

Abstract
Diffuse gliomas are a heterogenous group of neoplasms traditionally classified as grades II to IV based on histologic features, and with prognosis determined mainly by histologic grade and pretreatment clinical factors. Our understanding of the molecular basis of glioma initiation, tumor progression, and treatment failure is rapidly evolving. A molecular profile of diffuse gliomas is emerging. Studies evaluating gene expression and DNA methylation profile have found multiple glioma subtypes and an association between subtype and survival. The recent discovery of isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) mutations in glioma has provided reproducible prognostic biomarkers and novel therapeutic targets. Glioblastomas that exhibit CpG island hypermethylator phenotype, proneural gene expression, or IDH1 mutation identify a subset of patients with markedly improved prognosis. Accumulated evidence supports the stratification of both low-grade and anaplastic diffuse gliomas into prognostic groups using 1p/19q codeletion and IDH mutation status. A classification scheme incorporating clinical, pathologic, and molecular information may facilitate improved prognostication for patients treated in the clinic, the development of more effective clinical trials, and rational testing of targeted therapeutics.
Diffuse gliomas comprise the second most common primary CNS neoplasms, behind meningiomas, and account for 80% of primary, malignant brain tumors.1 WHO classification of diffuse gliomas is based on a grading scheme from II to IV based on histomorphology, proliferation, and the presence of microvascular proliferation or necrosis. Diffuse gliomas are traditionally separated by histology into 3 categories: astrocytomas, including glioblastoma (GBs), oligodendrogliomas, and a poorly reproducible group termed mixed oligoastrocytomas.2
GBs comprise 53.9% of all gliomas and are the most common primary CNS malignancy in adults.1 GBs are differentiated histologically from other diffuse astrocytomas by the presence of microvascular proliferation or necrosis. GBs can be partitioned into primary GB, which arise de novo, and secondary GB, which arise by progression from grade II or III astrocytomas. Primary GBs typically occur in patients over 50 years of age and are characterized by overexpression or mutation of EGFR, loss of heterozygosity (LOH) of chromosome 10q, and PTEN mutations. Secondary GBs usually occur in younger patients and are characterized by TP53 and isocitrate dehydrogenase 1 (IDH1) mutations, overexpression of platelet-derived growth factor receptor (PDGFR), and abnormalities of the retinoblastoma (Rb) pathway.3
Diffuse WHO grade II (low-grade glioma [LGG]) and WHO grade III (anaplastic glioma [AG]) include low-grade and anaplastic astrocytoma (LGA and AA), low-grade and anaplastic oligodendroglioma (LGO and AO), and low-grade and anaplastic mixed oligoastrocytoma (LGOA and AOA). Diffuse LGGs and AGs in aggregate comprise approximately 15% of gliomas.1 Accumulated evidence strongly suggests that astrocytomas and oligodendrogliomas are separate clinical and molecular entities with different prognoses and treatment responses.4–7
The WHO criteria for classification and grading of diffuse gliomas have limitations. In preceding decades, the principal determinants of prognosis were clinical, pathologic, and pretreatment factors. Using recursive partitioning analysis (RPA), tumor grade combined with age, performance score, and extent of surgical resection assigns diffuse glioma patients into RPA classes associated with survival.8,9 Although negative correlation exists between increasing WHO grade, RPA class, and survival, significant heterogeneity in clinical behavior among tumors with the same grade and clinical features is observed.
Accumulating evidence suggests that IDH mutation status and gene expression profiling provide prognostic information that extends beyond that provided by WHO classification and other prognostic biomarkers such as 1p/19q chromosomal codeletion and methylation of the promoter region of the methylguanine methyltransferase (MGMT) gene. It is becoming increasingly evident that diffuse gliomas can be meaningfully separated into prognostic groups based on molecular profiling. In this review, the clinical, diagnostic, and therapeutic implications of an emerging molecular classification of diffuse gliomas are provided.
1p/19q CODELETION
The pathologic criteria for classifying diffuse gliomas as “pure” astrocytoma oligodendroglioma or mixed oligoastrocytoma are not universally agreed upon, and are prone to significant subjectivity and interobserver variability.10 This can lead to poor reproducibility, diagnostic uncertainty, and perhaps explain the variability of treatment patterns observed in day-to-day clinical practice.11 Thus, there is an opportunity to integrate molecular classifiers to better delineate tumor subtypes with more uniform outcomes.
