

Abstract
In contrast to cholera toxin (CT), which is secreted solubly by Vibrio cholerae across the outer membrane, heat-labile enterotoxin (LT) is retained on the surface of enterotoxigenic Escherichia coli (ETEC) via an interaction with lipopolysaccharide (LPS). We examined the nature of the association between LT and LPS. Soluble LT binds to the surface of LPS deep-rough biosynthesis mutants but not to lipid A, indicating that only the Kdo (3-deoxy-D-manno-octulosonic acid) core is required for binding. Although capable of binding truncated LPS and Kdo, LT has a higher affinity for longer, more complete LPS species. A putative LPS binding pocket is proposed based on the crystal structure of the toxin. The ability to bind LPS and remain associated with the bacterial surface is not unique to LT, as CT also binds to E. coli LPS. However, neither LT nor CT is capable of binding to the surface of Vibrio. The core structures of Vibrio and E. coli LPS differ in that Vibrio contains a phosphorylated single Kdo-lipid A, and E. coli LPS contains unphosphorylated Kdo2-lipid A. We determined that the phosphate group on the Kdo core of Vibrio LPS prevents CT from binding, resulting in the secretion of soluble toxin. Because LT binds E. coli LPS, it remains associated with the extracellular bacterial surface and is released in association with outer membrane vesicles. We propose that difference in the extracellular fates of LT and CT contribute to the differences in disease caused by ETEC and Vibrio cholerae.
Pathogenic bacteria produce toxins that cause the symptoms of the diseases associated with infection. Several of these, including shiga toxin, pertussis toxin, cholera toxin (CT),1 and heat-labile enterotoxin (LT), belong to a family related by structural homology. These AB5 toxins consist of a catalytic A subunit (e.g. LTA, CTA) and a pentamer of receptor-binding B subunits (e.g. LTB, CTB) (1, 2). In addition to structural homology, the enterotoxigenic Escherichia coli (ETEC) toxin, LT, shares 80% nucleotide sequence identity with the Vibrio cholerae toxin, CT (3, 4). The ring-shaped B pentamers of both LT and CT mediate binding to the host cell receptor, GM1 (5–7). LTB is more promiscuous than CTB in that it can also bind other receptors containing a terminal galactose (6). After binding, the receptor/toxin complex is internalized by the host cell and the A subunit undergoes retrograde trafficking to the cytosol via the endoplasmic reticulum (ER) (6, 8, 9). Upon entry into the cytosol, the catalytic subunit constitutively activates adenylate cyclase, resulting in water and electrolyte efflux from the host cells (10).
Despite the equivalent activity CT and LT exhibit in cell culture, disease caused by ETEC is much less severe than that caused by V. cholerae (11). Several factors, including the toxins themselves, are likely to contribute to the severity of these diseases. Volunteers treated with 25 μg of CT lost an average of 20 liters of fluid, compared with only a 6 liter loss caused by the same amount of LT (12, 13). Hypotheses regarding the processing and host cell trafficking of the toxins have been proposed to explain this difference in toxicity. For example, the C terminus of CTA has a prototypical KDEL ER retrograde trafficking signal sequence whereas LTA has an RDEL sequence; however, this difference has not been shown to affect the intracellular trafficking of the toxins inside the host (14). Further, CTA secreted from V. cholerae is activated by a hemagglutinin/protease encoded by V. cholerae resulting in two polypeptides, A1 and A2, that remain linked by a disulfide bond until the toxin reaches the ER (15–17). LTA is nicked in vitro, but not when expressed and secreted by V. cholerae (16, 18). However, inefficient LTA processing is probably not a relevant factor since this result may be explained by an insufficient level of soluble hemagglutinin produced by the mutant used in those studies (19), and recently several serine proteases in the endocytic pathway have been demonstrated to proteolytically activate LTA (20). Finally, structural studies have shown that a difference in the A2 linker of CT and LT causes a difference in stability: the A2 portion of CT confers a greater stability, and thus a greater ability to survive intact within the host (14). This difference in stability, as well as host cell receptor binding differences, may explain some of the differing effects of purified LT and CT on human volunteers described above.
Other hypotheses to explain the difference in the severity of ETEC and V. cholerae infections focus on differences in the bacteria themselves. Certainly, many factors including colonization, persistence, adhesins, and other toxins, in addition to the processing, secretion, and presentation (e.g. soluble versus insoluble) of CT and LT are likely to play an enormous role in determining the severity of the diseases caused by ETEC and V. cholerae. Both CT and LT are secreted by the general secretory pathway (GSP) across the outer membrane (21–23), however, our group has shown that, in contrast to the secretion of soluble CT, LT remains associated with the cell surface and is transported to the host cell in association with outer membrane vesicles (21, 24). The association between LT and the bacterial surface was determined to be mediated by an interaction between LTB and LPS (21).
