

Abstract
The prediction of response or severe toxicity and therapy individualisation are extremely important in cancer chemotherapy. There are few tools to predict chemoresponse or toxicity in cancer patients. We investigated the correlation between the induction and repair of DNA double-strand breaks (DSBs) using constant-field gel electrophoresis (CFGE) and evaluating cell cycle progression and the sensitivity of four cancer cell lines to 5-fluorouracil (5FU). Using a sulphorhodamine-B assay, colon carcinoma cells (HCT116) were found to be the most sensitive to 5FU, followed by liver carcinoma cells (HepG2) and breast carcinoma cells (MCF-7). Cervical carcinoma cells (HeLa) were the most resistant. As measured by CFGE, DSB induction, but not residual DSBs, exhibited a significant correlation with the sensitivity of the cell lines to 5FU. Flow cytometric cell cycle analysis revealed that 14% of HCT116 or HepG2 cells and 2% of MCF-7 cells shifted to sub-G1 phase after a 96-h incubation with 5FU. Another 5FU-induced cell cycle change in HCT116, HepG2 and MCF-7 cells was the mild arrest of cells in G1 and/or G2/M phases of the cell cycle. In addition, 5FU treatment resulted in the accumulation of HeLa cells in the S and G2/M phases. Determination of Fas ligand (Fas-L) and caspase 9 as representative markers for the extrinsic and intrinsic pathways of apoptosis, respectively, revealed that 5FU-induced apoptosis in HCT116 and HepG2 results from the expression of Fas-L (extrinsic pathway). Therefore, the induction of DNA DSBs by 5FU, detected using CFGE, and the induction of apoptosis are candidate predictive markers that may distinguish cancer cells which are likely to benefit from 5FU treatment and the measurement of DSBs using CFGE may aid the prediction of clinical outcome.
Keywords: 5-fluorouracil, drug resistance, constant-field gel electrophoresis, induction of double-strand breaks
Introduction
For several decades, chemotherapy regimens based on the drug 5-fluorouracil (5FU) have been part of the treatment for high-risk stage II or III colon cancer (1).
In addition, 5FU is used in combination with other chemotherapy drugs to treat any stage of breast, ovarian, colon, head and neck and liver cancers. In mammalian cells, 5FU is converted to fluorodeoxyuridine monophosphate (FdUMP), which forms a stable complex with thymidylate synthase (TS) and thus inhibits deoxythymidine monophosphate (dTMP) production. dTMP is essential for DNA replication and repair and its depletion therefore causes cytotoxicity (2,3). Another mechanism of 5FU-induced cytotoxicity is its mis-incorporation into RNA and DNA in place of uracil or thymine. The interference with the normal biosynthesis or function of nucleic acids is therefore another possible mechanism of action for 5FU (4).
Understanding the mechanism of action of 5FU has led to the development of strategies that may increase its anticancer activity. Despite these advances, drug resistance remains a significant limitation to the clinical use of 5FU (5,6).
Resistance to 5FU occurs due to various causes, including alteration of drug influx and efflux, enhancement of drug inactivation and mutation of the drug target (7). A high level of expression of TS (8), increased activity of deoxyuridine triphosphatase (9), methylation of the MLH1 gene (10) and overexpression of Bcl-2, Bcl-XL (11,12) and Mcl-1 (13) proteins have all been reported to lead to resistance to 5FU, suggesting that multiple factors contribute to 5FU resistance (14,15).
Emerging technologies, such as DNA microarray profiling, have the potential to identify novel genes that are involved in mediating resistance to 5FU (16).
Alternative measures of sensitivity have been developed using different DNA damage assays, including the constant-field gel electrophoresis (CFGE), graded-field gel electrophoresis (CFGE), pulsed-field gel electrophoresis (PFGE) and the immuno-fluorescence or flow cytometric measurement of γ-H2AX techniques. These methods are used to quantify the apparent number of DNA double-strand breaks (DSBs) as well as DSB rejoining (17–19). DSBs are the most deleterious DNA lesions occurring in cells following treatment with chemotherapy and/or irradiation. The treatment of cancer cells with 5FU leads to indirect DSBs due to the misincorpration of FdUMP into DNA (20,21). There are three levels at which DSBs lead to mitotic cell death: initial induction of DSBs, the speed and efficiency of repair and fidelity of DSB repair. Also, the treatment of tumour cells with chemotherapy such as 5FU leads to omnipresent DNA damaging insults (e.g. DSBs) which activate the cellular DNA-damage response (DDR) machinery. This activation of the DDR network leads to cell cycle arrest, activation of DNA repair mechanisms or initiation of apoptosis if the damage is not repaired (22,23).
This study was designed to test the possibility of using parameters of the DDR machinery for determining the sensitivity of different cancer cells to 5FU. The main objective was to evaluate the correlation between chemosensitivity to 5FU and induction and repair of DSBs using CFGE, cell cycle progression and induction of cell death (apoptosis) to select predictive molecular markers for the measurement of the sensitivity of cancer cells to 5FU.
Materials and methods
Cell lines and culture conditions
Four different human cancer cell lines were used in this study: breast carcinoma (MCF-7), cervical carcinoma (HeLa), colon carcinoma (HCT116) and liver carcinoma (HepG2). These cell lines were selected to have different sensitivities to 5FU, as determined by a preliminary experiment using the multiple cell lines available in our lab. They were obtained frozen in liquid nitrogen from University Hospital (Hamburg, Germany) and the American Type Culture Collection (Manassas, VA, USA) and were maintained in the Cancer Biology Department, National Cancer Institute (Cairo, Egypt) by serial sub-culturing. The cells were grown as a monolayer culture in RPMI-1640 medium or DMEM supplemented with 10% foetal bovine serum, 100 U/ml penicillin and 2 mg/ml streptomycin. Mycoplasma was tested at 3-month intervals.
Drugs and chemicals
5FU (Calbiochem, Darmstadt, Germany) was dissolved in dimethyl sulfoxide (1 mmol/l stock solution) and was kept frozen in aliquots at −20°C.