Unbalanced translocation of chromosomes 1 and 19 with deletion of 1p and 19q (1p/19q codeletion) is present in 70% or more of oligodendroglial tumors. Using strict histologic criteria for classifying oligodendroglial and astrocytic tumors, the proportion of LGOs with 1p/19q codeletion may be over 90%.7 Historically, oligoastrocytomas have rates of TP53 mutation, 1p/19q codeletion, and survival outcomes intermediate between astrocytomas and oligodendrogliomas.12 However, due to the difficulty in reproducibly diagnosing oligoastrocytoma, 1p/19q codeletion is often considered to be the objective molecular definition of oligodendroglial lineage, with tumors that lack 1p/19q codeletion considered astrocytic. This approach is strengthened by the observation that TP53 mutation, a marker of astrocytic lineage, and 1p/19q codeletion are mutually exclusive in the vast majority of cases.13 GB with oligodendroglial features (GBO) is a WHO-recognized GB variant2; however, this entity remains controversial, and is poorly reproducible, similar to mixed oligoastrocytomas.14,15
1p/19q codeletion is associated with improved prognosis in LGGs and AGs regardless of treatment modality and is a reproducible prognostic biomarker.6,16–19 In a retrospective study, a trend toward improved survival outcomes in AOs with 1p/19q codeletion treated with PCV (procarbazine, CCNU, vincristine) compared to temozolomide (TMZ, Temodar, Merck & Co., NJ) was reported.4 Long-term follow-up data from the European Organisation for Research and Treatment of Cancer (EORTC)5 and Radiation Therapy Oncology Group (RTOG)20 trials testing radiotherapy vs radiotherapy plus adjuvant or neoadjuvant PCV in AOs were recently presented and the results suggest that 1p/19q codeletion is both prognostic and predictive of improved outcomes with PCV chemotherapy.21,22 Given the range of survival outcomes and challenge of reproducibly classifying astrocytomas, mixed oligoastrocytomas, and oligodendrogliomas, 1p/19q codeletion has become an important biomarker in the day-to-day management of LGGs and AGs.
MGMT PROMOTER METHYLATION
O6-methylguanine-DNA methyltransferase (MGMT) is a DNA repair enzyme that repairs O6 alkyl guanine adducts. The 5′ promoter region of MGMT contains a CpG island, and methylation of CpG islands in the MGMT promoter region results in epigenetic silencing of gene transcription. The DNA repair mechanism of MGMT and the cytotoxic effects of TMZ overlap as TMZ alkylates the O6 position on guanine.23
In a retrospective study of the EORTC-NCIC trial of concurrent chemoradiotherapy plus adjuvant TMZ vs radiotherapy, 45% of patients with available tissue had MGMT promoter methylation. These patients had significantly prolonged OS compared to patients with unmethylated MGMT promoter regions (18.2 vs 12.2 months). MGMT promoter methylation was associated with improved survival outcomes regardless of treatment arm. In the TMZ treatment arm, 46% of patients with a methylated MGMT promoter survived 2 years, vs 13% of unmethylated patients. In the radiotherapy arm, 22% of patients with a methylated MGMT promoter survived 2 years, vs less than 2% of patients with unmethylated tumors.24 A subsequent retrospective study found 30% of patients with GB with MGMT promoter methylation treated with upfront radiotherapy alone survived 2 years, vs only 16% for patients with unmethylated tumors treated similarly.25 In EORTC 26951 (radiotherapy vs radiotherapy plus procarbazine, CCNU, and vincristine chemotherapy), MGMT promoter methylation was associated with improved outcomes in patients with AO treated with radiotherapy alone or radiotherapy plus chemotherapy.26
MGMT promoter methylation was a stratification factor in RTOG 0525 and 0825; both trials tested standard upfront chemoradiotherapy plus adjuvant TMZ vs dose-intense TMZ or TMZ plus bevacizumab (Avastin, San Francisco, CA). In RTOG 0525, MGMT promoter methylation was prospectively confirmed as prognostic in patients treated with standard or dose-intense TMZ. As TMZ is an MGMT substrate, it was theorized that dose-intense regimens could overwhelm the DNA repair capacity of MGMT. However, MGMT promoter methylation was not associated with improved outcomes in the dose-intense TMZ treatment arm in patients with or without MGMT promoter methylation.27
In a nonrandomized, phase II trial of patients with newly diagnosed GB 70 years and older with Karnofsky performance scores 70 or less treated with standard dosing of TMZ without radiotherapy, MGMT promoter methylation was associated with significantly prolonged progression-free and overall survival.28 A phase III trial, NOA-08, randomized patients with GB over age 65 to either TMZ or radiotherapy alone after surgical resection. MGMT promoter methylation was associated with improved survival outcomes in the TMZ-treated patients and patients with unmethylated tumors had improved outcomes when treated with radiotherapy.29 These results suggest MGMT promoter methylation may be a predictive biomarker in certain patient subsets.
The role of MGMT promoter methylation as a predictive biomarker of temozolomide sensitivity is controversial. The accumulated evidence clearly supports MGMT promoter methylation as a prognostic marker of improved survival outcomes. MGMT promoter methylation is frequently associated with other prognostic biomarkers: 1p19q codeletion, IDH mutations, gene expression, and DNA methylation signatures. Promoter methylation may be an epiphenomenon related to these or other factors ultimately mediating improved survival outcomes.
