

Abstract
It has become evident that some of the natural or synthetic triterpenoids are natural proteasome inhibitors that have great potential for use in cancer prevention and treatment. However, the mechanisms for the antitumor activity of triterpenoids remain to be elucidated. In the present study, we investigated the anticancer activities of a natural triterpenoid, pristimerin, and the signaling pathways affected. Pristimerin was found to possess potent cytotoxic effects, inducing apoptosis and inhibiting proliferation in U87 human glioma cells. Hoechst 33258 staining and Annexin V/PI double staining exhibited the typical nuclear features of apoptosis and increased the proportion of apoptotic Annexin V-positive cells in a dose-dependent manner, respectively. Moreover, western blotting assay revealed that this apoptotic induction was associated with activated caspase-9, caspase-3, PARP cleavage and downregulation of Bcl-xl/Bax in a concentration-dependent manner. Pristimerin also increased the generation of reactive oxygen species and induced the subsequent release of cytochrome c from the mitochondria into the cytosol. Additionally, pristimerin downregulated EGFR protein expression and inhibited downstream signaling pathways in U87 cells. Our results suggest that pristimerin may have potential as a new targeting therapeutic strategy in the treatment of EGFR-overexpressing gliomas.
Keywords: pristimerin, glioma, apoptosis, reactive oxygen species, U87 cells
Introduction
Apoptosis is a highly regulated and organized cell death process, which controls the development and homeostasis of multicellular organisms. Activation of apoptosis pathways is an important mechanism by which anticancer drugs kill tumor cells (1). Two major pathways of apoptosis depending on different apoptotic stimuli are the mitochondrial pathway (intrinsic) and the death receptor pathway (extrinsic). In the intrinsic pathway, proapoptotic and antiapoptotic Bcl-2 family members converge on mitochondria in response to the death signal and compete to regulate the release of several mitochondrial intermembrane space proteins, including cytochrome c, which associates with Apaf-1 and procaspase-9 to form the apoptosome (2–4). In the extrinsic pathway, stimulation of death receptors, including Fas and tumor necrosis factor receptor-1, leads to clustering and formation of a death-inducing signaling complex, which includes the adapter protein, Fas-associated death domain and initiator caspases, such as caspase-8 (5,6). Caspase-8 drives its activation through self cleavage, and then either directly activates downstream caspase-3 or cleaves Bid, which then in turn triggers the mitochondrial pathway.
Plants are important sources of biologically active natural products which differ widely in terms of structure and biological properties. Many compounds that are used in cancer chemotherapy are derived from plant sources, such as paclitaxel, Vinca alkaloids, camptothecin and etoposide. Pristimerin is a natural quinonemethide triterpenoid compound that is isolated from the Celastraceae and Hippocrateaceae families. Pristimerin has shown anti-inflammatory, anti-oxidant, anti-malarial and insecticidal benefits (7,8).
It has been reported that pristimerin has promising clinical potential as both a therapeutic and chemopreventive agent for cancer (9). It is capable of inducing apoptotic cell death in certain human cancer cells, including breast and lung cancer (10) and human acute myeloid leukemia (11). Pristimerin has been shown to have several mechanisms of cell apoptosis induction, including proteosome inhibition (12), suppression of nuclear factor κB (NF-κB) activity (13) and cyclin D1 expression (14), and caspase activation. Although evidence demonstrates that the mode of cell death induced by pristimerin is caspase-dependent apoptotic cell death, the involved mechanisms of action, particularly crosstalk between the signaling pathway and apoptotic cell death machinery, are largely unknown.
Malignant gliomas account for approximately 50% of primary tumors in the brain. Currently, surgical resection, adjuvant chemotherapy and irradiation are still the main treatments, although the results are not always satisfactory (15,16). In this study we describe the potent anti-glioma activity of pristimerin. We show that pristimerin induces mitochondrial cell death in human glioma U87 cells and that reactive oxygen species (ROS)-dependent activation is critically required for mitochondrial dysfunction.