The outer membrane of Gram-negative bacteria is unique in its asymmetry. The inner leaflet is composed of the same phospholipids as are found in the cytoplasmic membrane: phosphatidylglycerol (PG), phosphatidylethanolamine (PE), and cardiolipin (25). The predominant outer leaflet lipid component is lipopolysaccharide (LPS), an amphiphilic molecule made up of lipid A (an acylated disaccharide of glucosamines), core sugars (including Kdo [3-deoxy-D-manno-octulosonic acid]) specific to each genus, and O-antigen sugars that define serotype (reviewed in Refs. 26 and 27). Under standard laboratory conditions, the minimum essential LPS structure in E. coli is Kdo2-lipid A. Two Kdo sugars are added to lipid A by a bifunctional E. coli Kdo transferase (28). The core structures of V. cholerae and H. influenzae LPS differ from E. coli in that they consist of Kdo-phosphate-lipid A, which are synthesized by a monofunctional Kdo transferase and a Kdo kinase (29 –31). Studies have shown that E. coli can survive with a single Kdo when its bifunctional Kdo transferase is replaced by a monofunctional Kdo transferase with or without coexpression of a Kdo kinase (32, 33).
In this work, we examine the molecular basis for the association between LT and LPS. We show that both CT and LT bind to E. coli LPS, but not Vibrio LPS, and that this binding requires only the Kdo component the inner core. We further demonstrate that LT and CT do not bind to Vibrio LPS due to the presence of the phosphate on the Kdo. Therefore, we propose that the LPS Kdo core and the respective toxin ability to bind to that LPS contribute to the difference between disease caused by ETEC and V. cholerae.
EXPERIMENTAL PROCEDURES
Strains and Media
Bacterial strains and plasmids used in this investigation are listed in Table I. Strains were grown in LB (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.0), and maintained on LB agar. Transformations were performed using a modified CaCl2 protocol (34). Antibiotics were added as required at the following concentrations: ampicillin (Amp), 100 μg/ml; kanamycin (Kan), 50 μg/ml; chloramphenicol (Cm), 35 μg/ml. Isothiogalactopyranoside (IPTG, 1.0 mM) was used to induce expression of LT and CT constructs. Unless specified, reagents were purchased from Fisher.
Table I.
Strains and plasmids
| Strain/plasmid | Genotype or alias | Relevant characteristics | Ref. |
|---|---|---|---|
| Strains | |||
| E9034P | ETEC cured of virulence plasmid (LT-deficient) | (54) | |
| E. coli CTB | MC4100 hns1001::kan/pCHAP4278/pLMP1 | E. coli K-12 with hns mutation, expressing GSP and CTB, KanR, CmR, AmpR | a |
| E. coli LTB | MC4100 hns1001::kan/pCHAP4278/pMMB68 | E. coli K-12 with hns mutation, expressing GSP and LTB, KanR, CmR, AmpR | (21) |
| K-12 | D21 | E. coli CGSC #5158, StrR | (55) |
| Rd | D21e7 | E. coli Rough mutant, rfa1, CGSC #5159, StrR | (55) |
| Re | D21f2 | E. coli Deep rough mutant, rfa1, rfa31, CGSC #5162, StrR | (55) |
| Vibrio/LTB | Vibrio sp. 60/pMMB68 | Vibrio expressing LTB, RifR, AmpR | (56) |
| Vibrio/LTB/KdtAEc | Vibrio sp. 60/pMMB68/pKdtAEc | Vibrio expressing LTB and bifunctional E. coli KdtA, Rif R, AmpR, CmR | a |
| CJB26 | E. coli kdtA::kan recA−/pKdtAEc | E. coli Kdo-transferase mutant expressing the bifunctional E. coli KdtA, KanR, CmR, ts | (28) |
| Kdo1 | E. coli kdtA::kan recA−/pKdtAHi | E. coli Kdo-transferase mutant expressing the monofunctional H. influenzae KdtA, KanR, CmR, ts | b |
| Kdo2 | WBB01 | E. coli heptosyltransferase I and II mutant (ΔwaaCF::tet6), TetR, ts | (33) |
| Kdo1P | WBB22 | E. coli ΔwaaCF::(tet6 kdkAHi+) kdtA::(kdtAHikan), TetR, KanR, ts | (33) |
| Plasmids | |||
| GSP | pCHAP4278 | gspAB and gspC-R, CmR | (57) |
| LTB | pMMB68 | IPTG-inducible LTB | (56) |
| CTB | pLMP1 | IPTG-inducible CTB | (58) |
| KdtAEc | pJSC2 | Bifunctional E. coli Kdo transferase, CmR, ts | (28) |
| KdtAHi | Monofunctional H. influenzae Kdo transferase, CmR, ts | b | |
This work.
K. White and C. Raetz, unpublished data.
LT-Bacterial Surface Binding Assay
Similar to the previously described assay (21), bacteria were grown to mid-log phase, 1.0 ml was pelleted, washed twice in cold HEPES (50 mM, pH 6.8), and resuspended in 0.5 ml of HEPES. LT (0.5 μg, 60 pmol) (List Biologicals) was preincubated (25 °C, 30 min.) with HEPES or with a 10-fold molar excess of sonicated E. coli LPS (O55, Ra, Rc, Rd, and Re), E. coli lipid A, Kdo, or V. cholerae LPS (Sigma) in HEPES. Washed bacteria (50 μl) were then incubated with the LT mixtures for 30 min on ice. Cells were pelleted, washed, and resuspended, and 20 μl applied to 15% SDS-PAGE. A standard immunoblotting protocol was performed (34) using an LT cross-reactive anti-CT antibody, anti-rabbit-horseradish peroxidase (Sigma), and the ECL detection reagents (Amersham Biosciences). NIH-Image was used for quantitative densitometry of the immunoblots.
BODIPY-GM1 Labeling
Labeling was performed as described (21).