Chemosensitivity assay
Cytotoxicity was determined using the sulphorhodamine-B (SRB) method as previously described by El-Awady et al(24). Cells were seeded in 96-well microtitre plates at a concentration of 5×104–105 cells/well in supplemented RPMI-1640 medium or DMEM. After 24 h, cells were incubated for 48 h with either drug-free medium (control cells) or increasing concentrations of the anticancer drug 5FU (0.05–50 μg/ml). Following the 48-h treatment, the cells were fixed with 50% trichloroacetic acid for 1 h at 4°C. The wells were washed five times with water, stained for 30 min at room temperature with 0.4% SRB dissolved in 1% acetic acid and then washed four times with 1% acetic acid. The plates were air-dried and the dye was solubilised with 100 ml/well of 10 mM Tris base (pH 10.5) for 5 min on a shaker at 1,600 rpm. The optical density (OD) of each well was measured spectrophotometrically at 564 nm with an ELISA microplate reader (Meter Tech. S960, Warminster, PA, USA). The IC60 values were calculated using sigmoidal concentration-response curve fitting models (Graph Pad, Prism Software, San Diego, CA, USA). IC60 showed the largest difference in the sensitivity of the four cell lines to 5FU; accordingly, IC60 values were selected for further experiments in our study.
CFGE of DNA
DSB induction and repair were measured by CFGE using exponentially growing cells as described by El-Awady et al(22). The cells were treated with 0.02–100 μg/ml 5FU at room temperature. Following treatment, the cells were incubated for 48 h at 37°C. The cells were detached with trypsin and the resulting cell suspension was mixed with an equal amount of 1.6% low melting point agarose (Bio-Rad, Munich, Germany), pipetted into a 180-μl mould and left on ice to solidify and form agarose cell plugs. The plugs were incubated in lysis buffer [0.4 M ethylene diamine tetraacetic acid, (EDTA), 2% N-lauryl sarcosine and 1 mg/ml proteinase K; Sigma, Deisenhofen, Germany] for 30 min on ice and then overnight at 37°C. The plugs were washed three times in Tris-EDTA buffer and a portion of the plug containing ∼105 cells was inserted into a preformed 0.8% agarose gel. The gel was then covered with a thin overlayer of 0.8% agarose to avoid light fraction artefacts upon optical imaging. Electrophoresis was performed for 36 h at 0.6 V/cm in 0.5X Tris-Borate-EDTA buffer (45 mM Tris base, 45 mM boric acid, 2 mM EDTA) in a conventional apparatus (Subcell, Bio-Rad). Following the completion of electrophoresis, the gel was stained overnight in 0.5 μg/ml ethidium bromide solution and destained overnight in distilled water. The fraction of DNA released (FDR) after each treatment was determined using a gel documentation system (Biometra, Gottingen, Germany).
The FDR into the gel, which corresponds to fragmented DNA, was calculated according to the equation:
where FDRrel is the FDR outside the well and FDRplug is the fraction of DNA remaining in the well. The FDR was calculated using one-site binding curve fitting models (Graph Pad, Prism Software).
Residual DNA DSBs
Residual DSBs were measured by CFGE using exponentially growing cells as described by El-Awady et al(22). Cells were treated with 100 μg/ml 5FU for 24 h at 37°C. The selection of this concentration was based on a preliminary experiment to measure residual DSBs after treatment with different concentrations of 5FU. It was found that 100 μg/ml 5FU is the lowest concentration that provides measurable bands after the specified repair time. The drug was then removed by washing the cells three times with drug-free medium followed by incubating the cells in drug-free medium at 37°C for 24 h to allow the repair of 5FU-induced DSBs. Monolayer cells were detached with trypsin and the resulting cell suspension was mixed with an equal amount of 1.6% low melting point agarose (Bio-Rad), pipetted into a 180-μl mould and allowed to solidify on ice. The plugs were incubated overnight in lysis buffer (0.4 M EDTA, 2% N-lauryl sarcosine and 1 mg/ml proteinase K; Sigma) and the DNA fragments were separated by electrophoresis on a 0.8% agarose gel using a constant field (0.6 V/cm, 36 h). The gel was stained with ethidium bromide and photographed under UV illumination. The FDR, which corresponds to unrepaired (residual) DNA fragments, was measured using a gel documentation system (Biometra) and was divided by the corresponding FDR obtained from cells treated with 100 μg/ml 5FU for 48 h without repair (obtained from the induction curve) to determine the percentage of DNA DSBs.
Cell cycle distribution analysis
The evaluation of cell cycle distribution was carried out according to the method of El-Awady et al(21). Cells were plated at a density of 5×106 cells/ml in supplemented medium. After 24 h, the medium was replaced with fresh medium containing the IC60 concentration of 5FU (μg/ml), with the exception of the control cells. All flasks were incubated at 37°C. After various time intervals (48, 72 and 96 h), the adherent cells were detached with trypsinisation, washed twice with phosphate-buffered saline (PBS) and fixed with 70% (v/v) ethanol. The sample was concentrated by removing the ethanol and was resuspended in 500 μl PBS and 500 μl RNase solution. The mixture was incubated at 37°C for 30 min. Propidium iodide solution (50 μl, 1 mg/ml) was added and flow cytometric DNA ploidy analysis was performed by acquiring a minimum of 200,000 nuclei. Cell cycle analysis was performed using the Coulter Epics XL software package (Beckman Coulter Corp., Miami, FL, USA). The percentages of cells with DNA content corresponding to sub-G1, G1, S and G2/M phases were computed by the planimetry of the histogram and then compared for the four cell lines.
