis or hypothenar; 3 patients). Quantitative sensory testing21 revealed heat-pain hyperalgesia (C fiber allodynia) in 8 of 19 tested pain patients (38%) compared with 0 of 5 nonpain patients (p < 0.001; table 2). Quantitative autonomic reflex test or thermoregulatory sweat test revealed sudomotor abnormalities in 29% of the pain cohort and <1% of the nonpain cohort (p < 0.001; table 2, figure 1). Pain commonly localized to the distribution of sudomotor sweat abnormalities (figure 1). Thermoregulatory sweat test revealed 15 patients with hyperhidrosis (i.e., early sweating) in addition to 17 who self-reported hyperhidrotic spells.
Figure 1. Neuropathic pain locations in 6 illustrative patients seropositive for voltage-gated potassium channel–complex-immunoglobulin G (IgG) correlated with sites of abnormal heat-induced sweat responses.

Both CASPR2-IgG and LGI1-IgG were detected in 2 patients. (A–F) Yellow skin areas in thermoregulatory sweat test23 indicate reduced or absent sweat output (revealed by alizarin red powder; face not tested [sphygmomanometer on arm of E]). (G) Indirect immunofluorescence. IgG in serum of patients B and E bound to HEK293 cells transfected with either LGI1 or CASPR2, but not to nontransfected cells (bar, 20 μm). The remaining 4 patients were seronegative for CASPR2-IgG and LGI1-IgG.
Serologic findings.
Only VGKC-complex-IgG correlated significantly with the occurrence of pain. Multivariate analysis revealed no pain association with coexisting neural autoantibodies or autoimmune disorders (organ-specific [thyroid] or nonorgan-specific [rheumatoid arthritis, inflammatory bowel disorder, systemic lupus, Sjögren syndrome]) or sarcoidosis. None among 126 healthy subjects was seropositive for VGKC-complex-IgG.
Serum VGKC-complex-IgG levels in the entire cohort did not differ significantly for patients with pain (median, 0.13 nmol/L; range, 0.03–4.34 nmol/L) and without pain (median, 0.15 nmol/L; range, 0.03–5.53 nmol/L). LGI1-IgG and CASPR2-IgG were detected in <20% of the total seropositive cohort (table e-1 on the Neurology® Web site at www.neurology.org). CASPR2-IgG associated significantly with occurrence of pain (p = 0.014), and LGI1-IgG associated significantly with higher VGKC-complex-IgG values (p = 0.0002).
VGKC-complex-IgG values and frequency of LGI1-IgG and CASPR2-IgG detection in the 45 patients with isolated pain (28%) were comparable to the entire cohort. Specifically, among 14 patients with low VGKC-complex-IgG values (0.03–0.09 nmol/L), 3 had LGI1-IgG and 4 had CASPR2-IgG; among 27 patients with medium VGKC-complex-IgG values (0.10–0.99 nmol/L), 2 had LGI1-IgG and 3 had CASPR2-IgG; and among 4 patients with high VGKC-complex-IgG values (>1.00 nmol/L), none had LGI1-IgG or CASPR2-IgG. Pain symptomatology was significantly less frequent in the disease control group (41 patients with PCA-1 autoimmunity [anti-Yo], 111 with ANNA-1 autoimmunity [anti-Hu], and 15 with amphiphysin autoimmunity) (9% prevalence) than in the VGKC-complex-IgG-positive group (50% prevalence; p < 0.01).
Pain outcome with immunotherapy.
All but 1 of 16 patients summarized in table 4 received multiple pain medications: nonsteroidal anti-inflammatory drugs, narcotics, or antiepileptics. The benefit from membrane stabilizing medications (e.g., gabapentin, pregabalin) was generally minimal unless accompanied by immunotherapy. The principal indication for immunotherapy in 14 patients was coexisting neurologic symptoms; in only 2 was intractable pain the principal indication (cases 1 and 2; table 4). Immunotherapy included oral prednisone (n = 7), IV methylprednisolone (n = 6), IV immune globulin (IVIg) (n = 2), methotrexate (n = 2), and hydroxychloroquine (n = 1). The median follow-up period was 18 weeks (range, 4–61 weeks). At last clinical evaluation, 81% reported pain improvement or resolution after initiating immunotherapy; 10 (63%) continued to receive low-dose steroid or a steroid-sparing immunosuppressant. Pain symptom improvement in 3 patients correlated with seizure improvement or resolution. VGKC-complex-IgG levels fell by more than 50% in 3 of 3 patients retested postimmunotherapy.
Table 4.
Pain responses in VGKC-complex-IgG-positive patients receiving immunotherapy

Abbreviations: ACh = acetylcholine; ANA = antinuclear autoantibody; GAD65 = glutamic acid decarboxylase-65-IgG; IgG = immunoglobulin G; IVIg = IV immunoglobulin; IVMP = IV methylprednisolone; TPO = thyroperoxidase antibody; VGKC = voltage-gated potassium channel.