RESULTS
LT Binds to the Core Sugars of LPS
In previous work we have determined that after secretion across the outer membrane, LT binds to LPS on the surface of ETEC. In addition, the addition of soluble, purified LT to an LT-deficient ETEC strain E9034P leads to LT binding and this interaction can be blocked with full-length purified LPS. However, GM1 was incapable of blocking the interaction between LT and the bacteria. These data demonstrated that LT has the capability to bind to LPS at a binding site distinct from the GM1 binding site (21). We were interested in determining the minimum LPS components that are required for LT binding and, in particular, whether LT binds the lipid or sugar moieties. As published previously, purified full-length O55 LPS and Ra LPS, which lacked O-antigen, blocked the binding of LT to the surface of ETEC ((21) and Table II). In fact, Ra LPS blocked binding more effectively than did O55, indicating that LT binding does not require O-antigen. The lipid core of LPS, lipid A, was not able to inhibit LT binding. We tested shorter truncated forms of LPS and found that Rc LPS, which lacks most of its outer core sugars, did block binding (Table II). LPS derived from mutants expressing fewer core sugars (Rd and Re) were unable to inhibit binding significantly. These data indicate that LT binds to LPS independent of O-antigen and complete outer core sugars.
Table II.
Inhibition of LT binding to ETEC strain E9034P
| Preincubation condition | % LT bounda | p valueb |
|---|---|---|
| Buffer | 100 | NAc |
| O55 E. coli LPS | 54.2 ± 8.9 | p ≤ 0.0005 |
| Ra E. coli LPS | 27.7 ± 4.5 | p ≤ 0.0002 |
| Rc E. coli LPS | 71.1 ± 8.1 | p ≤ 0.02 |
| Rd E. coli LPS | 69.6 ± 15.1 | NSd |
| Re E. coli LPS | 73.4 ± 6.8 | NS |
| Lipid A | 80.7 ± 19.2 | NS |
| Vibrio cholerae LPS | 106.0 ± 9.8 | NS |
Densitometric analysis of immunoblots with buffer control normalized to 100%. Values are ± S.E., n ≥ 3.
Determined by Student’s t test.
NA, not applicable.
NS, not significant (p ≥ 0.02).
LPS dispersed in aqueous solution may not possess the same conformation as native LPS presented on the surface of a bacterium. Although purified O55, Ra, and Rc LPS were able to block LT from binding to the surface of an ETEC cell expressing LPS with O-antigen, LT may not be able to bind to the surface of a K-12 strain (lacking O-antigen) or an LPS mutant (lacking core sugars). We therefore wanted to compare the ability of LT to bind bacteria expressing wild type and mutant forms of LPS. LT bound to the surface of K-12, as well as isogenic Rd and Re mutant strains, demonstrating that LT binding to LPS is, in fact, independent of all core sugars but Kdo (Fig. 1A). These results were unexpected, given that purified Rd and Re LPS were unable to block LT from binding to wild type cells (Table II). The fact that this binding was inhibited with wild type O55 LPS (Fig. 1A), suggested that LT may have a higher affinity for LPS with more sugars and a lower affinity for LPS with only core sugars. If this is the case, we predicted that, unlike the previous results with wild type LPS expressing bacteria (Table II), purified Re LPS would be able to block the binding of LT to Re mutant bacteria. Indeed, LT binding to the Re mutant was inhibited by purified Re LPS (Fig. 1B). Additionally, we found that preincubation with pure Kdo reduced LT binding to the Re mutant by 74 ± 8% (n = 3) (Fig. 1C), however Kdo did not inhibit LT binding to E9034P (data not shown). Together, these data suggest that LT binding to LPS relies minimally on the Kdo core however LT has a higher affinity for LPS with full-length core sugars.
Fig. 1. LT binds to the surface of E. coli LPS mutants.
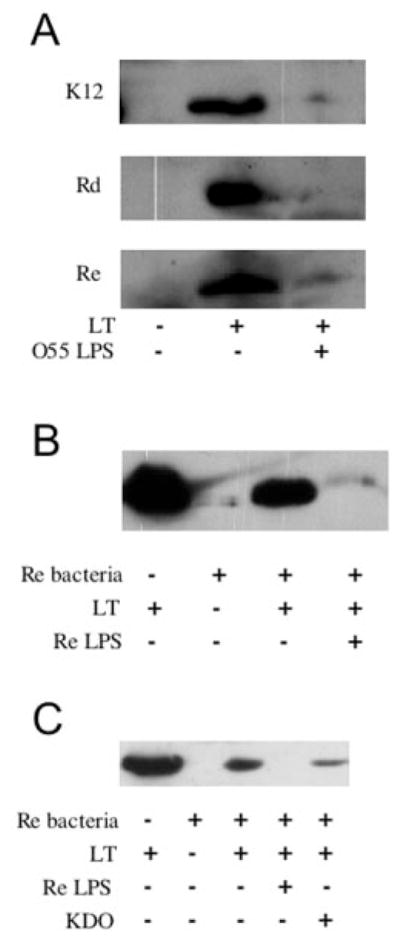
A, immunoblot of LT binding to wild type (K-12) and LPS mutant (Rd and Re) E. coli strains. Incubations of bacteria with buffer, LT, and LT preincubated with soluble O55 LPS are indicated. B, soluble Re LPS blocks LT from binding Re E. coli. Immunoblot of LT alone; and incubations of Re E. coli with buffer, LT, and LT preincubated with purified Re LPS, as indicated. C, Kdo inhibits LT binding to Re E. coli. Immunoblot of LT alone; and incubations of Re E. coli with buffer, LT, and LT preincubated with purified Re LPS or Kdo, as indicated.
