

Abstract
Gefitinib, the specific inhibitor of the epidermal growth factor receptor (EGFR), may cause growth delay in cancer cell lines. Thorough understanding of the downstream cellular signaling of gefitinib will facilitate the discovery of biomarkers for predicting outcomes and monitoring anti-EGFR therapies, and provide information for key targets for therapeutic intervention. In this study, we investigated the role of transducer of erbB2.1 (TOB1) in gefitinib therapy. Using the lung carcinoma cell lines A549 and NCI-H1975, the results suggested that gefitinib might mediate cell cycle arrest in lung cancer cells at least by targeting TOB1 expression. Gefitinib treatment caused cell cycle arrest predominantly at the G1 phase, which is associated with TOB1 nuclear translocation and its interaction with cyclin D1. We also showed that knockdown of TOB1 expression by RNAi rescued lung cancer cells from gefitinib-induced cell-proliferative arrest. These results suggest that TOB1 interaction with cyclin D1 and nuclear translocation is directly involved in the gefitinib-induced anti-proliferative cell cycle arrest.
Keywords: gefitinib, transducer of erbB2.1 (TOB1), cyclin D1, lung cancer
Introduction
Lung cancers, the leading cause of cancer-related mortality in the world, accounted for 30% of all cancer mortalities in 2010 (1). Clinically, there is no curative therapy for lung cancer; all treatment modalities, including combined chemotherapy and radiotherapy or intensive high-dose chemotherapy have failed to prevent disease recurrence and progression (2).
The recent availability of tyrosine kinase inhibitors (TKI) that specifically block the phosphorylation and function of epidermal growth factor receptor (EGFR) has provided a novel therapeutic strategy. One such inhibitor is gefitinib (Iressa), a selective EGFR-TKI (3). It acts through competitive inhibition of ATP binding to the receptor tyrosine kinase domain, resulting in growth delay in cancer cell lines (4). Recent phase II/III clinical studies showed that gefitinib was well tolerated and provided antitumor activity in patients with lung cancer as well as with other types of cancer when used as a monotherapy or in combination with other therapies (5). However, the clinical data also showed that only certain patients respond to gefitinib, indicating the existence of an intrinsic or de novo resistance to this type of TKI (6). In some clinical studies, the absence of a direct association between EGFR expression levels and response to the therapy with gefitinib was noted (7). Even high EGFR expression per se was not sufficient to determine sensitivity to gefitinib, due to molecular defects in downstream signaling pathways might impair transmission of the signal and the action of the drug (8). A thorough understanding of the downstream cellular targets of gefitinib will allow the discovery of markers that serve to identify the subset of patients likely to benefit from the treatment (9).
The transducer of erbB-2.1 (TOB1) gene was initially identified as a member of the anti-proliferative TOB/BTG (transducer of erbB-2/B-cell translocation gene) protein family, which was discovered in the 1990s (10). Recent studies in regard to TOB1 showed that TOB1 contributed to the genesis, transformation and progression of various malignancies, including lung cancer (11–13). It has been reported that TOB1 was upregulated following EGF stimulation in breast cancer cells; however, the role of TOB1 in gefitinib treatment is poorly understood (14).
In the present study, we investigated the function of TOB1 in gefitinib treatment and its potential as a novel therapeutic target in gefitinib lung cancer therapy.
Materials and methods
Cell culture
The human lung carcinoma cell lines NCI-H1975 and A549 were purchased from American Type Culture Collection (Manassas, VA, USA). Cells were maintained in DMEM supplemented with 10% fetal bovine serum, L-glutamine (5 mmol/l), non-essential amino acids (5 mmol/l), penicillin (100 U/ml), and streptomycin (100 U/ml) (Invitrogen, Carlsbad, CA, USA), at 37°C in a humidified 5% CO2 atmosphere. Gefitinib (Iressa) was purchased from AstraZeneca (Macclesfield, UK) and dissolved in DMSO to make 10 mmol/l stock solution. The study was approved by the Ethics Committee of Soochow University, Jiangsu, Suzhou, China.
RNA interfering
For gene silencing, cells were transiently transfected with 50 nmol/l of the small interfering RNA (siRNA) using Lipofectamine™ RNAiMAX reagent (Invitrogen). Twenty-four hours after transfection, the cells were treated with 10 μmol/l gefitinib or DMSO vehicle. The sequences for TOB1-siRNA #1 were GCUGUAAGCCCUACCUUCATT and UGAAGGUAG GGCUUACAGCTT (5’ to 3’).