Measurement of Fas ligand (Fas-L)
Fas-L-dependent cell death was evaluated using a colourimetric activity assay kit (Abcam, San Francisco, CA, USA). The assay was performed according to the manufacturer’s instructions. Cells were seeded in 96-well microtitre plates at a concentration of 2×103 cells/well in supplemented RPMI-1640 medium or DMEM. After 24 h, the cells were incubated with either drug-free medium (control cells) or with the IC60 concentration of 5FU for each cell line. Cells were incubated at 37°C in a 5% CO2 incubator. After various time intervals (48 or 72 h), the supernatants were collected and centrifuged for 10 min at 2,000 rpm and then stored at −70°C until use. The standards, samples and untreated controls (100 μl) were added to microwell strips, incubated for 2 h at room temperature and washed three times with distilled water. Diluted biotinylated anti-Fas-L antibody was added to each well, incubated for 1 h at room temperature and washed three times with distilled water. Horseradish peroxidase-conjugated streptavidin was added to the wells and the plates were incubated for 20 min at room temperature. After incubation, the wells were washed three times with distilled water. TMB then was added and the plates were incubated for 15–20 min for colour development. Stop reagent (H2SO4) was added to each well and the optical density (OD) of each well was measured spectrophotometrically at 450 nm with an ELISA microplate reader (Meter Tech. S960).
Measurement of caspase 9 activity
Caspase 9 activity in the control and 5FU-treated cells was measured colourimetrically using the caspase assay system purchased from BioVision (San Francisco, CA, USA). The assay was performed according to the manufacturer’s instructions. Briefly, the cells were seeded in T-175 (175 cm2) flasks and treated after 24 h with the corresponding IC60 concentration of 5FU. After various time intervals (48 or 72 h), the cells were collected by trypsinisation and the cell density was adjusted to 106 cells/ml. Cell pellets were kept on ice, washed once with ice-cold PBS and re-suspended in cell lysis buffer (supplied with the kit) at a concentration of 108 cells/ml. The cells were lysed by freezing and thawing and incubated on ice for 15 min. The supernatant was collected by centrifugation at 4°C. This cell extract was used to measure caspase activity by mixing in a 96-multiwell plate with the kit reagents (caspase assay buffer, DMSO, DTT and DEVD-pNA substrate). The plates were incubated at 37°C for 4 h and the absorbance of the wells was measured at 405 nm in an ELISA microplate reader (Meter Tech. S960) and the caspase activity was calculated.
Statistical analysis
Each experiment was repeated three times and the data are presented as the mean ± SEM. Multiple comparisons were carried out using a one-way analysis of variance (ANOVA) followed by LSD (least significant difference) for post hoc analysis. P<0.05 was considered to indicate a statistically significant result. Statistical analysis, data fitting and graphics were performed by the Prism computer program (GraphPad Software).
Results
Sensitivity of the cancer cell lines to 5FU
The sensitivity of the four cell lines to different concentrations of 5FU was determined by the SRB assay. Cell viability was expressed as the percentage of surviving treated cells compared with that of control cells. As shown in Fig. 1, 5FU reduced the survival of the four cell lines in a concentration-dependent manner. The colon carcinoma (HCT116) cells were the most sensitive, with IC60 approaching 2.2±0.03 μg/ml, whereas the other cell lines (HepG2, MCF-7 and HeLa) were less sensitive, with IC60 7±0.2, 24.5±0.6 and 50±1.30 μg/ml, respectively (Table I). This indicates that the HCT116 cells were 3, 11 and 23 times more sensitive to 5FU than the other three cell lines, respectively. The difference in sensitivity among the four cell lines was especially apparent at the highest 5FU concentration (50 μg/ml), which reduced the survival of HCT116 cells to 35% of that of the control untreated cells. The same 5FU concentration reduced the survival of the HepG2, MCF-7 and HeLa cells to 49, 55 and 60% of that of the control untreated cells, respectively.
Figure 1.
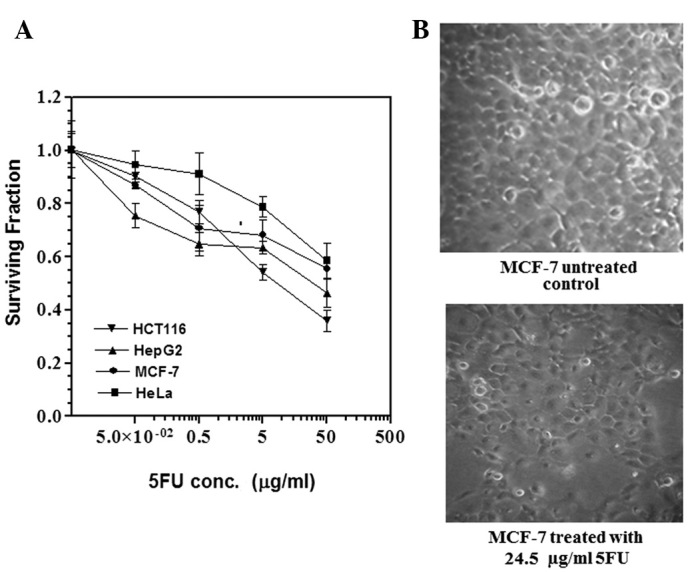
(A) 5FU sensitivity of four cancer cell lines. HCT116, HepG2, MCF-7 and HeLa were cultured in 96-well plates. After 24 h, the cells were treated with different concentrations of 5FU. After 48 h of exposure to the drug, the cells were fixed, stained with sulphorhodamine-B and the optical density (OD) was measured spectrophotometrically at 564 nm. Each point is the mean ± SEM of three separate experiments. Survival fraction = OD564 of drug-treated sample/OD564 of untreated control sample. aStatistical significance compared with the control using an unpaired t-test, P<0.01. (B) Viability of MCF-7 cells after treatment with a fixed 5FU concentration. MCF-7 cells were cultured in 75 cm2 tissue culture flasks and incubated for 48 h with either drug-free medium (control) or medium containing 24.5±0.6 μg/ml 5FU (IC60 for the MCF-7 cell line). Examination of the cells using an inverted microscope revealed that 40% of MCF-7 cells died compared with control untreated cells. 5FU, 5-fluorouracil; IC60, 5FU concentration that results in 40% cell death compared with control cell growth.
Table I.