The high frequency of LGI1 reflects the frequency of epilepsy as indication for immunotherapy.
Illustrative cases with isolated pain.
Case 1.
A man aged 80 years (table 4 and figure e-1) presented with bilateral foot pain that was burning/lancinating. It worsened over 2 weeks, extended to knees and elbows, and confined him to bed. Previous health was excellent. Pain, scored 10 of 10, was relieved minimally (8 of 10) by simultaneously administered methadone (10 mg, 3 times daily), oxycodone (10 mg, 4 times daily), and gabapentin 1 (800 mg, 3 times daily). The patient experienced episodic hyperhidrosis, 6.8 kg weight loss, and uncharacteristic anxiety, but had no other neuropsychiatric manifestations or complaints of cramping, fasciculations, or myokymia. Tests of mental status, strength, reflexes, and sensation were normal. MRI of the neuroaxis, serial nerve conduction studies, needle EMG, and autonomic testing (including cardiovagal, adrenergic, quantitative sudomotor, and whole body thermoregulatory sweat output) were normal apart from early sweat and heat-pain hyperalgesia (0.5, fifth percentile; quantitative sensory testing). A diagnosis of psychogenic pain was considered. Rheumatologic serology was negative (SSA, SSB, RNP, SM, SCL70). CSF showed isolated protein elevation (50 mg/dL; normal, <45 mg/dL). Serum VGKC-complex-IgG value was medium (0.16 nmol/L); LGI1-IgG was detected (figure e-1). Whole body PET–CT imaging was normal. Pain and anxiety improved within 2 weeks of completing a 5-day course of IV methylprednisolone, 1 g daily. Oxycodone, methadone, and methylprednisolone medications were discontinued by 3 months and methotrexate was initiated (20 mg weekly). Pain symptoms abated, weight was regained. By 6 months the patient had resumed normal activities.
Case 2.
A woman aged 55 years (table 4) experienced disabling total body pain of subacute onset, initially diagnosed as fibromyalgia and persisting 7 years. She had spells of entire body itching and sweating without hives. Pain was lancinating, burning, deep, and aching, and was typically scored 8 of 10. She was treated with oxycodone (15 mg twice daily) and gabapentin (600 mg twice daily). Motor, sensory, and reflex examinations were normal. There was no evidence of cramping, fasciculation, or myokymia. MRI of the entire neuroaxis, serial nerve conduction and needle electromyographic studies, autonomic testing (quantitative measures of cardiovagal, adrenergic, and sudomotor functions and whole body thermoregulatory sweat output) were all normal. Quantitative sensory testing showed heat-pain hyperalgesia. There was no serologic evidence of systemic autoimmunity. Serum VGKC-complex-IgG value was high (3.46 nm/L); neither LGI1-IgG nor CASPR2-IgG was detected. Whole body PET-CT imaging revealed no evidence of malignancy. Treatment with IV methylprednisolone (1 g every 4 days) conferred dramatic benefit, but pain returned when the interval between treatments was extended to 14 days. Oral prednisone therapy (60 mg daily) was partially effective, but was discontinued due to major weight gain. Plasma exchange, twice weekly for 2 months, reduced the self-reported pain score to 2 of 10, and VGKC-complex-IgG was no longer detectable. Pain symptoms were scored 2 of 10 at 24 weeks (receiving oral dexamethasone, 40 mg weekly).
DISCUSSION
Our findings implicate VGKC-complex autoimmunity as a cause of chronic pain. Pain was a declared symptom in 50% of seropositive patients, 5 times more common than in control patients with other neural autoantibodies. It was typically subacute in onset, chronic in nature, descriptively nociceptive or neuropathic. In all cases comprehensive serologic testing was requested to evaluate an unexplained neurologic presentation. The pain occurred in isolation or with recognized neurologic manifestations of VGKC-complex autoimmunity,11–13 with prominent morbidity, evidenced by medication requirements (including narcotics) and referrals to pain specialists. The 3-fold less frequent complaints of pain in patients with cognitive impairment suggest that pain is underestimated in patients potentially less able to verbalize symptoms, as is appreciated in cognitively impaired elderly populations.25
Documented hyperhidrosis, electromyographic hyperexcitability, and objectively measured heat-pain hyperalgesia support the concept of hyperexcitability within nociceptive pathways as a pathophysiologic basis of VGKC-complex autoimmune pain. Hyperexcitability of nociceptive pathways is more problematic to prove than the readily recordable manifestations of motor hyperexcitability: seizures, cramps, myokymia, and fasciculations. However, in support of our hypothesis, few patients had clinical or electrophysiologic evidence of neuronal loss and, when observed, pathways involved most commonly were sensory. Although cutaneous trophic and inflammatory changes characteristic of complex regional pain disorder26 were lacking, cutaneous small fiber abnormalities (sweating and heat-pain sensation) were often the only demonstrable abnormality. As in other chronic pain disorders, symptoms were disproportionate to objectively measured neuropathic dysfunction. Affirming the inadequacy of contemporary tests for establishing the etiology of chronic pain, fibromyalgia and psychogenic pain were commonly assigned as initial diagnoses.