Materials and methods
Chemicals and reagents
3-(4,5-Dimethylthiazol-2-yl) -2,5-diphenyltetrazolium bromide (MTT), 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) and Hoechst 33258 were purchased from Sigma (St. Louis, MO, USA). ApopNexin FITC Apoptosis Detection kit was purchased from Chemicon (Temecula, CA, USA). Antibodies against caspase-3, caspase-9, cytochrome c and Bcl-xl were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against EGFR (for western blot analysis) and Bax were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). PE-conjugated mouse monoclonal anti-human EGFR antibody and PE-labeled mouse IgG2bκ isotype control (for flow cytometry) were obtained from BD Biosciences (Franklin Lakes, NJ, USA). Antibodies against Akt, ERK1/2, p-ERK1/2, glyceraldehyde-3-phosphate dehydrogenase, anti-mouse IgG-horseradish peroxidase and anti-rabbit IgG-horseradish peroxidase were purchased from KangChen Biotechnology (Shanghai, China). All tissue culture supplies were purchased from Life Technologies (Carlsbad, CA, USA). Other routine laboratory reagents of analytical or high-performance liquid chromatography grade were obtained from Whiga Biotechnology (Guangzhou, China). Pristimerin (Fig. 1A) with a purity of >98% was purchased from the Guangzhou Xingda Biotechnology (Guangzhou, China).
Figure 1.

(A) Chemical structure of pristimerin; (B) Pristimerin showed potent cytotoxicity to human glioma U87 cells. Mean ± SD of at least triplicate determinations. Each experiment was performed in six replicate wells.
Cell lines and cell culture
U87 human glioma cell lines, kindly provided by Professor Zhong-Ping Chen, were cultured in Dulbecco’s modified Eagle’s medium (DMEM), which contained 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal bovine serum. All cells were cultured in a humidified atmosphere incubator of 5% CO2 and 95% air at 37°C. The study was approved by the Ethics Committee of Shanxi Datong University, Datong, China.
Cell viability assay
Cells were harvested during the logarithmic growth phase and seeded in 96-well plates at a density of 2×104/ml in a final volume of 190 μl/well. After 24 h incubation, 10 μl pristimerin at various concentrations was added to 96-well plates. After 68 h treatment, 10 μl MTT (10 mg/ml stock solution of saline) was added to each well for 4 h. Subsequently, the supernatant was removed, and MTT crystals were solubilized with 100 μl anhydrous DMSO in each well. Thereafter, cell viability was measured by Model 550 microplate reader (Bio-Rad, Hercules, CA, USA) at 540 nm, with 655 nm as reference filter. The 50% inhibitory concentration (IC50) was determined as the concentration causing 50% reduction in cell viability as calculated from the cytotoxicity curves (Bliss software). The cell survival rate was calculated using the following formula: Survival (%) = (mean experimental absorbance) / (mean control absorbance) × 100.
Assessment of apoptosis morphology by Hoechst 33258 staining
After treatment of the U87 cells with or without 2 to 8 μmol/l pristimerin for 24 h, both floating and trypsinized adherent cells were collected, washed once with ice-cold PBS, fixed with 1 ml 4% paraformaldehyde for 20 min, and washed once with ice-cold PBS. Then, the cells were incubated in 1 ml PBS containing 10 μmol/l Hoechst 33258 at 37°C for 30 min, washed twice, and observed using fluorescence microscopy with standard excitation filters (Leica Dmirb, Wetzlar, Germany) in random microscopic fields at ×400 magnification.
Annexin V/propidium iodide (PI) double-staining assay
Apoptosis was measured by flow cytometry using Annexin V/PI double staining. U87 cells were cultured in the presence of the indicated concentrations of pristimerin for 24 h, both floating and attached cells were collected, washed with ice-cold PBS twice, and resuspended in 200 μl 1X binding buffer containing Annexin V (1:50 according to the manufacturer’s instructions) and 40 ng/sample PI for 15 min at 37°C in the dark. Then, the number of viable, apoptotic and necrotic cells was quantified by flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA) and analyzed by the CellQuest software. Cells were excited at 488 nm and the emissions of Annexin V at 525 nm and PI were collected through 610-nm band-pass filters. At least 10,000 cells were analyzed for each sample. Apoptosis rate (%) = (number of apoptotic cells) / (number of total cells observed) × 100.
Expression of EGFR protein analyzed by flow cytometry
The expression of EGFR protein was determined using a detection kit. Cells (6×105) were seeded in 25-cm2 flasks and allowed to attach for 24 h. Following treatment with 2 to 8 μmol/l pristimerin for 24 h, both floating and attached cells were collected and washed with ice-cold PBS twice. Single-cell suspensions were prepared by the addition of 0.5 mmol/L EDTA followed by three washes with a PBS buffer [supplemented with 0.5% bovine serum albumin (BSA)]. cells (100 μl) were incubated at 4°C for 45 min with 20 μl of PE-conjugated anti-human EGFR antibody. Following this incubation, cells were washed twice with the above PBS buffer, and the supernatant was discarded. Then the cells were resuspended in 500 μl PBS buffer for flow cytometric analysis. Isotype control samples were treated in an identical manner with PE-labeled mouse IgG2bκ antibody.