ISOCITRATE DEHYDROGENASE MUTATIONS
Using genome-wide sequencing, IDH1 mutations were found in 18/149 (12%) of GB samples. These patients were noted to have prolonged overall survival compared to patients with wild-type IDH1.30 A subsequent analysis of 445 glioma tissue samples (grades I to IV), medulloblastomas, and 494 non-CNS tumor samples found IDH1 or IDH2 mutations in over 80% of grade II and III oligodendrogliomas and astrocytomas, IDH1 mutations in 80% of secondary GBs, and no IDH1 or IDH2 mutations in non-CNS solid tumors.31 IDH mutations, especially IDH1R132H, appear to be unique to gliomas. IDH1 or IDH2 mutations have also been discovered in a subset of cytogenetically normal acute myelogenous leukemia (AML), intrahepatic cholangiocarcinoma, and central cartilaginous neoplasms.32–34 The discovery of IDH mutations is novel as this metabolic pathway was not previously implicated in oncogenesis.35 IDH catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate, generating NADPH. Glial cells have high baseline levels of α-ketoglutarate due to uptake of glutamate and glutamine, which are ultimately metabolized to α-ketoglutarate.35,36
Whether mutant IDH results in a loss of tumor suppressor function or acts as an oncogene is a source of intense research. The exact mechanisms associated with tumorigenesis and improved prognosis are yet to be defined. Mutant IDH enzyme produces decreased cytoplasmic levels of α-ketoglutarate and NADPH. Resulting decreased cytosolic α-ketoglutarate may stabilize hypoxia inducible factor-1α (HIF-1α) facilitating cellular proliferation.35,36
Alternatively, mutant IDH1 may be oncogenic by a gain of neomorphic enzymatic activity and the ability to convert α-ketoglutarate to D-2-Hydroxyglutarate (2HG).35 2HG may act as an oncometabolite by antagonizing α-ketoglutarate-dependent enzymes such as ten-eleven-translocation 2 (TET2), which is responsible for histone and DNA methylation.37 IDH1 mutation appears to be a necessary molecular event to establish the glioma hypermethylation phenotype, discussed subsequently.38 2HG is found in micromolar cytoplasmic concentrations of mutated cells and provides a tumor-specific biomarker. Noninvasive means of diagnosis and monitoring treatment response may be possible as 2HG is detectable on magnetic resonance spectroscopy.39,40
Primary GBs make up the majority of grade IV tumors while secondary GBs account for 10% or less.3 IDH1 mutations are found in 73%–88% of secondary GBs, but only 3%–7% of primary GBs.3,41 GBs with IDH1 mutations are phenotypically and genotypically distinct.42 Over 60% of IDH1 mutant GBs are localized in the frontal lobe and the peak incidence occurs in the third decade of life. IDH1 mutant GBs share radiographic features with grade II and III gliomas. IDH1 mutant GBs are often nonenhancing, associated with a lesser extent of edema, and often have cystic or diffuse components more often than IDH1 wild-type GBs (figure 1). IDH1 mutant GBs also have a higher frequency of MGMT promoter methylation and p53 mutation. Overall survival ranges from 24 to 36 months in IDH1 mutant vs 9 to 15 months in IDH1 wild-type GBs.42–45 MGMT promoter methylation is 3 to 4 times more common than IDH1 mutation in GBs, and the relative contribution of each to prognosis has not been compared prospectively.
Figure 1. Representative MRI of an IDH1 mutant glioblastoma.

Fluid-attenuated inversion recovery sequence (A) of an IDH1 (R132H) mutant glioblastoma (mutation positive on immunohistochemistry and sequencing of IDH1 gene) which is located in the medial frontal lobe and does not enhance with gadolinium (B).
IDH1 or IDH2 mutations occur in 70% or more of LGGs and AGs.31 The majority of LGGs with TP53 mutations or 1p/19q codeletion have IDH mutations.46 IDH2 mutations are rare and occur mainly in oligodendrogliomas.31 A total of 90% to 100% of 1p/19q codeleted gliomas also harbor IDH1 or 2 mutations.46,47 IDH mutations are strongly associated with 1p19q codeletion and MGMT promoter methylation in AOs.19 IDH mutations have a robust statistical effect on survival outcome in LGGs and AGs.18,45,48 When controlling for factors such as age, extent of resection, MGMT promoter methylation, and 1p/19q codeletion, only IDH1 mutation was significantly associated with OS in a retrospective study of LGGs from the German Glioma Network.18
IDH wild-type LGGs and AGs have inferior survival outcomes compared to IDH-mutated tumors. Although comprising only a minority of LGGs, IDH wild-type tumors have a significantly worse prognosis compared to IDH-mutated LGGs, with a median PFS of only 1.4 years (vs 4.7 years), and with only 14% of patients surviving 5 years (vs 42%) in 1 study; a second study showed an OS of 150 vs 60 months for IDH1-mutated vs wild-type tumors.18,49 IDH wild-type LGGs are often “triple-negative,” lacking 1p/19q codeletion, TP53 mutation, and IDH mutation.49 In 1 study of 382 patients with AA or GB, IDH1 mutation was the most prominent prognostic factor, followed by age, histology (AA or GB), and MGMT promoter methylation status, suggesting that IDH1 mutation may be a more significant prognostic factor than histologic classification as either AA or GB.44
MULTIGENE SETS
The explosion of genomic datasets made available by high-throughput microarray technologies have revolutionized understanding of different types of cancer. Global genomic profiling is a promising approach for developing molecular subtype classifications, multigene clinical predictors, new target identification, and predictive markers for targeted therapeutics. The most well-known project, the Cancer Genome Atlas (TCGA), encompasses DNA, mRNA, microRNA, and epigenetic profiling (DNA methylation).50 Recent gene expression profiling has revealed multiple glioma subtypes with different clinical outcomes.