CASPR2-IgG significantly associated with pain. CASPR2, a member of the neurexin family that colocalizes with Kv1 channels in the paranodal axon, has been implicated in autism spectrum disorders27 and glioma tumor suppression.28 The link between CASPR2 autoimmunity and pain may involve disruption of VGKC channel localization at paranodes. Application of patient sera containing VGKC-complex-IgG to cultured neurons was reported to reduce the functional VGKC density at paranodes.7 The predicted outcome, impairment of repolarization with resultant channel hyperexcitability, is consistent with the reported association of CASPR2-IgG with peripheral motor hyperexcitability,15 and the nociceptive neuronal hyperexcitability that we postulate.
IgG in some patients' serum bound to the LGI1 component of the VGKC-complex. Autosomal recessive mutations of LGI1 are recognized to cause cortical hyperexcitability with seizures and auditory phenomena (buzzing and tinnitus).29 Although thought to be restricted to the CNS, both peripheral and central hypomyelination has been attributed to LGI1 defects.30 Contemporary concepts of chronic pain emphasize mechanisms increasing synaptic efficacy in central nociceptive pathways.31 IgG targeting LGI1 is a plausible contributor to central sensitization.
The treatment responses we observed in VGKC-complex-IgG seropositive patients with pain parallel experience documented in patients with other neurologic manifestations of VGKC-complex autoimmunity.12,13,32–35 Analogous to the seizure response in patients with VGKC-complex autoimmune encephalitis, membrane-stabilizing antiepileptic drugs partially benefited pain. Most remarkably, 81% of patients receiving immunotherapy experienced pain symptom improvement not attributable to changes in standard analgesic medication or dosing, and allowing narcotics to be discontinued in some cases.
Our findings add VGKC-complex autoimmune pain to the list of potentially treatable autoimmune channelopathies.36 Identification of antigenic VGKC-complex molecules relevant to idiopathic pain represents a new direction for pain-related research, a field where fundamental causative mechanisms have been elusive.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Vickie Mewhorter and Debby Cheung for technical assistance and Dr. Orna O'Toole for hyperglycemia review of the cohort.
GLOSSARY
- IASP
International Association for the Study of Pain
- IgG
immunoglobulin G
- IVIg
IV immune globulin
- NIS
Neuropathy Impairment Score
- VGKC
voltage-gated potassium channel
Footnotes
Editorial, p. 1080
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
C.J.K.: study concept or design, drafting and revising the manuscript, interpretation of data, statistical analysis, obtaining funding. P.A.A.: interpretation of data, acquisition of data. V.A.L.: study concept or design, revising the manuscript. S.J.P.: study concept or design, study supervision or coordination, drafting and revising the manuscript. A.M.: analysis or interpretation of data, revising the manuscript.
DISCLOSURE
C.J. Klein has received funds from Pfizer for helping in a study design for a drug trial in neuropathy. Serologic testing for VGKC-complex autoantibodies is offered on a service basis by Mayo Collaborative Service, Inc., an agency of Mayo Foundation, but no author benefits financially from this testing. V.A. Lennon, P.A. Aston, and A. McKeon report no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Na(V) 1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012;71:26–39 [DOI] [PubMed] [Google Scholar]
- 2. Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 2011;333:1462–1466 [DOI] [PubMed] [Google Scholar]
- 3. Takeda M, Tsuboi Y, Kitagawa J, Nakagawa K, Iwata K, Matsumoto S. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol Pain 2011;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ocana M, Cendan CM, Cobos EJ, Entrena JM, Baeyens JM. Potassium channels and pain: present realities and future opportunities. Eur J Pharmacol 2004;500:203–219 [DOI] [PubMed] [Google Scholar]
- 5. Shillito P, Molenaar PC, Vincent A, et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol 1995;38:714–722 [DOI] [PubMed] [Google Scholar]
- 6. Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997;41:238–246 [DOI] [PubMed] [Google Scholar]
- 7. Arimura K, Sonoda Y, Watanabe O, et al. Isaacs' syndrome as a potassium channelopathy of the nerve. Muscle Nerve 2002;(suppl 11):S55–S58 [DOI] [PubMed] [Google Scholar]
- 8. Josephs KA, Silber MH, Fealey RD, Nippoldt TB, Auger RG, Vernino S. Neurophysiologic studies in Morvan syndrome. J Clin Neurophysiol 2004;21:440–445 [DOI] [PubMed] [Google Scholar]