Sensitivity of the four cancer cell lines to 5FU expressed as IC60 values, p53 and MMR genes status.
| Cell line | p53 status | MMR status | IC60 (μg/ml) | Ref. |
|---|---|---|---|---|
| HCT116 | WT | Deficient | 2.2±0.30 | (25) |
| HepG2 | WT | Proficient | 7±0.20 | (26) |
| MCF-7 | WT | Proficient | 24.5±0.60 | (27) |
| HeLa | WT | Proficient | 50±1.30 | (28) |
p53, tumour suppressor 53 gene; MMR, DNA mismatch repair gene; WT, wild-type; IC60, 5FU concentration that results in 40% cell death compared with control cell growth.
Induction of DNA DSBs in the cancer cell lines by 5FU
The four cell lines were treated with different 5FU concentrations (0.2–100 μg/ml) for 48 h. The intensity of the DNA bands was quantified and the fraction of DNA released at each drug concentration is presented in Fig. 2A. The HCT116 cells showed the highest FDR (representing DNA DSBs) at all concentrations tested, followed by the HepG2 liver cancer cells, whereas the other two cell lines, MCF-7 and HeLa, showed low FDR at the low 5FU concentrations (0.2 and 2 μg/ml) but relatively high FDR at the high 5FU concentrations. Fig. 2B shows the correlation between cellular sensitivity to 5FU (expressed as IC60) and induction of DNA damage by 5FU (expressed as fraction of DNA released at 2 μg/ml 5FU). As shown in Fig. 2, a significant positive correlation (P<0.05) exists between the two parameters, such that cells which are most sensitive to 5FU (with low IC60) showed a high level of 5FU-induced DNA damage and vice versa.
Figure 2.
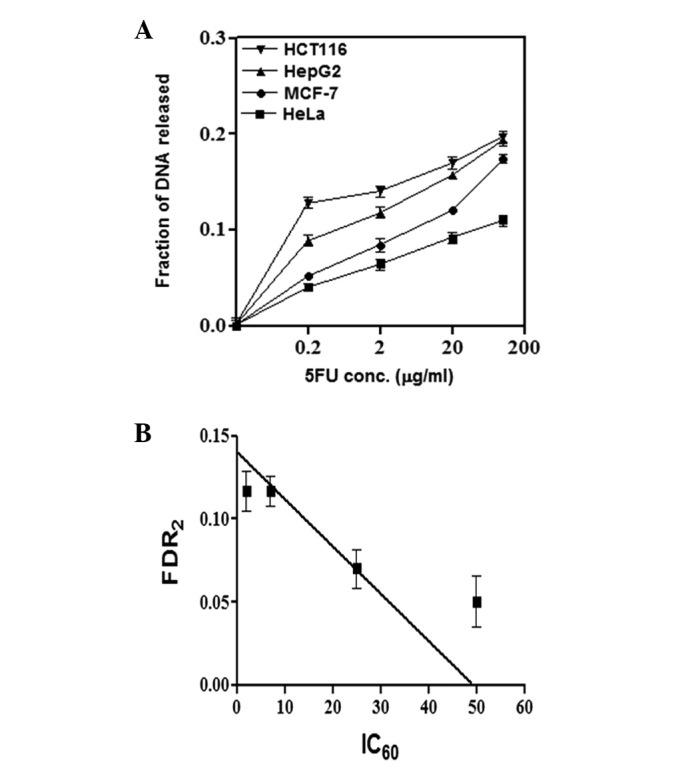
Induction of DNA double-strand breaks by 5FU and correlation with chemosensitivity. Cells were exposed for 48 h to either drug-free medium (control) or medium containing different concentrations of 5FU (0.2, 2, 20 or 100 μg/ml). Cells were collected and embedded in agarose plugs which were incubated overnight in lysis buffer (0.4 M EDTA, 2% N-lauryl sarcosine and 1 mg/ml proteinase K) and the DNA fragments were separated by electrophoresis in 0.8% agarose gel using a constant field (0.6 V/cm, 36 h). The band intensities from the gels were quantified and the FDR, which corresponds to fragmented DNA, was measured using a gel documentation system. (A) A curve was constructed for the FDR at each 5FU concentration. (B) The correlation between FDR2 and the 5FU IC60 value of the four cell lines was determined. The FDR was calculated according to the following equation: FDR = FDRrel / (FDRplug + FDRrel), where FDRrel is the FDR outside the well and FDRplug is the fraction of DNA remaining in the well. The data are the results of three independent experiments. FDR2, FDR following treatment with 2 μg/ml 5FU; 5FU, 5-fluorouracil; FDR, fraction of DNA released; IC60, 5FU concentration that results in 40% cell death compared with control cell growth.
DNA repair efficiency and correlation with 5FU sensitivity
To measure DNA repair efficiency in the four cancer cell lines, cells were treated with 100 μg/ml 5FU for 24 h and then incubated in fresh drug-free medium for another 24 h to allow the repair of 5FU-induced DNA damage. Fig. 3A shows the UV trans-illuminated gels of the four cancer cell lines showing the residual DNA damage after 24 h of repair. The intensity of the DNA bands was measured and the fraction of non-repaired (residual) DNA damage in the four cell lines was calculated by dividing the FDR from the cells allowed to repair by the corresponding FDR from cells treated with the same dose of 5FU without allowing time for repair. As shown in Fig. 3B, MCF-7 and HCT116 cells have higher residual DNA damage than HepG2 and HeLa cells.
Figure 3.