LT and CT Bind the Surface of E. coli but not Vibrio
Unlike E. coli, strains of Vibrio expressing CT, LT, and LTB secrete these products solubly, suggesting little to no association with the Vibrio membrane. Thus, we wanted to investigate the interaction of the toxins with Vibrio LPS and the surface of Vibrio using our in vitro assays. We observed that purified V. cholerae LPS was unable to block the binding of LT to ETEC (Table II). In case the native context of the LPS was critical to binding, we wanted to examine whether LT was able to bind to the surface of a Vibrio strain. We previously developed a quantitative fluorescence assay to demonstrate that expressed and secreted LTB bound to the surface of ETEC (21). Using this assay with Vibrio expressing LTB, we found little BODIPY-GM1 binding (Fig. 2A). Immunoblotting confirmed that LTB was expressed and secreted by this strain (data not shown). Thus, although LT binds to E. coli LPS, it does not bind to Vibrio LPS.
Fig. 2. LT does not bind Vibrio LPS, however CT binds E. coli LPS.

A, detection of surface-associated LTB and CTB expressed by Vibrio sp. 60 and secretion-competent E. coli by BODIPY-GM1 labeling. Error bars = S.E., n ≥ 3. B, immunoblot of E9034P incubated with buffer, CT, and CT preincubated with O55 LPS, as indicated.
Because CT and LT are structurally almost identical, we wondered whether CT would bind LPS. Interestingly, similar to LT, purified CT bound to the surface of ETEC and this association was blocked by soluble O55 LPS (Fig. 2B). Likewise, CTB expressed in a secretion-competent E. coli strain, which has a mutation in hns and overexpresses the GSP (21), was detected by BODIPY-GM1 binding, similar to LTB (Fig. 2A). Based on these data, we propose that CT and LT are secreted solubly from Vibrio because they cannot bind to Vibrio LPS, and that CT and LT remain associated with E. coli because they can bind E. coli LPS. These results further indicate that differences in the conformation or shape of E. coli and Vibrio LPS are critical to LT and CT binding.
Kdo Structure Dictates LT and CT Bacterial Surface Binding
Whereas E. coli LPS contains two Kdo sugars, Vibrio LPS contains only one Kdo which is phosphorylated (29, 35). Based on our data demonstrating that LT was unable to bind Vibrio LPS, we wanted to determine whether the second Kdo conferred binding or the phosphate group inhibited binding. We compared the ability of LT to bind to the surface of Re E. coli (CJB26), which produces Kdo2-lipid A, and to an isogenic strain (Kdo1), which expresses a monofunctional Kdo transferase and produces Kdo-lipid A. LT bound to the surface of bacteria expressing LPS with only one Kdo as well as it did to bacteria expressing LPS with two Kdos (Fig. 3A). Thus, the second Kdo on E. coli LPS is not the determinant of specificity for LT binding.
Fig. 3. Toxin binding is blocked by phosphorylation of the Kdo core of Vibrio LPS.

Immunoblots of LT binding to E. coli strains CJB26 and Kdo1 (A), and Kdo2 and Kdo1P (B), incubated with buffer and LT, as indicated. C, Detection by BODIPY-GM1 labeling of secreted LTB on the surface of Vibrio sp. 60 expressing LTB and Vibrio sp. 60 expressing LTB and Kdo2-Lipid A. Error bars = S.E., n ≥ 3.
Next we directly compared LT binding to isogenic strains of E. coli expressing Kdo2-lipid A and phosphorylated Kdo1-lipid A. We found that LT bound to Kdo2, the control E. coli strain, and not to E. coli strain Kdo1P, which expresses a monofunctional Kdo transferase and Kdo kinase (Fig 3B). This result leads us to propose that phosphorylation of the Kdo of LPS prevents LT and CT binding.
If the phosphate group on Vibrio LPS inhibits toxin binding, then LT should bind better to the surface of a Vibrio that displays unphosphorylated Kdo LPS. Since the Kdo kinase in Vibrio cannot use Kdo2-lipid A as a substrate (31), we constructed a Vibrio strain that expressed KdtAEc, the bifunctional E. coli transferase in addition to its endogenous Kdo transferase and kinase. This strain should display LPS with both Kdo-phosphate-lipid A and Kdo2-lipid A. We tested the ability of LT expressed and secreted from these strains to be associated with the bacterial cell surface by BODIPY-GM1 labeling (Fig. 3C). The expression of Kdo2-Lipid A caused a reproducible 2-fold increase in the amount of LT associated with the Vibrio cell surface. Because Vibrio does not naturally produce Kdo2-lipid A, its export was most likely very inefficient, leading to the modest increase in LT binding. Together, these results are consistent with a model of LT and CT binding to the Kdo or Kdo2 core portion of LPS, inhibited by Kdo phosphorylation.
DISCUSSION
This work describes the nature of the association between LT and the surface of the outer membrane of ETEC. Our prior work determined that LT binds LPS on the surface of E. coli independent of the O-antigen (21). LT also bound purified Ra LPS, which lacks O-antigen, and Rc LPS, which lacks outer core sugars, however, LT did not bind lipid A. Therefore, LT appeared to bind specifically the core sugars of LPS.