Whole-cell lysates and western blot analysis
After U87 cells were exposed to 2 to 8 μmol/l pristimerin for 24 h, whole cells were harvested and washed twice with ice-cold PBS, and the pellet was vortexed and 1X lysis buffer [50 mmol/l Tris-HCl (pH 6.8), 10% glycerol, 2% SDS, 0.25% bromophenol blue and 0.1 mol/l DTT] was added for 100 μl/5×106 cells. After heating at 95°C for 20 min, the lysates were centrifuged at 12,000 rpm for 10 min and the supernatant was collected. The protein concentration was determined by nucleic acid-protein analyzer (Beckman Coulter, Miami, FL, USA). Equal amounts of lysate protein were separated on 8 to 12% SDS-PAGE and transferred onto a BioTrace™ PVDF Transfer Membrane (Pall, NY, USA). The nonspecific binding sites were blocked with TBST buffer [150 mmol/l NaCl, 20 mmol/l Tris-HCl (pH 7.4) and 0.4% (v/v) Tween-20] containing 5% non-fat dried milk for 2 h. The membranes were incubated overnight at 4°C with specific primary antibodies. Then, the membranes were washed three times with TBST buffer and incubated at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibody. After three washes with TBST buffer, the immunoblots were visualized using the enhanced Phototope-Horseradish Peroxidase Detection kit purchased from Cell Signaling Technology and exposed to a Kodak medical X-ray processor (17).
Subcellular fractionation for western blot analysis of cytosolic cytochrome c
After U87 cells were exposed to 2 to 8 μmol/l pristimerin for 24 h, whole cells were harvested by centrifugation at 1,000 rpm for 5 min. The pellets were washed twice with ice-cold PBS, suspended with 5-fold volume of ice-cold cell extract buffer [20 mmol/l 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES-KOH; pH 7.5), 10 mmol/l KCl, 1.5 mmol/l MgCl2, 1 mmol/l EDTA, 1 mmol/l EGTA, 1 mmol/l DTT, 250 mmol/l sucrose, 0.1 mmol/l phenylmethylsulfonyl fluoride and 0.02 mmol/l aprotinin], and incubated for 40 min at 4°C. Then, the cells were centrifuged at 1,200 rpm for 10 min at 4°C; the supernatant was subsequently centrifuged at 12,000 rpm for 15 min at 4°C. The final supernatant was used as cytosolic fraction of cytochrome c. Then, 5X loading buffer [250 mmol/l Tris-HCl (pH 6.8), 50% (v/v) glycerol, 10% (w/v) SDS, 0.5% (w/v) bromophenol blue and 5% (w/v) DTT] was added to the above obtained supernatant and the mixture was boiled at 100°C for 15 min. Thus, the protein solution was used for identification of cytosolic cytochrome c by western blot analysis with 15% SDS-PAGE and blotting onto polyvinylidene difluoride membrane. The cytochrome c protein was detected by using anti-cytochrome c antibody at a ratio of 1:1,000 (18).
Measurement of ROS generation
DCFH-DA was used for ROS capture in the cells. It is cleaved intracellularly by nonspecific esterases to form 2′,7′-dichlorodihydrofluorescein (DCFH), which is further oxidized by ROS and becomes a highly fluorescent compound 2′,7′-dichlorofluorescein (DCF). Thus, the fluorescence intensity of DCF is proportional to the amount of ROS produced by the cells. After U87 cells were exposed to 2 to 8 μmol/l pristimerin for 24 h, 6×105 cells were harvested, washed once with ice-cold PBS, and incubated with 50 μmol/l DCFH-DA at 37°C for 20 min in the dark. Then, the cells were washed twice and maintained in 1 ml PBS. ROS generation was assessed from 10,000 cells each sample by FACSCalibur flow cytometer at the excitation wavelength of 488 nm and emission wavelength of 530 nm. The DCF fluorescence intensity data were evaluated by CellQuest software and expressed as mean fluorescence intensity. The experiment was performed at least three times (19).
Statistical analysis
For each protocol, three independent experiments were performed. Results were expressed as the mean ± standard error of the mean (SEM). Statistical calculations were performed by using SPSS 16.0 software. Differences in measured variables between experimental and control groups were assessed by the Student’s t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Pristimerin exerted potent cytotoxicity against U87 cells
MTT assay measures the activity of mitochondrial dehydrogenase enzymes based on its ability to cleave the tetrazolium ring to produce formazan. Thus, the assay may be used as an index of cell viability. The cytotoxicity of pristimerin to U87 cells was measured by MTT assay. The IC50 of pristimerin was 3.94±0.80 μmol/l for U87 cells (Fig. 1B). The data suggested that pristimerin exhibited potent cytotoxicity against U87 cells.