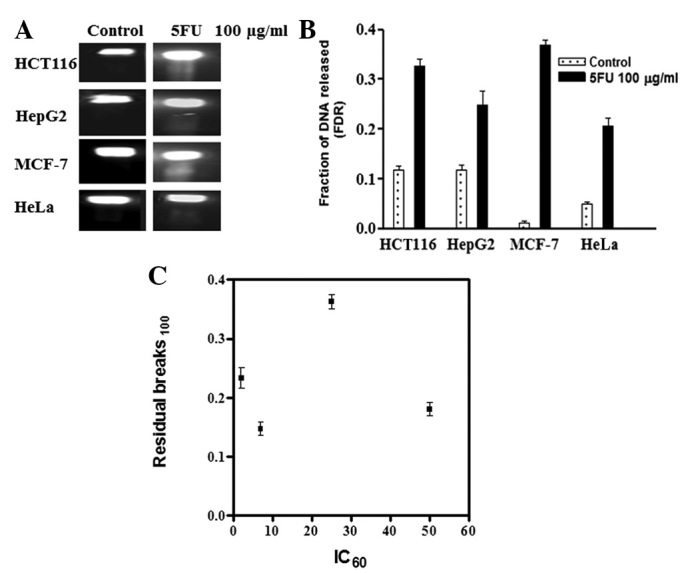
DNA repair efficiency and correlation with 5FU sensitivity. Cells were treated with 100 μg/ml 5FU for 24 h and then incubated in fresh drug-free medium for an additional 24 h to allow the repair of 5FU-induced DNA damage. Cells were collected and embedded in agarose plugs which were incubated overnight in lysis buffer (0.4 M EDTA, 2% N-lauryl sarcosine and 1 mg/ml proteinase K) and the DNA fragments were separated by electrophoresis in 0.8% agarose gel using a constant field (0.6 V/cm, 36 h). (A) The gel was stained in ethidium bromide and photographed under UV illumination. (B) FDR, which corresponds to unrepaired (residual) DNA fragments, in the four cell lines was measured using a gel documentation system. (C) Correlation between the unrepaired (residual) DNA fragments and the 5FU IC60 value of the four cell lines was determined. FDR100, FDR after treatment with 100 μg/ml 5FU; 5FU, 5-fluorouracil; FDR, fraction of DNA released; IC60, 5FU concentration that results in 40% cell death when compared with control cell growth.
To test whether differences in DNA repair efficiency explains sensitivity differences in the four cell lines, we tested the correlation between cellular sensitivity to 5FU (expressed as IC60) and the residual DNA damage. As shown in Fig. 3C, although the most sensitive cells (HCT116) showed a higher fraction of residual DNA damage, the correlation was not significant (P>0.05). This is mainly due to the MCF-7 cells, which had the highest level of residual DNA damage but were less sensitive to 5FU than the HCT116 cells.
Evaluation of cell cycle distribution after 5FU treatment
To examine other parameters that may explain the differences in sensitivity to 5FU, we measured changes in cell cycle distribution after treating the four cell lines with 5FU. DNA histograms from cells incubated with 5FU (IC60) for various time intervals (48, 72 and 96 h) revealed induction of apoptosis in HCT116 and HepG2 cells at all tested time points. This is indicated by the appearance of ∼5, 6 and 14% sub-G1 cells in the DNA histogram of HCT116 and HepG2 cells at 48, 72 and 96 h, respectively (Fig. 4A and B).
Figure 4.
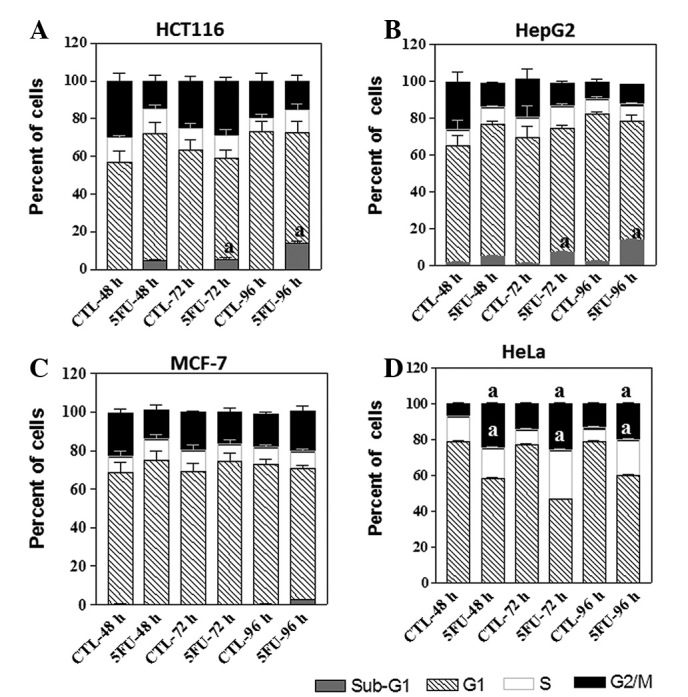
Cell cycle distribution analysis of four cancer cell lines after treatment with 5FU. HCT116, HepG2, MCF-7 and HeLa cells were treated with the IC60 of each cell line (2.2, 7.0, 24.5 and 50 μg/ml 5FU, respectively) for different time intervals (48, 72 or 96 h). The cells (control and 5FU-treated) were collected by trypsinisation at the indicated time, washed, incubated with RNase and treated with propidium iodide; the DNA content was then measured by flow cytometry. (A and B) 5FU treatment led to induction of apoptosis in both HCT116 and HepG2 cells as indicated by the appearance of sub-G1 cells in the flow cytometric chart and mild arrest at G1-phase of the cell cycle after 48 h of drug treatment. (C) MCF-7 cells showed mild arrest in both G1 after 48 h and G2/M phase after 96 h of drug treatment. (D) HeLa cells blocked in both S and G2/M phases after 5FU treatment for different time intervals (48, 72 or 96 h). Graphs show the percentage of cells at each cell cycle phase at the indicated time intervals. The data are from three experiments. aStatistical significance compared with the control group, P<0.05. 5FU, 5-fluorouracil; IC60, 5FU concentration that results in 40% cell death compared with control cell growth.
The percentage of sub-G1 cells in the histogram of MCF-7 cells after treatment with 5FU was low compared with the HCT116 and HepG2 cells, ∼2% sub-G1 MCF-7 cells were found after 96 h of 5FU treatment (Fig. 4C). Another 5FU-induced cell cycle change in HCT116, HepG2 and MCF-7 cells was a mild arrest of cells in G1 and/or G2 phases of the cell cycle, especially after 48 and 72 h of 5FU treatment (Fig. 4A, B and C). In addition, 5FU treatment resulted in the accumulation of HeLa cells in both S and G2/M phases after different time intervals of 5FU treatments (48, 72 and 96 h; Fig. 4D).