The first study using multigene sets to define molecular subtypes of high grade gliomas discovered 3 subtypes: proneural, mesenchymal, and proliferative.51 A similar analysis of the TCGA dataset confirmed the proneural and mesenchymal subtypes, and also identified neural and classic subtypes.52 The proneural subtype is associated with PDGFR amplification, IDH1 mutations, and overexpression of genes related to neural and glial development. The mesenchymal subtype is associated with increased expression of angiogenic peptides, neurofibromatosis 1 gene (NF1) loss or mutation, and overexpression of genes related to motility, the extracellular matrix, and cell adhesion. The TCGA neural and classic subtypes are associated with epidermal growth factor receptor (EGFR) mutation or amplification, and the classic subtype is associated with PTEN loss. Tumor grade is associated with the proneural and mesenchymal subtypes. Grade II and III diffuse gliomas are predominantly proneural while GBs represent a mix of all subtypes. At recurrence, some proneural tumors undergo a proneural to mesenchymal gene expression transformation.51
The proneural and mesenchymal subtypes are the best characterized across studies and are differentiated by clinical outcomes. Proneural GBs have improved survival outcomes compared to GBs with mesenchymal signatures. The other subtypes (e.g., neural, classic) are not clearly associated with different survival outcomes. However, the importance of differentiating among these subtypes may lie in the enrichment of characteristic gene mutations or amplified signaling pathways. For example, the enrichment of NF1 mutations in mesenchymal GBs and EGFR mutations in classic GBs may be important for designing personalized clinical trials in molecularly selected patient populations.51,52
GBs with a mesenchymal genetic signature overexpress genes associated with a glioma stem cell (GSC) phenotype.53 GSCs are intrinsically resistant to radio- and cytotoxic chemotherapy; a stem-cell like phenotype may account for a proportion of the patients with poor response to the current standard of care in newly diagnosed GB.54 Targeting pathways that regulate mesenchymal and GSC phenotypes, such as STAT3, TGF-β, and CEBP-β/δ,55,56 may be more effective strategies in mesenchymal GBs.
Changes in DNA methylation are a hallmark of some cancers with global hypomethylation alternating with hypermethylation of CpG islands located in gene promoter regions. A study from the TCGA evaluated CpG island methylation patterns in gliomas. A distinct CpG island hypermethylator phenotype (G-CIMP) was found (figure 1). G-CIMP tumors are seen in younger patients and make up the majority of WHO grade II and III gliomas. These patients experience significantly improved outcomes compared to non-G-CIMP tumors.57
G-CIMP GBs have improved survival outcomes.42,57 IDH1 mutant and proneural GBs appear to segregate almost exclusively into the G-CIMP phenotype (table 1, figure 2). G-CIMP status further separates clinical outcomes in proneural GBs; G-CIMP, proneural GBs have improved survival outcomes compared to non-G-CIMP, proneural GBs, which have similar survival outcomes to other GB subtypes.57 G-CIMP, proneural GBs with IDH1 mutations appear to maintain their G-CIMP status at recurrence and do not undergo proneural to mesenchymal transformation.42,57 A few G-CIMP GBs have nonproneural genetic signatures or are IDH1 wild-type; these rare exceptions are challenging with regard to both classification and prognostication. Given the clustering of positive prognostic biomarkers in G-CIMP GBs (table 1, figure 2), G-CIMP status may be the most robust prognostic biomarker in GB; this assertion requires prospective evaluation.
Table 1.
Gene expression profile, characteristic mutations, and survival of glioblastoma subtypesa

Abbreviations: DNA meth = DNA methylation profile; EGFR mut = epidermal growth factor receptor gene mutation; G-CIMP = CpG hypermethylator phenotype; GB = glioblastoma; gene exp = genetic expression profile; IDH1 mut = isocitrate dehydrogenase 1 gene mutation; NF-1 mut = neurofibromatosis type 1 gene mutation; OS = overall survival.
Glioblastoma subtypes separated into 3 groups based on DNA methylation and gene expression profile. Characteristic gene mutations enriched within each group are annotated. Group 1 has markedly improved survival outcomes compared to groups 2 and 3.
A small percentage of G-CIMP (+) GBs have non-proneural genetic signatures or are IDH1 wild-type.
Non-mesenchymal, G-CIMP (−) GBs including proneural, neural, and classical genetic subtypes have similar outcomes to mesenchymal GBs.
Figure 2. Identification of the CpG island methylator phenotype in glioblastoma.

DNA methylation from 91 TCGA glioblastomas was profiled and subjected to unsupervised clustering (statistical methodology, reference 57). Three distinct methylation clusters were identified, with cluster 1 (red top bar) designated as G-CIMP+ due to a high frequency of DNA methylation. The gene expression profile and mutation status of selected genes (EGFR, IDH1, NF1, PTEN, and p53) are shown in the bars below each methylation cluster. IDH1 mutations were found exclusively in G-CIMP+ tumors, and nearly all G-CIMP+ tumors had a proneural gene expression profile. Adapted from Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010;17:510–522; with permission from Elsevier. EGFR = epidermal growth factor receptor; G-CIMP = CpG hypermethylator phenotype; GBM = glioblastoma multiforme; IDH1 = isocitrate dehydrogenase 1 gene; NF1 = neurofibromatosis 1 gene; PTEN = phosphatase and tensin homologue gene; TCGA = The Cancer Genome Atlas; TP53 = p53 gene.