To examine the LT binding site on LPS, the ability of LT to bind to the surface LPS biosynthesis mutant strains expressing different truncations of LPS was tested. We were surprised to find that LT bound to rough (Rd) and deep rough (Re) E. coli. This result apparently contradicted the previous data that showed that purified Rd and Re LPS were unable to inhibit LT binding the surface of E. coli expressing full O-antigen. Therefore, we considered the possibility that LT may have a lower affinity for truncated LPS compared with longer LPS. The ability of purified Re LPS to block LT from binding to Re E. coli proved that LT binds to E. coli Kdo2-lipid A but has a higher affinity for less truncated forms of LPS.
The minimal LT binding site on Re LPS was defined with further experiments. LT binding to Re LPS was significantly inhibited by preincubation with Kdo, implicating Kdo as the minimal portion of LPS required for LT binding. Purified lipid A did not inhibit LT binding to ETEC, suggesting that lipid A did not contain the LT binding site. However, it was possible that the affinity for purified lipid A was too low to compete with full length LPS in its native conformation. The fact that the Re-truncated LPS is the minimal requirement for E. coli viability prevented us from testing this directly. However, if lipid A is sufficient for LT binding, then every Gram-negative bacterial surface would be expected to bind LT, and we found that this was not the case. LT did not bind Vibrio. Thus toxin binding depends on the Kdo core of LPS. This result also suggested that LT discriminated between Kdo2-lipid A and Kdo1-phosphate-lipid A. Indeed, LT could not bind E. coli expressing phosphorylated Kdo-lipid A, whereas LT bound to E. coli expressing Kdo-lipid A. Additionally, LT could bind better to Vibrio expressing the E. coli Kdo-transferase which results in Kdo2-lipid A. Therefore, we conclude that LT binding to LPS depends on the unphosphorylated Kdo core.
Both LT and CT expressed in E. coli bind to the surface of E. coli. This indicates that LPS binding is not a characteristic unique to LT and cannot be explained by the 20% difference in amino acid sequence between LT and CT. Although the toxins LT and CT are almost identical, the diseases caused by ETEC and V. cholerae are very dissimilar. Whereas cholera is a disease of pandemic proportions, diarrhea caused by ETEC is much less severe. This difference was proposed to be due to the mode of toxin delivery: CT was secreted extracellularly and purportedly LT remained periplasmic. Work from our group and others, however, has disproven that theory since extracellular LT secretion can occur in E. coli by the GSP (21–23). Based on the results in this paper, we propose that a factor contributing to the difference between the diseases is the fate of the secreted toxin. LT and CT do not associate with the surface of Vibrio, nor does V. cholerae LPS block binding of LT to E. coli LPS. Therefore, perhaps the difference in LPS structure, rather than in the toxins of ETEC and V. cholerae significantly contributes to the difference in disease. LT and CT secreted by Vibrio is liberated into the medium because it cannot bind to Vibrio LPS which contains Kdo-phosphate (29). LT and CT secreted by ETEC remain associated with the surface of ETEC because E. coli LPS contains a non-phosphorylated Kdo core (35). Thus, the difference between the extracellular localization of these two similar toxins is most likely due to the differing LPS core structures of E. coli and V. cholerae and this difference in the affinity of LPS for the toxins could significantly contribute to the difference in activity and spread of the toxin in the host.
As a consequence of the LT-LPS interaction, one would predict LT to remain associated with the outer surface of ETEC and never be released into the extracellular milieu. However, ETEC, similar to all Gram-negative bacteria, produce outer membrane vesicles (21, 36, 37). We have shown that ETEC vesicles harbor LT both in the lumen and bound to their surface (21, 24). The secretion of LT on the surface of vesicles effectively decreases the number of functional toxin units secreted when compared with the release of soluble CT by V. cholerae, thereby potentially decreasing the number of host cells affected by the toxin. In addition, LT and CT have been shown to inhibit cytokine release (38, 39).2 Thus the association of LT with vesicles also affects the host immune response.
Because all Gram-negative bacteria produce vesicles, we do not believe that ETEC evolved vesiculation exclusively for the transport of LT. Nor do we suggest that the core of E. coli LPS evolved for the sole purpose of binding to LT. It is far more likely that LT, acquired by E. coli from V. cholerae via horizontal gene transfer (40), bound E. coli LPS by chance and therefore was retained on the cell and vesicle surface. Regardless of the manner by which ETEC came to transport LT via vesicles, vesicles are the final step in LT secretion and LT transported on vesicles is toxic. Therefore vesicles should be considered a specific secretion mechanism for virulence factors.
Several recent observations in other Gram-negative bacteria have revealed that host cell delivery of toxin bound to the surface of outer membrane vesicles is a strategy not unique to ETEC. Kolling et al. (41) demonstrated that shiga toxin is present in vesicles produced by O157:H7. Vesicle-associated toxin was partially protease sensitive, which we interpret as demonstrating surface association of some of the toxin. Leukotoxin has been detected on the surface of Actinobacillus actinomycetemcomitans cells (42), and that toxin has been found in association with the membrane of the vesicles produced by that organism (43). Immunogold electron microscopy was used to detect a protease on the surface of Apx-containing Actinobacillus pleuropneumoniae vesicles (44) and Helicobacter pylori vacuolating toxin, VacA, closely associated with the membrane of H. pylori vesicles (45). These vesicles all deliver physiologically active toxin to host cells.