Pristimerin induced apoptosis in U87 cells
To observe the morphological characteristics of apoptosis, U87 cells were stained with Hoechst 33258 following exposure to 2 to 8 μmol/l pristimerin for 24 h, and detected by fluorescence microscopy. Control cells showed an even distribution of the stain and round homogeneous nuclei. Apoptotic cells increased gradually in a dose-dependent manner and displayed typical changes including reduction of cellular volume, bright staining and condensed or fragmented nuclei (Fig. 2A). For a further assessment of apoptosis induced by pristimerin, we examined the exposure of phosphatidylserine on the cell surface by using Annexin V/PI double staining. Flow cytometry analysis revealed that the percentage of apoptotic cells with Annexin V-positive but PI-negative cells increased gradually with concentration in pristimerin-treated cells (Fig. 2B). The early apoptosis rate was 4.6±0.5, 10.8±0.7, 19.7±1.5 and 25.5±1.7% in U87 controls and cells treated with 2, 4 and 8 μmol/l pristimerin, respectively (Fig. 2C).
Figure 2.

(A) Pristimerin-mediated cell apoptosis morphological changes were examined by Hoechst 33258 staining and observed under fluorescence microscope at ×400 magnification. (B) Apoptosis in U87 cells was assessed by Annexin V/PI double staining. (C) Apoptosis rate. Following treatment with pristimerin, early apoptotic cell population with Annexin V-positive but PI-negative U87 cells increased in a dose-dependent manner. Mean ± SD of three assays. *P<0.05, **P<0.01, versus control.
Release of cytochrome c and activation of caspases were involved in the apoptosis induced by pristimerin
A variety of signaling pathways may be involved in apoptosis, and the mitochondrial pathway is one of the major apoptosis pathways. Release of cytochrome c from the mitochondria to the cytosol is the limiting factor in the mitochondrial pathway, and mitochondrial dysfunction has been suggested to cause the release of cytochrome c. Subsequently, it causes apoptosis by activation of caspase-9 in the presence of Apaf-1 and in turn results in the activation of downstream caspase-3, which can cleave PARP (20,21). Additionally, the Bcl-2 protein family and death receptor pathway are increasingly believed to be associated with mitochondrial dysfunction (22). To elucidate the interactions between the above apoptotic proteins, we detected cytochrome c in cytosol, caspases and other apoptosis-related proteins of whole-cell lysates by western blot analysis after U87 cells were exposed to 2 to 8 μmol/l pristimerin for 24 h. The results showed that the release of cytochrome c increased following treatment of pristimerin in a concentration-dependent manner (Fig. 4B). Moreover, the activation of caspase-9 and caspase-3 and the cleavage of PARP in a dose-dependent manner were observed. Additionally, pristimerin downregulated the expression of Bcl-xl and upregulated the expression of Bax (Fig. 4A). These results revealed that the mitochondrial apoptosis pathway was involved in the apoptosis induced by pristimerin in U87 cells.
Figure 4.

(A) Pristimerin augmented the instability of EGFR and Akt protein and induced changes of apoptotic proteins related to the mitochondrial pathway in U87 cells in a dose-dependent manner. (B) Release of cytochrome c was involved in the apoptosis induced by pristimerin in U87 cells.
Pristimerin downregulated EGFR protein expression and inhibited downstream signaling pathways in U87 cells
EGFR-overexpressing cancer cells have been shown to activate downstream MAPK (mitogen-activated protein kinase) and PI3K (phosphatidylinositol 3-kinase)/Akt signaling pathway. As shown in Figs. 3 and 4A, pristimerin significantly downregulated EGFR protein expression in a dose-dependent manner in U87 cells. Furthermore, Akt kinase has been shown to phosphorylate several key substrates that regulate protein translation, apoptosis and cellular proliferation (23,24). Our data showed that pristimerin inhibited Akt and p-ERK1/2, whereas it had no effect on total ERK1/2 proteins (Fig. 4A).
Figure 3.

Pristimerin induced decrease of EGFR membrane protein in U87 cells. Mean ± SD of three assays. *P<0.05, **P<0.01, versus control.
Increase of intracellular ROS level in U87 cells by pristimerin
To investigate whether the mitochondrial dysfunction was mediated by increased ROS generation, we detected the intracellular ROS level by flow cytometry. DCFH-DA) was used as the fluorescent probe. After U87 cells were exposed to 2 to 8 μmol/l pristimerin for 24 h, ROS increased in a concentration-dependent manner in U87 cells (Fig. 5A). The intracellular ROS levels were 122.8±0.1, 302.9±0.2 and 386.4±0.2% with 2, 4 and 8 μmol/l pristimerin, respectively (Fig. 5B). The results indicated that mitochondrial dysfunction was ROS-dependent.