Similar to GBs, DNA methylation and genetic signature can identify LGGs and AGs with improved prognoses. In 2 retrospective studies, proneural AOs and LGOs had improved outcomes compared to oligodendrogliomas with nonproneural gene expression.58,59 The accumulated evidence suggests that oligodendrogliomas with 1p/19q codeletion and IDH mutations typically also have MGMT promoter methylation, a proneural genetic signature, and a CpG island hypermethylator phenotype.47,60 A retrospective analysis of EORTC 26951 (radiotherapy plus or minus adjuvant chemotherapy in AOs) found G-CIMP status to be the most robust prognostic factor when considering clinical features, 1p/19q codeletion, IDH mutation status, and MGMT promoter methylation status.60
Important practical considerations limiting widespread application of gene expression profiling include feasibility outside of a research setting and cost effectiveness. A 38-gene profile consistently associated with patient outcome was further refined to a 9-gene set prognostic of outcome in GB after controlling for clinical factors and MGMT promoter methylation.53 The 9-gene profile can be performed on formalin-fixed paraffin-embedded tissue and conceptually represents a multigene tool with broad applicability. An important question to answer prospectively is the relative contributions of G-CIMP status, genetic subtype, IDH1 mutational status, and MGMT promoter methylation to prognosis.
MOLECULAR CLASSIFICATION OF DIFFUSE GLIOMAS
The accumulated data support molecular stratification of LGGs and AGs using first 1p19q codeletion and then IDH mutation status in 1p/19q intact patients. This separates both LGGs and AGs into 3 groups with different prognoses (table 2). Molecular stratification in prospective trials using 1p/19q codeletion status, IDH mutation status, and possibly other molecular biomarkers (G-CIMP status, genetic subtype) would be the ideal means of evaluating a classification scheme including these biomarkers.
Table 2.
Molecular classification and survival of low-grade and anaplastic gliomasa
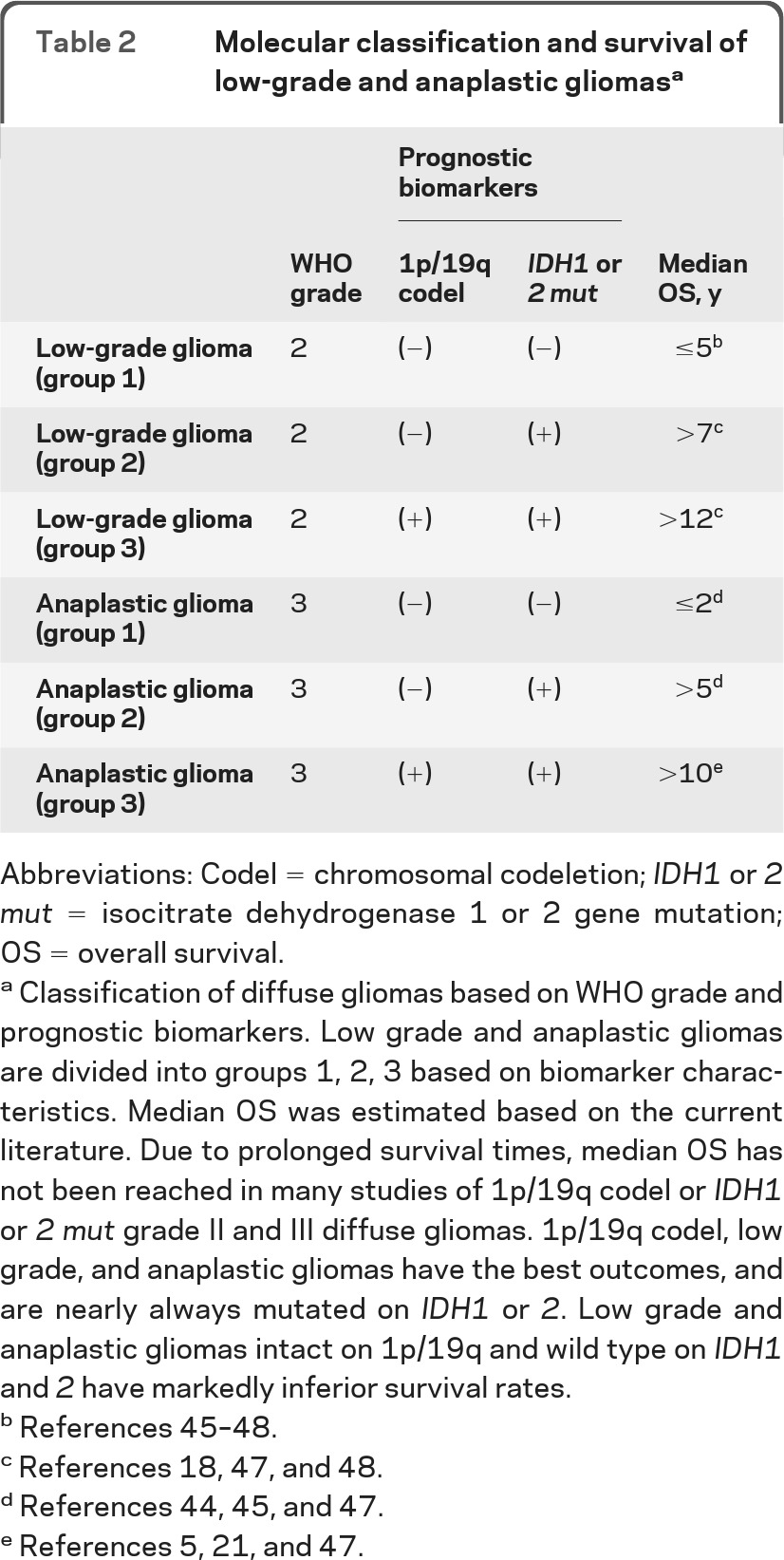
Abbreviations: Codel = chromosomal codeletion; IDH1 or 2 mut = isocitrate dehydrogenase 1 or 2 gene mutation; OS = overall survival.