Cell cycle analysis
Cell cycle analysis was performed using propidium iodide staining. For this, cells were trypsinized, washed once with 1X phosphate-buffered saline (PBS), and then fixed in 75% ethanol for at least 4 h. Fixed cells were centrifuged and washed with 1X PBS and stained with 10 μg/ml propidium iodide in PBS containing 300 μg/ml RNase (Sigma-Aldrich, St. Louis, MO, USA) for 1 h, and then filtered with a 53 μm nylon mesh. The cell cycle distribution was calculated from 10,000 cells using ModFit LT software (Becton Dickinson, CA, USA) using FACSCalibur (Becton Dickinson).
Immunofluorescence
Cells were grown on sterile coverslips and fixed in 4% formaldehyde before being permeabilized in 0.25% Triton X-100. Coverslips were blocked in PBS containing 3% BSA, and antibody recognizing TOB1 was added at 10 μg/ml. Specific staining was visualized with a secondary antibody conjugated to FITC 488 and analyzed on a Zeiss confocal microscope.
Preparation of nuclear and cytoplasmic extracts
Following treatment, cells were collected by scraping and were washed three times with ice-cold PBS. Cells were vortexed for 5 sec in CER II, and then the cells were lysed on ice for 1 min. Nuclei were sedimented by 5 min centrifugation at 16,000 x g, and the supernatant containing the cytoplasmic fraction was snap frozen. The pellet, containing the nuclei, was resuspended in Nuclear Extraction reagent and rotated at maximum speed on a wheel at 4°C for 40 min. The samples were then centrifuged at 4°C for 5 min at 16,000 x g. The supernatant containing the nuclear fraction was snap frozen and kept at −80°C.
Western blot and immunoprecipitation (IP)/immunoblot analyses
Western blot and IP analyses were performed as previously described (15). For western blot assay, the following primary antibodies were used for immunoblotting (Santa Cruz Biotech, Santa Cruz, CA, 1:500 dilution): β-actin (C-4), TOB1 (H-18) and cyclin D1 (A-12). For the IP/western blot, the anti-TOB1 (H-18) and anti-cyclin D1 (A-12) antibodies were applied for immunoblot. The protein bands were visualized using an enhanced chemiluminescence system (Union Bioscience Corporation, Hangzhou, China) with prestained markers as molecular size standards.
Statistical analysis
The data are presented as the means and standard deviations. Statistical comparisons of the experimental results between the treated group and the control group were made using a two-tailed Student’s t-test. All statistical tests were performed using SPSS version 17.0. P≤0.05 was considered to indicate a statistically significant result.
Results
Gefitinib causes G0/G1 arrest and TOB1 overexpression in lung cancer cell lines
To examine the effect of gefitinib on cell proliferation, we analyzed the cell cycle phase distribution by flow-cytometric analysis of propidium iodide-stained cells after treatment with 10 μmol/l gefitinib for 0, 12, 24 and 48 h. As shown in Fig. 1A, gefitinib induced G0/G1 arrest in both lung cancer cell lines A549 and NCI-H1975, of which A549 cells are EGFR wild-type and NCI-H1975 are EGFR mutated. To investigate the possible effect of TOB1 in mediating cell cycle arrest response to gefitinib, we compared the expression of TOB1 as well as cyclin D1 in the two lung cancer cell lines after gefitinib treatment. The result showed that gefitinib caused a significant reduction in cyclin D1 expression (P<0.05), and an obvious increase in TOB1 protein expression (P<0.05; Fig. 1B and C). This finding suggests that TOB1 may be a candidate indicator for gefitinib activity and response.
Figure 1.

Effect of gefitinib on cell cycle distribution, TOB1 and cyclin D1 expression in two lung cancer cell lines. (A) A549 and NCI-H1975 cells were treated with 10 μmol/l gefitinib for 0, 12, 24 and 48 h, fixed in ethanol, and stained with propidium iodide, and DNA content was determined by flow cytometry. The percentage of cells in each phase is indicated. (B and C) Protein lysates were prepared from A549 and NCI-H1975 cells after treatment with 10 µmol/l gefitinib, and protein expression levels were analyzed by western blotting using specific antibodies. Quantitative analysis of TOB1 and cyclin D1 protein expression was shown after normalizing the data to β-actin. All the experiments were performed independently at least three times. Statistical analysis was performed using Student’s t-test. *P<0.05.