Fas-L and caspase 9 activity after 5FU treatment
To understand the apoptotic pathways stimulated by 5FU, we measured the activity of the Fas-L and caspase 9 proteins in the four cell lines after treatment with 5FU (IC60). The two proteins represent two different apoptotic pathways (Fas-L is involved in the extrinsic pathway whereas caspase 9 is involved in the intrinsic pathway). As shown in Fig. 5A, no increase (over the control) in the level of caspase 9 was detected in any of the four cancer cell lines following treatment with 5FU. On the other hand, the level of Fas-L in HCT116 and HepG2 cells was increased ∼2–3 times over the control cells after treatment with 5FU for 48 and 72 h. However, in MCF-7 and HeLa cells no increase in the level of Fas-L over the control cells was detected at the two time points (48 and 72 h; Fig. 5B).
Figure 5.
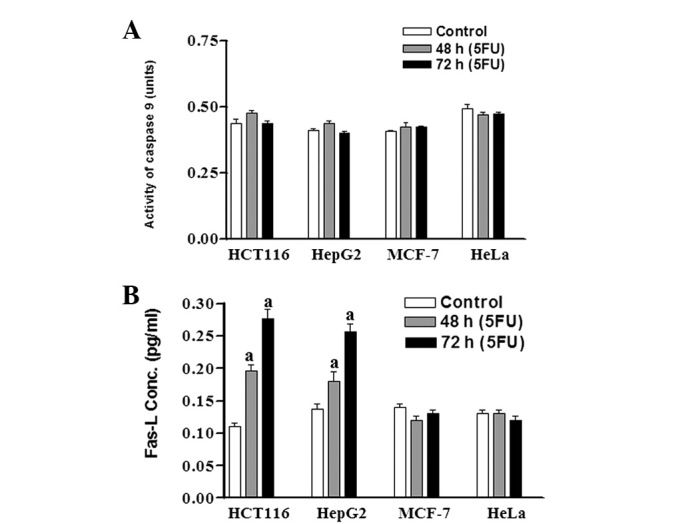
Caspase 9 and Fas-L activities in the four cancer cell lines after treatment with 5FU. HCT116, HepG2, MCF-7 and HeLa cells were treated with IC60 for each cell line (2.2, 7.0, 24.5 and 50 μg/ml 5FU, respectively). Control and 5FU-treated cells were collected after 48 and 72 h and cell lysates were prepared. Fas-L and caspase 9 activities in the four cell lines served as markers for the extrinsic and intrinsic pathways of apoptosis induction, respectively, and were monitored colourimetrically using the CaspASE assay system. Each point is the mean ± SEM of three separate experiments. aStatistical significance compared with the control group, P<0.01. Fas-L, Fas ligand; 5FU, 5-fluorouracil; IC60, 5FU concentration that results in 40% cell death compared with control cell growth.
Discussion
The use of 5FU and other fluorinated pyrimidines for different types of malignancies started approximately 50 years ago (25). To date, their exact mechanism of action has not been completely understood, making it difficult to establish predictive markers to aid the identification of tumours with intrinsic or acquired drug resistance.
In mammalian cells, 5FU is converted into FdUMP which inhibits the TS enzyme, leading to nucleotide pool imbalance and/or uridine incorporation into DNA (26). 5FU is also converted to fluoronucleotides that are incorporated into DNA and RNA, modifying their function and stability. These fluoronucleotides and uridine are recognised as false nucleotides in the DNA and stimulate DNA repair proteins to eliminate these lesions and restore DNA integrity.
Different predictive markers for the response to fluoropyrimidines have been tested during the last 20 years (27) and mismatch repair (MMR) genes were the only marker reported to have some correlation with cellular sensitivity to this class of anticancer drugs (28–30). Most of these studies were designed based on the evidence that the effects of fluoropyrimidines are primarily due to mis-incorporation into nucleic acids and/or stopping DNA synthesis by depleting the cellular thymine content. The recent demonstration that 5FU may induce DNA DSB opens the avenue for new predictive testing for cellular response to fluoropyrimidines (21).
Studies have shown that 5FU induces indirect DNA DSBs in tumour cells, probably during the repair and elimination of the false fluoronucleotides or FdUTP incorporated into the DNA (21,31). Among the DNA lesions induced by anticancer agents, DSBs are the most deleterious in terms of cell death (32). They stimulate many cellular proteins that signal these lesions to effector proteins, resulting in cell cycle delay or arrest, repair of the lesion or cell death. If left unrepaired or incorrectly repaired, DSBs lead to cell death or tumourigenesis (33).
The aim of the present study was to evaluate new parameters that may predict the sensitivity of cancer cells to fluorinated pyrimidines which may aid the individualisation of this type of therapy. We investigated the induction and repair of DNA DSBs using CFGE in four cancer cell lines after treatment with 5FU. In addition, cell cycle changes and induction of apoptosis were also investigated.
In the present study, we found that the HCT116 cell line with the lowest IC60 (i.e the most sensitive to 5FU) was less sensitive to 5FU than MCF-7 and HepG2 at low 5FU concentrations (up to 0.5 μg/ml) but more sensitive at the higher 5FU concentrations. We suggest that at low 5FU concentrations, the number of mis-incroporated false bases (mismatches) is low and the cells tolerate this low level of mismatches and continue cycling and proliferation, but at higher 5FU concentrations, the number of mismatches increases, stimulating different repair mechanisms (MMR, BER and NER) that remove these mismatches and lead to the generation of DSBs which are lethal to the cells. Although the HCT116 cells are MMR-deficient and have intact p53, they are capable of removing these mismatches using the p53-regulated BER and NER and accumulating the highest number of DSBs, which may account for their high sensitivity to 5FU. A similar finding was reported by Adamsen et al(34) who suggested that BER (or NER) is sufficient to repair the bolus-5FU-induced DNA damage (FdUTP/dUTP mis-incorporation) in the absence of functional MMR in the TP53-proficient HCT116 cell line. In addition, Li et al(35) have shown that MMR-deficient cells incorporate higher levels of fluoropyrimidine metabolites than MMR-proficient cells.