Classification of diffuse gliomas based on WHO grade and prognostic biomarkers. Low grade and anaplastic gliomas are divided into groups 1, 2, 3 based on biomarker characteristics. Median OS was estimated based on the current literature. Due to prolonged survival times, median OS has not been reached in many studies of 1p/19q codel or IDH1 or 2 mut grade II and III diffuse gliomas. 1p/19q codel, low grade, and anaplastic gliomas have the best outcomes, and are nearly always mutated on IDH1 or 2. Low grade and anaplastic gliomas intact on 1p/19q and wild type on IDH1 and 2 have markedly inferior survival rates.
Molecular classification of LGGs and AGs has become standard at our institution; diffuse glioma cases are reported by our neuropathologists using this general format:
DIFFUSE GLIOMA
WHO GRADE II
IDH: IDH1 R132H MUTANT PROTEIN-POSITIVE
1p/19q status: CO-DELETED
Other pathologic features, including morphologic characteristics such as oligodendroglial or astrocytic phenotype, the results of other relevant immunohistochemical (IHC) stains or diagnostic molecular tests (e.g., p53, EGFR amplification, proliferation indices), and the method of molecular feature determination (e.g., FISH, IHC, CGH) are included in a Comments section. Receiving this data routinely from our neuropathologists informs both clinical decision-making and patient counseling. This format also allows for continued evolution of molecular classification as new robust markers are verified.
Results from ongoing phase II and III clinical trials in 1p/19q codeleted and 1p/19q intact LGGs and AGs are anxiously awaited to determine the best therapeutic approach for astrocytic vs oligodendroglial tumors with or without 1p/19q codeletion. Despite inferior outcomes in 1p/19q intact, IDH wild-type tumors (table 2), appropriate treatment has not been established. Combined treatment regimens, including radiotherapy, concurrent chemoradiotherapy, or adjuvant chemotherapy, have not been proven to improve survival or have acceptable rates of toxicity.
Figure 3 (see also table 1) demonstrates the relative frequency, overlap, and association with survival outcomes between gene expression profile, DNA methylation signature, IDH1 mutation, and MGMT promoter methylation in GB. Incorporating these biomarkers into clinical trials may prevent over- or underestimating benefit of a particular agent by balancing GBs with different molecular profiles in different treatment arms. This paradigm may also allow determination of benefit in specific subgroups of patients with a particular molecular profile. Extrapolation from the genes and pathways differentially activated will facilitate rational clinical trial design of targeted therapeutics in molecularly defined patient subgroups. In an application of this molecular-based paradigm, the 9-gene profile53 was integrated into the stratification of the ongoing phase III clinical trial RTOG 0825 (TMZ vs TMZ plus bevacizumab) in newly diagnosed GB.
Figure 3. Frequency, overlap, and relative survival glioblastomas (GBs) (includes GB and all GB variants including gliosarcoma) based on molecular profile.

Relative frequency of G-CIMP status, gene expression profiles, IDH1 mutation, and MGMT promoter methylation in GBs. IDH1 mutation and G-CIMP are depicted as discrete categories while gene expression and MGMT methylation status are depicted as a continuum. GBs with PN gene expression profiles and IDH1 mutations cluster almost exclusively within G-CIMP GBs and have improved clinical outcomes (left side of figure, see also table 1). Mesenchymal GBs are exclusively non-G-CIMP and IDH1 wild-type and have inferior clinical outcomes (see also table 1). G-CIMP = CpG hypermethylator phenotype; IDH1 mut = isocitrate dehydrogenase 1 gene mutation; IDH1 wt = isocitrate dehydrogenase 1 wild-type gene.
A minority, approximately 10%, of GBs have a favorable biomarker profile, including proneural gene expression, G-CIMP phenotype, IDH1 mutation, and MGMT promoter methylation (table 1). The majority of GBs, however, are non-G-CIMP, nonproneural, IDH1 wild-type, and are not separated by clinical outcomes (table 1). There is no alternative treatment for patients with a favorable or unfavorable molecular profile. The current standard of care should be routinely administered to all patients with GB unless within the confines of a clinical trial. At present, the technology to profile gene expression and DNA methylation is expensive and not widely available. Further development and prospective validation of molecular profiles capable of classifying GB with relevance to both prognosis and treatment is necessary before widespread application is justified.
DISCUSSION
Molecular profiling, including gene expression, DNA methylation, key mutations, and cytogenetic events can separate diffuse gliomas into prognostic groups. Incorporation of this information when classifying diffuse gliomas will better account for the clinical, pathologic, and molecular heterogeneity observed among all tumor grades. Targeting patients with poor prognosis or specific molecular profiles will become increasingly feasible if such a classification can be standardized. Significant challenges remain. Further stratification by molecular means (e.g., separating LGGs into 3 groups as in table 2) will potentially shrink the number of patients available for clinical trials. Prospective studies with large groups of patients requiring multi-institution collaboration will be necessary to effectively validate prognostic biomarkers before more widespread inclusion into a classification scheme is warranted. Collaboration will be even more important to design the next generation of clinical trials in select groups of patients with similar molecular profiles.