Figure 5.
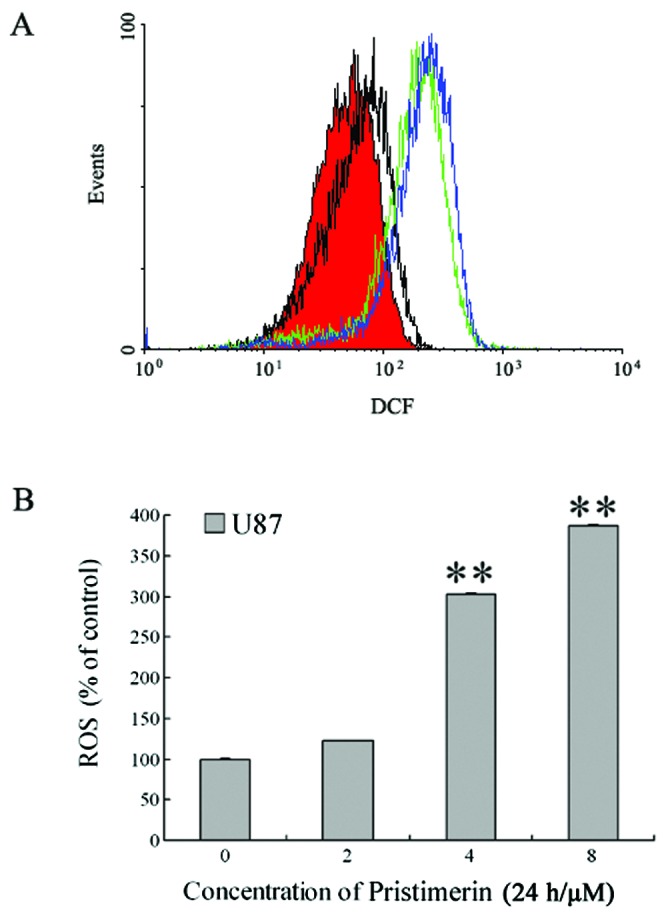
Pristimerin induced increase of ROS generation in U87 cells. After U87 cells were exposed to 2 to 8 μmol/l pristimerin for 24 h, intracellular ROS levels were analyzed by flow cytometry. Significant increases of ROS production were observed after pristimerin treatment. (A) ROS levels in U87 cells. Red area and black, green and blue lines indicate ROS levels for control and 2, 4 and 8.0 μmol/l pristimerin treatment, respectively. (B) ROS levels of U87 cells, expressed as units of mean fluorescence intensity, were calculated as percentage of control. Mean ± SD of at least three determinations. **P<0.01, versus control.
Discussion
Malignant gliomas are the most common primary tumor in the central nervous system (CNS). Even when a combination of surgery, chemotherapy and radiotherapy is used, the median survival time of patients with glioblastoma multiforme (GBM), the most malignant type of glioma, remains <1 year from diagnosis (25). Resistance to apoptosis, a characteristic of many types of cancer, particularly malignant glioma, underlies not only tumorigenesis but also the inherent resistance of cancer cells to radiotherapy and chemotherapy (26). Therefore, novel strategies are essential to improve the prognosis of patients.
Traditional Chinese medicine, used in China for thousands of years, is still widely used to prevent and cure diseases. With the development of modern science and technology, study has begun into its mechanisms, even at the molecular and genetic levels, in order to find new chemical drug entities and expand the scope of its use in treatment. In recent years, screening for active anticancer drugs found in Chinese medicine has become a hot spot in anticancer research. We hope that new and more effective anticancer drugs obtained from Chinese herbs can be developed to improve the prognosis of cancer patients. Certain plants, such as Maytenus chuchuhuasca and Maytenus laevis, have traditionally been used in the treatment of arthritis and skin cancer in South America (27). Pristimerin is a quinonemethide triterpenoid compound which has been found in various species belonging to the Celastraceae and Hippocrateaceae families. It is known that pristimerin exhibits antimicrobial, anti-inflammatory and antitumor effects (28). A number of studies have shown that pristimerin is a potent and broad-spectrum antitumor agent, with activity against a wide range of different human cancers (9–11). However, the mechanism for the antiglioma activity of pristimerin was never explored. Our experimental results showed that pristimerin displayed potent cytotoxicity to human glioma U87 cells and the IC50 was 3.94±0.80 μmol/l (Fig. 1B).