Gefitinib induces nuclear translocation of TOB1
To investigate further the mechanism of action of gefitinib, we next studied the subcellular localization of TOB1 in response to gefitinib treatment by confocal microscopy following staining with a TOB1-specific antibody. In the parental A549 and NCI-H1975 cells, endogenous TOB1 resided both in the cytoplasm and nucleus, whereas 48 h after gefitinib treatment, TOB1 relocated predominantly to the nucleus (Fig. 2A and B). The change in TOB1 localization was further confirmed by western blot analysis of nuclear/cytoplasmic lysates. As shown in Fig. 2C and D, treatment of A549 and NCI-H1975 cells with gefitinib resulted in a decrease in the TOB1 level in the cytoplasm and a parallel increase in TOB1 in the nucleus. Together, these results suggest that gefitinib induces TOB1 overexpression and nuclear translocation in A549 and NCI-H1975 lung cancer cells to mediate G0/G1 cell cycle arrest.
Figure 2.

Gefitinib affects nuclear translocation of TOB1 in A549 and NCI-H1975 cells. (A and B) TOB1 was visualized with a mouse monoclonal antibody followed by the addition of Alexa 488 (green)-labeled anti-mouse antisera. 4,6-diamidino-2-phenylindole (blue) was also applied to visualize the nuclei. (C and D) Nuclear and cytoplasmic lysates were prepared after treatment with 10 µmol/l gefitinib, and changes in TOB1 protein level were analyzed by western blotting. The protein expression was expressed as fold changes in band density, with β-actin and lamin as the loading control. All the experiments were performed independently at least three times.
TOB1 knockdown suppresses gefitinib-induced proliferative arrest
To certify the potential effect of TOB1 in response to gefitinib treatment, we transfected the two lung cancer cell lines with either a TOB1-specific siRNA or a nonspecific control siRNA pool and studied the proliferation of these cells after gefitinib treatment. Consistent with this, cell cycle analysis showed that silencing of TOB1 by siRNA rescued cells from gefitinib-induced proliferative arrest (Fig. 3A and B). This is evident from the increase in the fraction of cells in S and G2/M phases in TOB1-siRNA-transfected A549 and NCI-H1975 cells compared with control siRNA transfected cells. The results suggest that gefitinib mediates proliferative arrest in lung cancer cells, partly through targeting TOB1 expression.
Figure 3.

TOB1 knockdown partly rescues cell proliferation in response to gefitinib treatment. (A and B) Comparison of proliferating cells, as judged by total cells in S/G2/M phases between cells transfected with TOB1-specific or control siRNA during gefitinib treatment. Statistical evaluation was performed using Student’s t-test. *P<0.05, statistically significant differences. All the experiments were performed independently at least three times.
TOB1 interacts with cyclin D1
Western blot analysis was utilized to evaluate the expression level of cyclin D1 in TOB1-siRNA transfected A549 cells and NCI-H1975 cells compared with control siRNA transfected cells. The result showed that TOB1 knockdown slightly increased cyclin D1 expression (Fig. 4A). The potential interaction between cyclin D1 and TOB1 was examined through an IP/immunoblot assay. For the first time we confirmed the presence of cyclin D1 protein in the TOB1 IP of A549 and NCI-H1975 cells (Fig. 4B).
Figure 4.
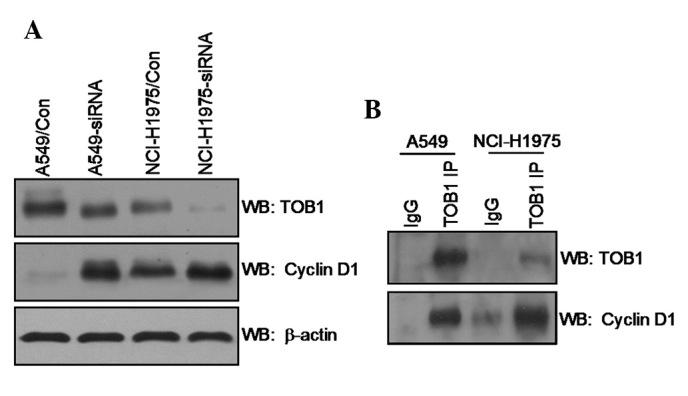
(A) TOB1 regulates cyclin D1 expression. Protein lysates were prepared from A549, A549-siRNA, NCI-H1975 and NCI-H1975-siRNA cells. (B) TOB1 forms protein-protein complex with cyclin D1 in A549 and NCI-H1975 cells. Whole cell lysate (1 mg) was immunoprecipitated with anti-TOB antibodies. The immunoprecipitated proteins were subsequently separated by SDS-PAGE, followed by immunoblotting with anti-cyclin D1 antibody, and then reprobing with TOB1 antibodies. All the experiments were performed independently at least three times.