GLOSSARY
- AA
anaplastic astrocytoma
- AG
anaplastic glioma
- AML
acute myelogenous leukemia
- AO
anaplastic oligodendroglioma
- AOA
anaplastic mixed oligoastrocytoma
- EGFR
epidermal growth factor receptor
- EORTC
European Organisation for Research and Treatment of Cancer
- G-CIMP
CpG island hypermethylator phenotype
- GB
glioblastoma
- GBO
glioblastoma with oligodendroglial features
- GSC
glioma stem cell
- IHC
immunohistochemical
- LGA
low-grade astrocytoma
- LGG
low-grade glioma
- LGO
low-grade oligodendroglioma
- LGOA
low-grade mixed oligoastrocytoma
- LOH
loss of heterozygosity
- PDGFR
platelet-derived growth factor receptor
- Rb
retinoblastoma
- RPA
recursive partitioning analysis
- RTOG
Radiation Therapy Oncology Group
- TCGA
the Cancer Genome Atlas
Footnotes
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
AUTHOR CONTRIBUTIONS
Brett J. Theeler wrote and revised the manuscript. Gregory N. Fuller revised the manuscript. W.K. Alfred Yung revised the manuscript. John F. De Groot wrote and revised the manuscript.
DISCLOSURE
B.J. Theeler reports no disclosures. W.K.A. Yung reports grant/research support from Novartis and consultant/Data Safety Monitoring Board participation for Novartis, Merck, and Eden. G.N. Fuller reports no disclosures. J.F. De Groot reports grant/research support from AstraZeneca, Adnexus, Sanofi-Aventis, advisory board participation for Genentech, and consultant/Data Safety Monitoring Board participation for VBL Pharmaceuticals. Go to Neurology.org for full disclosures.
REFERENCES
- 1. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2004–2008. In: Central Brain Tumor Registry of the United States. Hinsdale, IL: CBTRUS; 2012. [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD. The 2007 WHO Classification of Tumors of the Central Nervous System. Lyon, France: IARC Press; 2007. [Google Scholar]
- 3.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res 2004; 64: 6892– 6899 [DOI] [PubMed] [Google Scholar]
- 4.Lassman AB, Iwamoto FM, Cloughesy TF, et al. International retrospective study of over 1000 adults with anaplastic oligodendroglial tumors. Neuro-Oncol 2011; 13: 649– 659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 2006; 24: 2715– 2722 [DOI] [PubMed] [Google Scholar]
- 6.Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 1998; 90: 1473– 1479 [DOI] [PubMed] [Google Scholar]
- 7.Sanai N, Chang S, Berger MS. Low-grade gliomas in adults. J Neurosurg 2011; 115: 948– 965 [DOI] [PubMed] [Google Scholar]
- 8.Curran WJ, Jr, Scott CB, Horton J, et al. Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. J Natl Cancer Inst 1993; 85: 704– 710 [DOI] [PubMed] [Google Scholar]
- 9.Pignatti F, van den Bent M, Curran D, et al. Prognostic factors for survival in adult patients with cerebral low-grade glioma. J Clin Oncol 2002; 20: 2076– 2084 [DOI] [PubMed] [Google Scholar]
- 10.Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol 2003; 62: 1118– 1128 [DOI] [PubMed] [Google Scholar]
- 11.Abrey LE, Louis DN, Paleologos N, et al. Survey of treatment recommendations for anaplastic oligodendroglioma. Neuro-Oncol 2007; 9: 314– 318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okamoto Y, Di Patre PL, Burkhard C, et al. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol 2004; 108: 49– 56 [DOI] [PubMed] [Google Scholar]
- 13.Molinari C, Iorio P, Medri L, et al. Chromosome 1p and 19q evaluation in low-grade oligodendrogliomas: a descriptive study. Int J Mol Med 2010; 25: 145– 151 [PubMed] [Google Scholar]
- 14.Klink B, Schlingelhof B, Klink M, Stout-Weider K, Patt S, Schrock E. Glioblastomas with oligodendroglial component - common origin of the different histological parts and genetic subclassification. Anal Cell Pathol 2010; 33: 37– 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinto LW, Araujo MB, Vettore AL, et al. Glioblastomas: correlation between oligodendroglial components, genetic abnormalities, and prognosis. Virchows Arch 2008; 452: 481– 490 [DOI] [PubMed] [Google Scholar]
- 16.Pouratian N, Gasco J, Sherman JH, Shaffrey ME, Schiff D. Toxicity and efficacy of protracted low dose temozolomide for the treatment of low grade gliomas. J Neurooncol 2007; 82: 281– 288 [DOI] [PubMed] [Google Scholar]
- 17.Kaloshi G, Benouaich-Amiel A, Diakite F, et al. Temozolomide for low-grade gliomas: predictive impact of 1p/19q loss on response and outcome. Neurology 2007; 68: 1831– 1836 [DOI] [PubMed] [Google Scholar]
- 18.Hartmann C, Hentschel B, Tatagiba M, et al. Molecular markers in low-grade gliomas: predictive or prognostic? Clin Cancer Res 2011; 17: 4588– 4599 [DOI] [PubMed] [Google Scholar]
- 19.Wick W, Hartmann C, Engel C, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol 2009; 27: 5874– 5880 [DOI] [PubMed] [Google Scholar]
- 20.Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 2006; 24: 2707– 2714 [DOI] [PubMed] [Google Scholar]
- 21.Cairncross G, Wang M, Shaw E, et al. Chemotherapy plus radiotherapy (CT-RT) versus RT alone for patients with anaplastic oligodendroglioma: long-term results of the RTOG 9402 phase III study. J Clin Oncol 2012; (suppl 30): abstract 2008b [Google Scholar]
- 22.Van Den Bent M, Hoang-Xuan K, Brandes A, et al. Long-term follow-up results of EORTC 26951: a randomized phase III study on adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors (AOD). J Clin Oncol 2012; (suppl 30): abstract 2 [Google Scholar]