The ability to bind both LPS and GM1 enables LT to act as an adhesin, promoting an interaction between the vesicle and host cell, resulting in internalization of the vesicle and all its contents.3 We previously demonstrated that the GM1 and LPS binding sites on LT are distinct (21), and we wondered if a LPS binding site could be elucidated from the solved crystal structures of LT and CT (2, 46 –50). Based on examination of the crystal structures of many different lectins (bacterial, mammalian, and plant), a general ligand binding motif emerges. Sugar-binding proteins interact with their receptors at the periphery or surface of the protein, often “snuggled” under an exterior loop. We identified such a region in the crystal structure of CT (and similarly, LT) bound to GM1 pentasaccharide, directly opposite the GM1 binding site on the B-pentamer (Fig. 4A, star). This could be the region that LT/CT bind to E. coli LPS. Mutations in this putative LPS binding pocket of LT and CT were found to confer binding to A and B blood group determinants (51). Intriguingly, molecular dynamics simulations conducted in that work defined exactly the same binding pocket for blood group antigens as we have proposed for LPS. As a result of these insights, we propose a model in which LT/CT binds E. coli LPS in a manner that allows LT/CT to bind simultaneously to one or more receptors (but not five) on the epithelial cell (Fig. 4B).
Fig. 4. Proposed location of LPS binding site on the toxin.

A, view from the top of an isolated LTB/CTB subunit, as if looking down into central pore of the pentamer. Star denotes putative binding site. B, oblique side view of a LTB/CTB-pentamer. Green, GM1 pentasaccharide; blue, putative LPS binding site. Adapted from PDB file 2CHB of CT, using Swiss PDBviewer.
When the structure of CT was solved with the GM1 pentasaccharide, Merrit et al. (47) presented a model of how CT (and LT) would interact with the host cell. In this model, the toxin “lands” flat on the surface of the host cell, binding multiple GM1 receptors and filling all of the five binding pockets. Based on this model, however, it is difficult to envision how LT could be bound to LPS on the surface of a vesicle, and still “land” on the surface of an epithelial cell. We propose that the geometry of LT/CT binding to GM1 is more flexible than previously described, in that the toxin may bind GM1 at an angle other than 90° and have freedom to rotate and bend. This model does not preclude the possibility that after this “docking event,” the toxin follows the model proposed by Merritt, filling all five of its GM1 binding sites. This may bring the vesicle in closer proximity to the host cell, allowing secondary adhesins in the vesicle membrane to bind their receptors, increasing the intimacy of the interaction between host and vesicle.
To date, no other protein has been demonstrated to interact with LPS in the bacterial membrane in the same manner as LT. The interaction of leukotoxin to the surface of A. actinomycetemcomitans appears to be mediated by nucleic acids bound to the bacterial surface (52). Adenylate cyclase toxin binds the surface of Bordetella pertussis via filamentous hemagglutinin (53). Because shiga toxin, a member of the AB5 toxin family, is most likely on the surface of EHEC vesicles, examination of the association between shiga toxin and the outer membrane of EHEC may show that LPS plays a role. In addition, further study of the previously described toxins that are transported via vesicles may yield more proteins that bind to LPS in a lectin-like fashion.
Our studies have disproven the theories that bacterial retention of LT or a lack of the GSP are the reasons for the reduced toxicity of ETEC compared with V. cholerae. We have proposed that the difference between the diseases caused by V. cholerae and ETEC are in part due to the difference in the affinity of their toxins to their LPS. One may question the evolutionary perpetuation of the attenuation of the toxicity of LT bound to LPS, and thus the reliance on delivery via vesicles. To ETEC, the gut may not be a particularly hospitable environment, but it does provide a constant source of nutrients that may not be readily available outside of the host. By adopting a less toxigenic phenotype than V. cholerae, ETEC has increased the amount of time it inhabits the host and therefore increased the amount of nutrients it may access. Thus, the strategy of binding a toxin to the bacterial surface may be a general virulence property that is beneficial to a bacterial pathogen.
Acknowledgments
We thank M. Rosser for critical reading of the manuscript and the following generous contributions: J. Giron for E9034P; O. Francetic for MC4100Δhns/GSP; R. Holmes for Vibrio sp. 60, pMMB68, and pLMP1; C. Raetz for CJB26, Kdo1, and pKdtAEc; and H. Brade for WBB01 and WBB22. We are especially thankful to Kim White and Chris Raetz for the use of strain Kdo1 prior to publication.
Footnotes
The abbreviations used are: CT, cholera toxin; LT, heat-labile enterotoxin; ETEC, enterotoxigenic E. coli; LPS, lipopolysaccharide; Kdo, 3-deoxy-D-manno-octulosonic acid; ER, endoplasmic reticulum.
A. Horstman, K. Mason, and M. Kuehn, unpublished data.
N. Kesty, K. Mason, and M. Kuehn, submitted manuscript.
This work was supported by a Burroughs Wellcome Fund Investigator in Pathogenesis of Infectious Disease Award (to M. J. K.) and the National Institutes of Health.