Discussion
EGFR plays a central role in the development and progression of lung cancer. As a consequence, EGFR has emerged as a notable molecular target for lung cancer therapy, with monoclonal antibodies and small-molecule tyrosine kinase inhibitors (TKIs), such as gefitinib, being developed to specifically block EGFR signaling (16). These anti-EGFR agents have shown promising outcomes in initial preclinical and clinical settings; however, there is a high rate of de novo and acquired resistance, which limits the efficacy of such treatments (17). Several investigators have reported results from retrospective analyses of associations between EGFR gene mutations and EGFR-TKI sensitivity (18). These analyses indicated that approximately 70–80% of mutation positive cases are EGFR-TKI sensitive whereas in wild-type patients the response rate is 10–20%, but these mutations are uncommon (19).
The biological function of TOB1 in carcinogenesis became apparent when it was observed that mice lacking TOB1 spontaneously developed tumors, primarily in lung, liver and lymph nodes (20). In human lung cancers, decreased TOB1 expression and increased phosphorylation-triggered TOB1 inactivation are observed compared to that of normal epithelial tissues (21). Overexpression of TOB1 negatively regulates the cell cycle by inhibiting G1 progression (22). This evidence suggests that TOB1 is involved in the negative regulation of cell growth. In the present study, using immunofluorescence studies and western blot analysis of nuclear/cytoplasmic cellular protein fractions, we observed that endogenous TOB1 resided both in the cytoplasm and nucleus, whereas TOB1 relocated predominantly to the nucleus following gefitinib treatment. Gefitinib treatment causes cell cycle arrest predominantly at the G0/G1 phase, which is associated with TOB1 overexpression and nuclear translocation. Furthermore, silencing of the endogenous TOB1 expression by siRNA at least partially rescued A549 and NCI-H1975 cells from undergoing proliferative arrest in response to gefitinib, suggesting that the induction of TOB1 expression has a direct role in mediating the effect of gefitinib on cell proliferation.
The well-known function ascribed to cyclin D1 is the positive regulation of cell cycle distribution (23). In lung cancer cells, constitutive cyclin D1 activation maintains retinoblastoma (RB) protein in a phosphorylated state and promotes cell proliferation, thus likely initiating tumorigenesis (24). Previous studies have reported that TOB1 regulated proliferation by affecting cyclin D1 activity (25). Based on these findings, we investigated the mechanisms responsible for the inhibited therapeutic efficacy of gefitinib in lung cancer cells following TOB1 inhibition. Notably, using western blot analysis, we found that cyclin D1 was increased after TOB1 silencing. Further analysis demonstrated that TOB1 can interact with cyclin D1 in A549 and NCI-H1975 cells. These results suggest that TOB1 interacting with cyclin D1 and nuclear translocation have a direct role in mediating the gefitinib-induced proliferative arrest. Taken together, our data showed that TOB1 may be a potential target of anti-EGFR therapy in lung cancer. However, since silencing of TOB1 expression in A549 and NCI-H1975 cells did not completely rescue the cells from proliferation suppression, it is reasonable to conclude that other mechanisms may be involved in the response to gefitinib treatment.
In summary, the present study helps to define the mechanism by which gefitinib mediates cell cycle arrest in lung cancer. It also reveals that TOB1 could be a promising biomarker for monitoring anti-EGFR therapy.
Acknowledgments
This study was supported by grants from the Doctoral Fund of the Ministry of Education of China (Grant No. K512602110), the College Nature Science Foundation of Jiangsu Province (Grant No. SZ126821), the Social Development Projects of Kunshan City (Grant No. KS1224) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- 1.Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA Cancer J Clin. 2011;2:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Inamura K, Ishikawa Y. Lung cancer progression and metastasis from the prognostic point of view. Clin Exp Metastasis. 2010;6:389–397. doi: 10.1007/s10585-010-9313-4. [DOI] [PubMed] [Google Scholar]
- 3.Wakeling AE, Guy SP, Woodburn JR, et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–5754. [PubMed] [Google Scholar]
- 4.Ciardiello F. Epidermal growth factor receptor inhibitors in cancer treatment. Future Oncol. 2005;1:221–234. doi: 10.1517/14796694.1.2.221. [DOI] [PubMed] [Google Scholar]