- 23.Weller M, Stupp R, Reifenberger G, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol 2010; 6: 39– 51 [DOI] [PubMed] [Google Scholar]
- 24.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352: 997– 1003 [DOI] [PubMed] [Google Scholar]
- 25.Rivera AL, Pelloski CE, Gilbert MR, et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro-Oncol 2010; 12: 116– 121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Bent MJ, Dubbink HJ, Sanson M, et al. MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: a report from EORTC Brain Tumor Group Study 26951. J Clin Oncol 2009; 27: 5881– 5886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilbert M, Wang M, Aldape K, al. E. RTOG 0525: a randomized phase III trial comparing standard adjuvant temozolomide (TMZ) with a dose-dense (dd) schedule in newly diagnosed glioblastoma (GBM). J Clin Oncol 2011; (suppl 29): abstract 2006 [Google Scholar]
- 28.Gallego Perez-Larraya J, Ducray F, Chinot O, et al. Temozolomide in elderly patients with newly diagnosed glioblastoma and poor performance status: an ANOCEF phase II trial. J Clin Oncol 2011; 29: 3050– 3055 [DOI] [PubMed] [Google Scholar]
- 29.Wick W, Platten M, Meisner C, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol Epub 2012 May 10 [DOI] [PubMed] [Google Scholar]
- 30.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807– 1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009; 360: 765– 773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marcucci G, Maharry K, Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2010; 28: 2348– 2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012; 17: 72– 79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011; 224: 334– 343 [DOI] [PubMed] [Google Scholar]
- 35.Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med 2009; 360: 813– 815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ. Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? Lancet Oncol 2011; 12: 83– 91 [DOI] [PubMed] [Google Scholar]
- 37.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011; 19: 17– 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012; 483: 479– 483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elkhaled A, Jalbert LE, Phillips JJ, et al. Magnetic resonance of 2-hydroxyglutarate in IDH1-mutated low-grade gliomas. Sci Transl Med 2012; 4: 116ra115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi C, Ganji SK, Deberardinis RJ, et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med 2012; 18: 624– 629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 2009; 15: 6002– 6007 [DOI] [PubMed] [Google Scholar]
- 42.Lai A, Kharbanda S, Pope WB, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol 2011; 29: 4482– 4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weller M, Felsberg J, Hartmann C, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 2009; 27: 5743– 5750 [DOI] [PubMed] [Google Scholar]
- 44.Hartmann C, Hentschel B, Wick W, et al. Patients with IDH1 wild type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: implications for classification of gliomas. Acta Neuropathol 2010; 120: 707– 718 [DOI] [PubMed] [Google Scholar]
- 45.Sanson M, Marie Y, Paris S, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009; 27: 4150– 4154 [DOI] [PubMed] [Google Scholar]
- 46.Kim YH, Nobusawa S, Mittelbronn M, et al. Molecular classification of low-grade diffuse gliomas. Am J Pathol 2010; 177: 2708– 2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Labussiere M, Idbaih A, Wang XW, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology 2010; 74: 1886– 1890 [DOI] [PubMed] [Google Scholar]
- 48.Dubbink HJ, Taal W, van Marion R, et al. IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology 2009; 73: 1792– 1795 [DOI] [PubMed] [Google Scholar]
- 49.Metellus P, Coulibaly B, Colin C, et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol 2010; 120: 719– 729 [DOI] [PubMed] [Google Scholar]
- 50. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061– 1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9: 157– 173 [DOI] [PubMed] [Google Scholar]
- 52.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17: 98– 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Colman H, Zhang L, Sulman EP, et al. A multigene predictor of outcome in glioblastoma. Neuro-Oncol 2010; 12: 49– 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murat A, Migliavacca E, Gorlia T, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 2008; 26: 3015– 3024 [DOI] [PubMed] [Google Scholar]
- 55.Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010; 463: 318– 325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anido J, Saez-Borderias A, Gonzalez-Junca A, et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 2010; 18: 655– 668 [DOI] [PubMed] [Google Scholar]
- 57.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17: 510– 522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ducray F, Idbaih A, de Reynies A, et al. Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer 2008; 7: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooper LA, Gutman DA, Long Q, et al. The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PLoS One 2010; 5: e12548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van den Bent MJ, Gravendeel LA, Gorlia T, et al. A hypermethylated phenotype is a better predictor of survival than MGMT methylation in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. Clin Cancer Res 2011; 17: 7148– 7155 [DOI] [PubMed] [Google Scholar]