Pristimerin has recently been shown to induce apoptosis in certain cancer cells. Currently, it is still not completely clear how pristimerin induces proteasome inhibition and apoptosis. Pristimerin-triggered caspase activation was confirmed in human breast cancer cells (10). MDA-MB-231 cells treated with pristimerin showed rapid induction of apoptosis through caspase activation, which could be completely prevented by pretreatment with pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (z-VAD-fmk). In our study, apoptosis assay showed that apoptotic cells induced by pristimerin displayed condensed and fragmented nuclei by Hoechst 33258 staining (Fig. 2A). Annexin V/PI double staining assay further confirmed the results of Hoechst 33258 staining, demonstrating that the important membrane alterations relating to apoptosis in U87 cells and the apoptosis rate increased in a dose-dependent manner (Fig. 2B and C). Additionally, we showed that pristimerin induces caspase-dependent apoptosis in the human glioma U87 cells, showing that pristimerin activated caspase-9 and caspase-3, and was followed by the cleavage of PARP (Fig. 4A). Taken together, these results suggest that pristimerin is able to decrease the viability of U87 cells through the induction of caspase-dependent apoptosis.
A variety of signaling pathways may be involved in apoptosis, and the mitochondrial pathway is one of the major apoptosis pathways. Mitochondria have been shown to play a central role in the apoptotic process, since both the intrinsic pathway and the extrinsic pathway converge at the mitochondrial level and trigger mitochondrial membrane permeabilization (29,30). Release of cytochrome c and other proapoptotic proteins, including apoptosis-inducing factor, SMAC/Diablo and EndoG, from the mitochondria to the cytosol is the limiting factor in the mitochondrial pathway, and mitochondrial dysfunction has been suggested to cause the release of the above proapoptotic proteins. Recent studies showed that treatment of breast tumor cells with pristimerin resulted in a rapid release of cytochrome c from mitochondria, which preceded caspase activation and the decrease of mitochondrial membrane potential (10). This process did not depend on the Bcl-2 family (Bcl-2, Bcl-xl and Bax) protein levels and did not require translocation of Bax to the mitochondria. In the present study, treatment of glioma U87 cells with pristimerin resulted in a rapid release of cytochrome c from mitochondria, which preceded caspase activation and cell apoptosis. This process also depended on Bcl-2 family (Bcl-2, Bcl-xl and Bax) protein levels, showing that the ratio of Bcl-xl and Bax was downregulated (Fig. 4A). Taken together, our results revealed that intrinsic pathways took part in pristimerin-induced apoptosis in U87 cell lines.
In more recent years, it has become apparent that ROS play an important role during apoptosis induction (31). Numerous studies have indicated that certain anticancer agents (e.g., Adriamycin, epirubicin and daunomycin) induce apoptosis in part with the generation of ROS and the disruption of redox homeostasis (32). Intracellular production of ROS may lead directly to activation of the mitochondrial permeability transition and to loss of mitochondrial membrane potential (33). Pristimerin-induced mitochondrial cell death in cervical cancer cells was reported through ROS generation (34). However, a recent study using pristimerin in breast cancer cells convey a different viewpoint. The generation of ROS in MDA-MB-231 cells was not affected by pristimerin (10). Our research showed that pristimerin induced production of ROS and subsequent mitochondrial cell death in U87 cells (Fig. 5). We attempted to explain how pristimerin chemically and biologically influenced ROS generation. It is a known fact that most prooxidants/antioxidants behave differently depending on the experimental conditions, and it is possible that such a phenomenon also applies for pristimerin. This effect may be enhanced when cell lines of different tissue origin are used for analysis; it is known that cells vary with respect to their antioxidant status. Therefore, the exact mechanisms by which pristimerin increases the intracellular ROS level and subsequent mitochondrial cell death remain to be elucidated. These results indicate that ROS-dependent activation by pristimerin is critically required for mitochondrial dysfunction in U87 cells.
The ubiquitin-proteasome pathway is essential for many fundamental cellular processes, including cell cycle, apoptosis, angiogenesis and differentiation (35). In addition, the proteasome contributes to the pathologic state of several human diseases including cancer and AIDS, in which certain regulatory proteins are either stabilized due to decreased degradation or lost owing to accelerated degradation (36). Therefore, targeting disease-associated proteins for ubiquitination and degradation represents a promising alternative therapeutic strategy in cancer. Pristimerin is a natural analog of celastrol and also targets the proteasome. Nucleophilic susceptibility and in silico docking studies demonstrate that C6 of pristimerin is highly susceptible to a nucleophilic attack by the hydroxyl group of Thr1 of the proteasomal chymotrypsin subunit (12). EGFR-overexpressing cancer cells have been shown to activate downstream MAPK and PI3K/Akt signaling pathways. As shown in Figs. 3 and 4A, we discovered that pristimerin also inhibited growth in glioma U87 cells. Pristimerin blocked ERK1/2 phosphorylation and expression of EGFR and Akt. EGFR and Akt inhibition by pristimerin may occur through the proteasome pathway. Studies will be performed in near future to explore this further.