- 5.Maione P, Gridelli C, Troiani T, et al. Combining targeted therapies and drugs with multiple targets in the treatment of NSCLC. Oncologist. 2006;11:274–284. doi: 10.1634/theoncologist.11-3-274. [DOI] [PubMed] [Google Scholar]
- 6.Polychronis A, Sinnett HD, Hadjiminas D, et al. Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growt h-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial. Lancet Oncol. 2005;6:383–391. doi: 10.1016/S1470-2045(05)70176-5. [DOI] [PubMed] [Google Scholar]
- 7.Montemurro F, Valabrega G, Aglietta M. Lapatinib: a dual inhibitor of EGFR and HER2 tyrosine kinase activity. Expert Opin Biol Ther. 2007;7:257–268. doi: 10.1517/14712598.7.2.257. [DOI] [PubMed] [Google Scholar]
- 8.Ranson M, Hammond LA, Ferry D, et al. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: results of a phase I trial. J Clin Oncol. 2002;20:2240–2250. doi: 10.1200/JCO.2002.10.112. [DOI] [PubMed] [Google Scholar]
- 9.Krol J, Francis RE, Albergaria A, et al. The transcription factor FOXO3a is a crucial cellular target of gefitinib (Iressa) in breast cancer cells. Mol Cancer Ther. 2007;6:3169–3179. doi: 10.1158/1535-7163.MCT-07-0507. [DOI] [PubMed] [Google Scholar]
- 10.Matsuda S, Kawamura-Tsuzuku J, Ohsugi M, et al. Tob, a novel protein that interacts with p185erbB2, is associated with anti-proliferative activity. Oncogene. 1996;4:705–713. [PubMed] [Google Scholar]
- 11.Winkler GS. The mammalian anti-proliferative BTG/Tob protein family. J Cell Physiol. 2010;1:66–72. doi: 10.1002/jcp.21919. [DOI] [PubMed] [Google Scholar]
- 12.Wang XM, Gao X, Zhang XH, et al. The negative cell cycle regulator, Tob (transducer of ErbB-2), is involved in motor skill learning. Biochem Biophys Res Commun. 2006;4:1023–1027. doi: 10.1016/j.bbrc.2005.12.125. [DOI] [PubMed] [Google Scholar]
- 13.Jia S, Meng A. Tob genes in development and homeostasis. Dev Dyn. 2007;4:913–921. doi: 10.1002/dvdy.21092. [DOI] [PubMed] [Google Scholar]
- 14.Helms MW, Kemming D, Contag CH, et al. TOB1 is regulated by EGF-dependent HER2 and EGFR signaling, is highly phosphorylated, and indicates poor prognosis in node-negative breast cancer. Cancer Res. 2009;12:5049–5056. doi: 10.1158/0008-5472.CAN-08-4154. [DOI] [PubMed] [Google Scholar]
- 15.Jiao Y, Sun KK, Zhao L, et al. Suppression of human lung cancer cell proliferation and metastasis in vitro by the transducer of ErbB-2.1 (TOB1) Acta Pharmacol Sin. 2012;33:250–260. doi: 10.1038/aps.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Astsaturov I, Cohen RB, Harari P. Targeting epidermal growth factor receptor signaling in the treatment of head and neck cancer. Expert Rev Anticancer Ther. 2006;6:1179–1193. doi: 10.1586/14737140.6.9.1179. [DOI] [PubMed] [Google Scholar]
- 17.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12:5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 18.Johnston JB, Navaratnam S, Pitz MW, et al. Targeting the EGFR pathway for cancer therapy. Curr Med Chem. 2006;13:3483–3492. doi: 10.2174/092986706779026174. [DOI] [PubMed] [Google Scholar]
- 19.Uchida A, Hirano S, Kitao H, et al. Activation of downstream epidermal growth factor receptor (EGFR) signaling provides gefitinib-resistance in cells carrying EGFR mutation. Cancer Sci. 2007;98:357–363. doi: 10.1111/j.1349-7006.2007.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwanaga K, Sueoka N, Sato A, et al. Alteration of expression or phosphorylation status of tob, a novel tumor suppressor gene product, is an early event in lung cancer. Cancer Lett. 2003;1:71–79. doi: 10.1016/j.canlet.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 21.Ito Y, Suzuki T, Yoshida H, et al. Phosphorylation and inactivation of Tob contributes to the progression of papillary carcinoma of the thyroid. Cancer Lett. 2005;2:237–242. doi: 10.1016/j.canlet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 22.Yanagie H, Tanabe T, Sumimoto H, et al. Tumor growth suppression by adenovirus-mediated introduction of a cell-growth-suppressing gene tob in a pancreatic cancer model. Biomed Pharmacother. 2009;4:275–286. doi: 10.1016/j.biopha.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 24.Felsher DW. Cancer revoked: oncogenes as therapeutic targets. Nat Rev Cancer. 2003;3:375–380. doi: 10.1038/nrc1070. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, K-Tsuzuku J, Ajima R, et al. Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras-mediated cell proliferation and transformation. Genes Dev. 2002;11:1356–1370. doi: 10.1101/gad.962802. [DOI] [PMC free article] [PubMed] [Google Scholar]