By contrast, the HeLa cells, which are proficient in MMR but do not express p53, accumulated the lowest number of DSBs at all the tested 5FU concentrations, confirming the assumption that p53 is more important than MMR in removing the fluoropyrimidines and the FdUTP mismatches and results in more DSBs. The other two cell lines (MCF-7 and HepG2) accumulated intermediate number of DSBs, although they are MMR proficient and express wild-type p53. We speculate that competition between p53-regulated BER and MMR may decrease the efficiency of BER in removing mismatches, thereby decreasing the number of DSBs.
Differences between the four cell lines in the conversion of 5FU to its active metabolites and/or its catabolism by dihydropyrimidine dehydrogenase to inactive metabolites is another possible explanation for the different number of 5FU-induced DSBs in the four cell lines at the same drug concentration [reviewed by Koopman et al(27) and Li et al(35)].
The significant positive correlation (P<0.05) reported in the present study between sensitivity to 5FU and induction of DSBs measured by CFGE in the four tested cell lines is a promising novel finding for the use of DSB induction as a predictive marker for the response of tumours to fluoropyrimidine therapy. This finding needs to be confirmed using clinical samples from cancer patients.
Understanding the repair of lesions induced by genotoxic anticancer agents should facilitate the identification of predictive markers for response prediction and aid the identification of tumours with intrinsic or acquired drug resistance. Using CFGE, we studied the efficiency of the four cell lines in repairing 5FU-induced DSBs by measuring the number of DSBs left unrepaired 24 h after removing 5FU. Unrepaired or incorrectly repaired DSBs are thought to be the most important initiators of cell death after DNA damaging agents. Although the breast carcinoma cell line MCF-7 had the highest level of residual (unrepaired) DSBs, it was not the most sensitive to 5FU; the colon carcinoma (HCT116) cell line was the most sensitive, but showed fewer unrepaired DSBs than the MCF-7 cells. This discrepancy was clarified through the results of cell cycle distribution and Fas-L activity where a fraction of the HCT116 cells was found in the sub-G1 phase of the cell cycle and Fas-L activity was 2–3 times higher following incubation with 5FU, indicating the induction of apoptosis. By contrast, the MCF-7 and HeLa cells did not show any sub-G1 fraction or activation of Fas-L. This finding indicates that the presence of non-repaired DSBs is not the final parameter that determines the sensitivity of cancer cells to 5FU, but the ability of these breaks to stimulate death pathways (apoptosis) and the genetic readiness of the cells to execute these death pathways also have roles to play.
In conclusion, our results indicate that the induction, but not the repair, of DNA DSBs as measured by CFGE is an important predictive molecular marker for the sensitivity of cancer cells to the cytotoxic effects of 5FU. However, this finding needs to be confirmed using clinical samples from cancer patients before being integrated into routine clinical practice.
Acknowledgments
We thank all the members of the Cancer Biology Department, ENCI for their stimulating ideas and practical support.
Abbreviations:
- 5FU
5-fluorouracil;
- DSBs
double-strand breaks;
- Fas-L
Fas ligand;
- SRB
sulphorhodamine-B;
- CFGE
constant-field gel electrophoresis;
- DDR
DNA-damage response
References
- 1.Sargent D, Sobrero A, Grothey A, O’Connell MJ, Buyse M, Andre T, Zheng Y, Green E, Labianca R, O’Callaghan C, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009;27:872–877. doi: 10.1200/JCO.2008.19.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parker WB, Cheng YC. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol Ther. 1990;48:381–395. doi: 10.1016/0163-7258(90)90056-8. [DOI] [PubMed] [Google Scholar]
- 3.Longley DB, Latif T, Boyer J, Allen WL, Maxwell PJ, Johnston PG. The interaction of thymidylate synthase expression with p53-regulated signaling pathways in tumor cells. Semin Oncol. 2003;30:3–9. doi: 10.1016/s0093-7754(03)00119-2. [DOI] [PubMed] [Google Scholar]
- 4.An Q, Robins P, Lindahl T, Barnes DE. 5-fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007;67:940–945. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Cassidy J, O’Brien V, Ryan KM, Collie-Duguid E. Mechanistic and predictive profiling of 5-fluorouracil resistance in human cancer cells. Cancer Res. 2004;64:8167–8176. doi: 10.1158/0008-5472.CAN-04-0970. [DOI] [PubMed] [Google Scholar]
- 6.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 7.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005;205:275–292. doi: 10.1002/path.1706. [DOI] [PubMed] [Google Scholar]
- 8.van Triest B, Pinedo HM, van Hensbergen Y, Smid K, Telleman F, Schoenmakers PS, van der Wilt CL, van Laar JA, Noordhuis P, Jansen G, Peters GJ. Thymidylate synthase level as the main predictive parameter for sensitivity to 5-fluorouracil, but not for folate-based thymidylate synthase inhibitors, in 13 nonselected colon cancer cell lines. Clin Cancer Res. 1999;5:643–654. [PubMed] [Google Scholar]
- 9.Grem JL. Screening for dihydropyrimidine dehydrogenase deficiency. Clin Cancer Res. 2005;11:5067–5068. doi: 10.1158/1078-0432.CCR-05-0769. [DOI] [PubMed] [Google Scholar]
- 10.Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer. 2003;106:66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]