In summary, our findings demonstrated that pristimerin induced ROS-dependent mitochondrial cell apoptosis in human glioma cells. The mechanisms may involve inhibition of expression of EGFR and Akt protein. We conclude that pristimerin may be a leading compound for further drug development to overcome resistance in glioma patients.
Acknowledgments
This study was supported by the Scientific Research Foundation for Ph.D. of Shanxi Datong University (No. 2010-16-1) and the Natural Science Foundation of Shanxi Province (No. 2008011082-1).
References
- 1.Debatin KM. Activation of apoptosis pathways by anticancer drugs. Adv Exp Med Biol. 1999;457:237–244. doi: 10.1007/978-1-4615-4811-9_25. [DOI] [PubMed] [Google Scholar]
- 2.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 3.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 4.Gulbins E, Dreschers S, Bock J. Role of mitochondria in apoptosis. Exp Physiol. 2003;88:85–90. doi: 10.1113/eph8802503. [DOI] [PubMed] [Google Scholar]
- 5.Friesen C, Fulda S, Debatin KM. Cytotoxic drugs and the CD95 pathway. Leukemia. 1999;13:1854–1858. doi: 10.1038/sj.leu.2401333. [DOI] [PubMed] [Google Scholar]
- 6.Sharma K, Wang RX, Zhang LY, et al. Death the Fas way: regulation and pathophysiology of CD95 and its ligand. Pharmacol Ther. 2000;88:333–347. doi: 10.1016/s0163-7258(00)00096-6. [DOI] [PubMed] [Google Scholar]
- 7.Gao JM, Wu WJ, Zhang JW, Konishi Y. The dihydro-beta-agarofuran sesquiterpenoids. Nat Prod Rep. 2007;24:1153–1189. doi: 10.1039/b601473a. [DOI] [PubMed] [Google Scholar]
- 8.Brinker AM, Ma J, Lipsky PE, Raskin I. Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae) Phytochemistry. 2007;68:732–766. doi: 10.1016/j.phytochem.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salminen A, Lehtonen M, Suuronen T, Kaarniranta K, Huuskonen J. Terpenoids: natural inhibitors of NF-kappaB signaling with anti-inflammatory and anticancer potential. Cell Mol Life Sci. 2008;65:2979–2999. doi: 10.1007/s00018-008-8103-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR, Wu YC. Pristimerin induces caspase-dependent apoptosis in MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer Ther. 2005;4:1277–1285. doi: 10.1158/1535-7163.MCT-05-0027. [DOI] [PubMed] [Google Scholar]
- 11.Nagase M, Oto J, Sugiyama S, Yube K, Takaishi Y, Sakato N. Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci Biotechnol Biochem. 2003;67:1883–1887. doi: 10.1271/bbb.67.1883. [DOI] [PubMed] [Google Scholar]
- 12.Yang H, Landis-Piwowar KR, Lu D, et al. Pristimerin induces apoptosis by targeting the proteasome in prostate cancer cells. J Cell Biochem. 2008;103:234–244. doi: 10.1002/jcb.21399. [DOI] [PubMed] [Google Scholar]
- 13.Lu Z, Jin Y, Chen C, Li J, Cao Q, Pan J. Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-kappaB signaling and depleting Bcr-Abl. Mol Cancer. 2010;9:112. doi: 10.1186/1476-4598-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tiedemann RE, Schmidt J, Keats JJ, et al. Identification of a potent natural triterpenoid inhibitor of proteosome chymotrypsin-like activity and NF-kappaB with antimyeloma activity in vitro and in vivo. Blood. 2009;113:4027–4037. doi: 10.1182/blood-2008-09-179796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shao CJ, Fu J, Shi HL, Mu YG, Chen ZP. Activities of DNA-PK and Ku86, but not Ku70, may predict sensitivity to cisplatin in human gliomas. J Neurooncol. 2008;89:27–35. doi: 10.1007/s11060-008-9592-7. [DOI] [PubMed] [Google Scholar]
- 16.Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncology. 2010;12:328–340. doi: 10.1093/neuonc/nop005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan YY, Zheng LS, Zhang X, et al. Blockade of Her2/neu binding to Hsp90 by emodin azide methyl anthraquinone derivative induces proteasomal degradation of Her2/neu. Mol Pharm. 2011;8:1687–1697. doi: 10.1021/mp2000499. [DOI] [PubMed] [Google Scholar]