References
- 1.Merritt EA, Hol WG. Curr Opin Struct Biol. 1995;5:165–171. doi: 10.1016/0959-440x(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 2.Sixma TK, Kalk KH, van Zanten BA, Dauter Z, Kingma J, Witholt B, Hol WG. J Mol Biol. 1993;230:890–918. doi: 10.1006/jmbi.1993.1209. [DOI] [PubMed] [Google Scholar]
- 3.Mekalanos JJ, Swartz DJ, Pearson GD, Harford N, Groyne F, de Wilde M. Nature. 1983;306:551–557. doi: 10.1038/306551a0. [DOI] [PubMed] [Google Scholar]
- 4.Dallas WS, Falkow S. Nature. 1980;288:499–501. doi: 10.1038/288499a0. [DOI] [PubMed] [Google Scholar]
- 5.Fukuta S, Magnani JL, Twiddy EM, Holmes RK, Ginsburg V. Infect Immun. 1988;56:1748–1753. doi: 10.1128/iai.56.7.1748-1753.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishman PH, Pacuszka T, Orlandi PA. Adv Lipid Res. 1993;25:165–187. [PubMed] [Google Scholar]
- 7.Holmgren J, Fredman P, Lindblad M, Svennerholm AM, Svennerholm L. Infect Immun. 1982;38:424– 433. doi: 10.1128/iai.38.2.424-433.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lencer WI, de Almeida JB, Moe S, Stow JL, Ausiello DA, Madara JL. J Clin Investig. 1993;92:2941–2951. doi: 10.1172/JCI116917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lencer WI, Hirst TR, Holmes RK. Biochim Biophys Acta. 1999;1450:177–190. doi: 10.1016/s0167-4889(99)00070-1. [DOI] [PubMed] [Google Scholar]
- 10.Verlinde CL, Merritt EA, Van den Akker F, Kim H, Feil I, Delboni LF, Mande SC, Sarfaty S, Petra PH, Hol WG. Protein Sci. 1994;3:1670–1686. doi: 10.1002/pro.5560031006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunkel SL, Robertson DC. Infect Immun. 1979;25:586–596. doi: 10.1128/iai.25.2.586-596.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine MM. Scand J Gastroenterol Suppl. 1983;84:121–134. [PubMed] [Google Scholar]
- 13.Bowman CC, Clements JD. Infect Immun. 2001;69:1528–1535. doi: 10.1128/IAI.69.3.1528-1535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodighiero C, Aman AT, Kenny MJ, Moss J, Lencer WI, Hirst TR. J Biol Chem. 1999;274:3962–3969. doi: 10.1074/jbc.274.7.3962. [DOI] [PubMed] [Google Scholar]
- 15.Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 16.Booth BA, Boesman-Finkelstein M, Finkelstein RA. Infect Immun. 1983;42:639– 644. doi: 10.1128/iai.42.2.639-644.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gill DM. J Supramol Struct. 1979;10:151–163. doi: 10.1002/jss.400100205. [DOI] [PubMed] [Google Scholar]
- 18.Hirst TR, Sanchez J, Kaper JB, Hardy SJ, Holmgren J. Proc Natl Acad Sci U S A. 1984;81:7752–7756. doi: 10.1073/pnas.81.24.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spangler BD. Microbiol Rev. 1992;56:622– 647. doi: 10.1128/mr.56.4.622-647.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lencer WI, Constable C, Moe S, Rufo PA, Wolf A, Jobling MG, Ruston SP, Madara JL, Holmes RK, Hirst TR. J Biol Chem. 1997;272:15562–15568. doi: 10.1074/jbc.272.24.15562. [DOI] [PubMed] [Google Scholar]
- 21.Horstman AL, Kuehn MJ. J Biol Chem. 2002;277:32538–32545. doi: 10.1074/jbc.M203740200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tauschek M, Gorrell RJ, Strugnell RA, Robins-Browne RM. Proc Natl Acad Sci U S A. 2002;99:7066–7071. doi: 10.1073/pnas.092152899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandkvist M, Michel LO, Hough LP, Morales VM, Bagdasarian M, Koomey M, DiRita VJ. J Bacteriol. 1997;179:6994–7003. doi: 10.1128/jb.179.22.6994-7003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horstman AL, Kuehn MJ. J Biol Chem. 2000;275:12489–12496. doi: 10.1074/jbc.275.17.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DiRienzo JM, Nakamura K, Inouye M. Annu Rev Biochem. 1978;47:481–532. doi: 10.1146/annurev.bi.47.070178.002405. [DOI] [PubMed] [Google Scholar]
- 26.Raetz CR. J Bacteriol. 1993;175:5745–5753. doi: 10.1128/jb.175.18.5745-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raetz C. In: Escherichia coli and Salmonella. Neidhardt F, editor. Vol. 1. ASM Press; Washington, D. C: 1996. pp. 1035–1063. [Google Scholar]
- 28.Belunis CJ, Clementz T, Carty SM, Raetz CR. J Biol Chem. 1995;270:27646–27652. doi: 10.1074/jbc.270.46.27646. [DOI] [PubMed] [Google Scholar]
- 29.Cox AD, Brisson JR, Varma V, Perry MB. Carbohydr Res. 1996;290:43–58. doi: 10.1016/0008-6215(96)00135-8. [DOI] [PubMed] [Google Scholar]