- 11.Violette S, Poulain L, Dussaulx E, Pepin D, Faussat AM, Chambaz J, Lacorte JM, Staedel C, Lesuffleur T. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of Bcl-2 and Bcl-X(L) in addition to Bax and p53 status. Int J Cancer. 2002;98:498–504. doi: 10.1002/ijc.10146. [DOI] [PubMed] [Google Scholar]
- 12.Liu R, Page C, Beidler DR, Wicha MS, Núñez G. Overexpression of Bclx(L) promotes chemotherapy resistance of mammary tumors in a syngeneic mouse model. Am J Pathol. 1990;155:1861–1867. doi: 10.1016/S0002-9440(10)65505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi X, Liu S, Kleeff J, Friess H, Büchler MW. Acquired resistance of pancreatic cancer cells towards 5-Fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology. 2002;62:354–362. doi: 10.1159/000065068. [DOI] [PubMed] [Google Scholar]
- 14.Miyashita T. Tumor suppressor p53 is a regulator of Bcl-2 and Bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 15.Petak I, Tillman DM, Houghton JA. p53 dependence of Fas induction and acute apoptosis in response to 5-fluorouracil-leucovorin in human colon carcinoma cell lines. Cancer Res. 2000;6:4432–4441. [PubMed] [Google Scholar]
- 16.Zhang N, Yin Y, Xu S, Chen W. Fluorouracil: mechanisms of resistance and reversal strategies. Molecules. 2008;13:1551–1569. doi: 10.3390/molecules13081551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iliakis GE, Cicilioni O, Metzger L. Measurement of DNA double-strand breaks in CHO cells at various stages of the cell cycle using pulsed field gel electrophoresis: calibration by means of 1251 decay. Int J Radiat Biol. 1991;59:343–357. doi: 10.1080/09553009114550321. [DOI] [PubMed] [Google Scholar]
- 18.Dahm-Daphi J, Dikomey E. Separation of DNA fragments induced by ionizing irradiation using a graded-field gel electrophoresis. Int J Radiat Biol. 1995;67:161–168. doi: 10.1080/09553009514550201. [DOI] [PubMed] [Google Scholar]
- 19.Redon CE, Nakamura AJ, Martin OA, Parekh PR, Weyemi US, Bonner WM. Recent developments in the use of γ-H2AX as a quantitative DNA double-strand break biomarker. Aging (Albany NY) 2011;3:168–174. doi: 10.18632/aging.100284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Angelis PM, Svendsrud DH, Kravik KL, Stokke T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Cancer. 2006;5:20. doi: 10.1186/1476-4598-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Awady RA, Saleh EM, Dahm-Daphi J. Targeting DNA double-strand break repair: is it the right way for sensitizing cells to 5-fluorouracil? Anticancer Drugs. 2010;21:277–287. doi: 10.1097/CAD.0b013e328334b0ae. [DOI] [PubMed] [Google Scholar]
- 22.El-Awady RA, Dikomey E, Dahm-Daphi J. Radiosensitivity of human tumour cells is correlated with the induction but not with the repair of the DNA double-strand breaks. Br J Cancer. 2003;89:593–601. doi: 10.1038/sj.bjc.6601133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saleh EM, El-Awady RA, Abdel Alim MA, Abdel Wahab AA. Altered expression of proliferation-inducing and proliferation-inhibiting genes might contribute to acquired doxorubicin resistance in breast cancer cells. Cell Biochem Biophys. 2009;55:95–105. doi: 10.1007/s12013-009-9058-3. [DOI] [PubMed] [Google Scholar]
- 24.El-Awady RA, Ali MM, Saleh EM, Ghaleb FM. Apoptosis is the most efficient death-pathway in tumor cells after topoisomerase II inhibition. Saudi Med J. 2008;29:558–564. [PubMed] [Google Scholar]
- 25.Miura K, Kinouchi M, Ishida K, Fujibuchi W, Naitoh T, Ogawa H, Ando T, Yazaki N, Watanabe K, Haneda S, et al. 5-FU metabolism in cancer and orally-administrable 5-FU drugs. Cancers. 2010;2:1717–1730. doi: 10.3390/cancers2031717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pettersen HS, Visnes T, Vågbø CB, Svaasand EK, Doseth B, Slupphaug G, Kavli B, Krokan HE. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011;39:8430–8444. doi: 10.1093/nar/gkr563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koopman M, Venderbosch S, Nagtegaal ID, van Krieken JH, Punt CJ. A review on the use of molecular markers of cytotoxic therapy for colorectal cancer, what have we learned? Eur J Cancer. 2009;45:1935–1949. doi: 10.1016/j.ejca.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Valeri N, Gasparini P, Braconi C, Paone A, Lovat F, Fabbri M, Sumani KM, Alder H, Amadori D, Patel T, et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog2 (hMSH2) Proc Natl Acad Sci USA. 2010;107:21098–21103. doi: 10.1073/pnas.1015541107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pino MS, Chung DC. Microsatellite instability in the management of colorectal cancer. Expert Rev Gastroenterol Hepatol. 2011;5:385–399. doi: 10.1586/egh.11.25. [DOI] [PubMed] [Google Scholar]
- 30.Sinicrope FA, Yang ZJ. Prognostic and predictive impact of DNA mismatch repair in the management of colorectal cancer. Future Oncol. 2011;7:467–474. doi: 10.2217/fon.11.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Z, Xue J, Sowin TJ, Rosenberg SH, Zhang H. A novel mechanism of checkpoint abrogation conferred by Chk1 downregulation. Oncogene. 2005;24:1403–1411. doi: 10.1038/sj.onc.1208309. [DOI] [PubMed] [Google Scholar]
- 32.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 33.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 34.Adamsen BL, Kravik KL, De Angelis PM. DNA damage signaling in response to 5-fluorouracil in three colorectal cancer cell lines with different mismatch repair and TP53 status. Int J Oncol. 2011;39:673–682. doi: 10.3892/ijo.2011.1080. [DOI] [PubMed] [Google Scholar]
- 35.Li LS, Morales JC, Veigl M, Sedwick D, Greer S, Meyers M, Wagner M, Fishel R, Boothman DA. DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets. Br J Pharmacol. 2009;158:679–692. doi: 10.1111/j.1476-5381.2009.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]