- 18.Yan Y, Su X, Liang Y, et al. Emodin azide methyl anthraquinone derivative triggers mitochondrial-dependent cell apoptosis involving in caspase-8-mediated Bid cleavage. Mol Cancer Ther. 2008;7:1688–1697. doi: 10.1158/1535-7163.MCT-07-2362. [DOI] [PubMed] [Google Scholar]
- 19.Zhang JY, Wu HY, Xia XK, et al. Anthracenedione derivative 1403P-3 induces apoptosis in KB and KBv200 cells via reactive oxygen species-independent mitochondrial pathway and death receptor pathway. Cancer Biol Ther. 2007;6:1413–1421. doi: 10.4161/cbt.6.9.4543. [DOI] [PubMed] [Google Scholar]
- 20.Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 21.Barry M, Heibein JA, Pinkoski MJ, et al. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol Cell Biol. 2000;20:3781–3794. doi: 10.1128/mcb.20.11.3781-3794.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavrik IN, Golks A, Baumann S, Krammer PH. Caspase-2 is activated at the CD95 death-inducing signaling complex in the course of CD95-induced apoptosis. Blood. 2006;108:559–565. doi: 10.1182/blood-2005-07-007096. [DOI] [PubMed] [Google Scholar]
- 23.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 346 Pt. 2000;3:561–576. [PMC free article] [PubMed] [Google Scholar]
- 24.Marte BM, Downward J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22:355–358. doi: 10.1016/s0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 25.Surawicz TS, Davis F, Freels S, Laws ER, Jr, Menck HR. Brain tumor survival: results from the National Cancer Data Base. J Neurooncol. 1998;40:151–160. doi: 10.1023/a:1006091608586. [DOI] [PubMed] [Google Scholar]
- 26.Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12:1395–1403. doi: 10.1634/theoncologist.12-12-1395. [DOI] [PubMed] [Google Scholar]
- 27.Shirota O, Morita H, Takeya K, Itokawa H. Cytotoxic aromatic triterpenes from Maytenus ilicifolia and Maytenus chuchuhuasca. J Nat Prod. 1994;57:1675–1681. doi: 10.1021/np50114a009. [DOI] [PubMed] [Google Scholar]
- 28.Chang FR, Hayashi K, Chen IH, et al. Antitumor agents. 228 five new agarofurans, Reissantins A–E, and cytotoxic principles from Reissantia buchananii. J Nat Prod. 2003;66:1416–1420. doi: 10.1021/np030241v. [DOI] [PubMed] [Google Scholar]
- 29.Li SS, Tang QL, Wang SH, Chen YH, Liu JJ, Yang XM. Simultaneously targeting Bcl-2 and Akt pathways reverses resistance of nasopharyngeal carcinoma to TRAIL synergistically. Tumori. 2011;97:762–770. doi: 10.1177/030089161109700614. [DOI] [PubMed] [Google Scholar]
- 30.Yoo NJ, Kim MS, Park SW, et al. Expression analysis of caspase-6, caspase-9 and BNIP3 in prostate cancer. Tumori. 2010;96:138–142. doi: 10.1177/030089161009600122. [DOI] [PubMed] [Google Scholar]
- 31.Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- 32.Xia Z, Lundgren B, Bergstrand A, DePierre JW, Nassberger L. Changes in the generation of reactive oxygen species and in mitochondrial membrane potential during apoptosis induced by the antidepressants imipramine, clomipramine, and citalopram and the effects on these changes by Bcl-2 and Bcl-X(L) Biochem Pharmacol. 1999;57:1199–1208. doi: 10.1016/s0006-2952(99)00009-x. [DOI] [PubMed] [Google Scholar]
- 33.Chan WH, Wu CC, Yu JS. Curcumin inhibits UV irradiation-induced oxidative stress and apoptotic biochemical changes in human epidermoid carcinoma A431 cells. J Cell Biochem. 2003;90:327–338. doi: 10.1002/jcb.10638. [DOI] [PubMed] [Google Scholar]
- 34.Byun JY, Kim MJ, Eum DY, et al. Reactive oxygen species-dependent activation of Bax and poly(ADP-ribose) polymerase-1 is required for mitochondrial cell death induced by triterpenoid pristimerin in human cervical cancer cells. Mol Pharmacol. 2009;76:734–744. doi: 10.1124/mol.109.056259. [DOI] [PubMed] [Google Scholar]
- 35.Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol. 2005;23:4776–4789. doi: 10.1200/JCO.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 36.Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]