- 30.White KA, Kaltashov IA, Cotter RJ, Raetz CR. J Biol Chem. 1997;272:16555–16563. doi: 10.1074/jbc.272.26.16555. [DOI] [PubMed] [Google Scholar]
- 31.White KA, Lin S, Cotter RJ, Raetz CR. J Biol Chem. 1999;274:31391–31400. doi: 10.1074/jbc.274.44.31391. [DOI] [PubMed] [Google Scholar]
- 32.Isobe T, White KA, Allen AG, Peacock M, Raetz CR, Maskell DJ. J Bacteriol. 1999;181:2648–2651. doi: 10.1128/jb.181.8.2648-2651.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brabetz W, Muller-Loennies S, Brade H. J Biol Chem. 2000;275:34954–34962. doi: 10.1074/jbc.M005204200. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1989. pp. 1.82–1.84. [Google Scholar]
- 35.Raetz CR. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- 36.Wai SN, Takade A, Amako K. Microbiol Immunol. 1995;39:451– 456. doi: 10.1111/j.1348-0421.1995.tb02228.x. [DOI] [PubMed] [Google Scholar]
- 37.Beveridge TJ. J Bacteriol. 1999;181:4725– 4733. doi: 10.1128/jb.181.16.4725-4733.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leal-Berumen I, Snider DP, Barajas-Lopez C, Marshall JS. J Immunol. 1996;156:316–321. [PubMed] [Google Scholar]
- 39.Bacon KB, Camp RD. Biochem Biophys Res Commun. 1990;169:1099–1104. doi: 10.1016/0006-291x(90)92008-n. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto T, Gojobori T, Yokota T. J Bacteriol. 1987;169:1352–1357. doi: 10.1128/jb.169.3.1352-1357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kolling GL, Matthews KR. Appl Environ Microbiol. 1999;65:1843–1848. doi: 10.1128/aem.65.5.1843-1848.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lally ET, Golub EE, Kieba IR, Taichman NS, Rosenbloom J, Rosenbloom JC, Gibson CW, Demuth DR. J Biol Chem. 1989;264:15451–15456. [PubMed] [Google Scholar]
- 43.Kato S, Kowashi Y, Demuth DR. Microb Pathog. 2002;32:1–13. doi: 10.1006/mpat.2001.0474. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Cuellar C, Montanez C, Tenorio V, Reyes-Esparza J, Duran MJ, Negrete E, Guerrero A, de la Garza M. Can J Vet Res. 2000;64:88–95. [PMC free article] [PubMed] [Google Scholar]
- 45.Keenan J, Day T, Neal S, Cook B, Perez-Perez G, Allardyce R, Bagshaw P. FEMS Microbiol Lett. 2000;182:259–264. doi: 10.1111/j.1574-6968.2000.tb08905.x. [DOI] [PubMed] [Google Scholar]
- 46.Merritt EA, Pronk SE, Sixma TK, Kalk KH, van Zanten BA, Hol WG. FEBS Lett. 1994;337:88–92. doi: 10.1016/0014-5793(94)80635-7. [DOI] [PubMed] [Google Scholar]
- 47.Merritt EA, Sarfaty S, van den Akker F, L’Hoir C, Martial JA, Hol WG. Protein Sci. 1994;3:166–175. doi: 10.1002/pro.5560030202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merritt EA, Sixma TK, Kalk KH, van Zanten BA, Hol WG. Mol Microbiol. 1994;13:745–753. doi: 10.1111/j.1365-2958.1994.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 49.Merritt EA, Sarfaty S, Jobling MG, Chang T, Holmes RK, Hirst TR, Hol WG. Protein Sci. 1997;6:1516–1528. doi: 10.1002/pro.5560060716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sixma TK, Pronk SE, Kalk KH, van Zanten BA, Berghuis AM, Hol WG. Nature. 1992;355:561–564. doi: 10.1038/355561a0. [DOI] [PubMed] [Google Scholar]
- 51.Angstrom J, Backstrom M, Berntsson A, Karlsson N, Holmgren J, Karlsson KA, Lebens M, Teneberg S. J Biol Chem. 2000;275:3231–3238. doi: 10.1074/jbc.275.5.3231. [DOI] [PubMed] [Google Scholar]
- 52.Ohta H, Hara H, Fukui K, Kurihara H, Murayama Y, Kato K. Infect Immun. 1993;61:4878– 4884. doi: 10.1128/iai.61.11.4878-4884.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zaretzky FR, Gray MC, Hewlett EL. Mol Microbiol. 2002;45:1589–1598. doi: 10.1046/j.1365-2958.2002.03107.x. [DOI] [PubMed] [Google Scholar]
- 54.Giron JA, Gomez-Duarte OG, Jarvis KG, Kaper JB. Gene (Amst) 1997;192:39– 43. doi: 10.1016/s0378-1119(97)00039-5. [DOI] [PubMed] [Google Scholar]
- 55.Havekes L, Tommassen J, Hoekstra W, Lugtenberg B. J Bacteriol. 1977;129:1– 8. doi: 10.1128/jb.129.1.1-8.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nashar TO, Webb HM, Eaglestone S, Williams NA, Hirst TR. Proc Natl Acad Sci U S A. 1996;93:226–230. doi: 10.1073/pnas.93.1.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Francetic O, Belin D, Badaut C, Pugsley AP. EMBO J. 2000;19:6697– 6703. doi: 10.1093/emboj/19.24.6697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jobling MG, Palmer LM, Erbe JL, Holmes RK. Plasmid. 1997;38:158–173. doi: 10.1006/plas.1997.1309. [DOI] [PubMed] [Google Scholar]