- 9. Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M, Vernino S. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology 2004;62:1177–1182 [DOI] [PubMed] [Google Scholar]
- 10. Liguori R, Vincent A, Clover L, et al. Morvan's syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain 2001;124:2417–2426 [DOI] [PubMed] [Google Scholar]
- 11. Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008;70:1883–1890 [DOI] [PubMed] [Google Scholar]
- 12. McKnight K, Jiang Y, Hart Y, et al. Serum antibodies in epilepsy and seizure-associated disorders. Neurology 2005;65:1730–1736 [DOI] [PubMed] [Google Scholar]
- 13. Geschwind MD, Tan KM, Lennon VA, et al. Voltage-gated potassium channel autoimmunity mimicking Creutzfeldt-Jakob disease. Arch Neurol 2008;65:1341–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia Brain 2010;133:2734–2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lancaster E, Huijbers MG, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Traka M, Goutebroze L, Denisenko N, et al. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol 2003;162:1161–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med 2009;15:1208–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. International Association for the Study of Pain Pain definitions: IASP.org: cited February 2nd 2012; derived from Bonica JJ. The need of a taxonomy. Pain 1979;6:247–248460931 [Google Scholar]
- 20. Dyck PJ, Sherman WR, Hallcher LM, et al. Human diabetic endoneurial sorbitol, fructose, and myo-inositol related to sural nerve morphometry. Ann Neurol 1980;8:590–596 [DOI] [PubMed] [Google Scholar]
- 21. Dyck PJ, O'Brien PC, Johnson DM, Klein CJ, Dyck PJ. Quantitative sensation testing. In: Dyck PJ. ed. Peripheral Neuropathy. Philadelphia: Elsevier; 2005: 1063–1093 [Google Scholar]
- 22. Low PA. Quantitation of autonomic function. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF. eds. Peripheral Neuropathy, 3rd ed. Philadelphia: W.B. Saunders; 1993: 729–745 [Google Scholar]
- 23. Fealey RD, Low PA, Thomas JE. Thermoregulatory sweating abnormalities in diabetes mellitus. Mayo Clin Proc 1989;64:617–628 [DOI] [PubMed] [Google Scholar]
- 24. Pittock SJ, Kryzer TJ, Lennon VA. Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann Neurol 2004;56:715–719 [DOI] [PubMed] [Google Scholar]
- 25. Collett B, O'Mahoney S, Schofield P, Closs SJ, Potter J. The assessment of pain in older people. Clin Med 2007;7:496–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kohr D, Singh P, Tschernatsch M, et al. Autoimmunity against the beta(2) adrenergic receptor and muscarinic-2 receptor in complex regional pain syndrome. Pain 2011;152:2690–2700 [DOI] [PubMed] [Google Scholar]
- 27. Arking DE, Cutler DJ, Brune CW, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet 2008;82:160–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bralten LB, Gravendeel AM, Kloosterhof NK, et al. The CASPR2 cell adhesion molecule functions as a tumor suppressor gene in glioma. Oncogene 2010;29:6138–6148 [DOI] [PubMed] [Google Scholar]
- 29. Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet 2002;30:335–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silva J, Sharma S, Hughes B, Yu YE, Cowell JK. Homozygous inactivation of the LGI1 gene results in hypomyelination in the peripheral and central nervous systems. J Neurosci Res 2010;88:3328–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain 2011;152:S2–S15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alonso-Navarro H, Fernandez-Diaz A, Martin-Prieto M, Ruiz-Ezquerro JJ, Lopez-Alburquerque T, Jimenez-Jimenez FJ. Tremor associated with chronic inflammatory demyelinating peripheral neuropathy: treatment with pregabalin. Clin Neuropharmacol 2008;31:241–244 [DOI] [PubMed] [Google Scholar]
- 33. Ishii A, Hayashi A, Ohkoshi N, et al. Clinical evaluation of plasma exchange and high dose intravenous immunoglobulin in a patient with Isaacs' syndrome. J Neurol Neurosurg Psychiatry 1994;57:840–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Newsom-Davis J, Mills KR. Immunological associations of acquired neuromyotonia (Isaacs' syndrome): report of five cases and literature review. Brain 1993;116:453–469 [DOI] [PubMed] [Google Scholar]
- 35. Sinha S, Newsom-Davis J, Mills K, Byrne N, Lang B, Vincent A. Autoimmune aetiology for acquired neuromyotonia (Isaacs' syndrome). Lancet 1991;338:75–77 [DOI] [PubMed] [Google Scholar]
- 36. Vincent A, Irani SR, Lang B. The growing recognition of immunotherapy-responsive seizure disorders with autoantibodies to specific neuronal proteins. Curr Opin Neurol 2010;23:144–150 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.