

Abstract
Antibody drug conjugates (ADCs) can deliver potent drugs to cancer cells by employing the specificity of monoclonal antibodies (mAbs). ADCs have demonstrated significant anticancer activity and, in 2011, brentuximab vedotin has been approved by the FDA for the treatment of Hodgkin's and anaplastic large cell lymphomas. CD22 is an ideal target for ADC against B-cell malignancies because of its lineage-specific expression and rapid internalization upon antibody binding. In this study, we evaluated the anti-CD22 mAb HB22.7 as a vehicle for the targeted delivery of the potent toxin saporin (SAP). In vitro, HB22.7-SAP was cytotoxic against a panel of non-Hodgkin's lymphoma (NHL) cell lines representing the most common types of the disease. Moreover, in a xenograft model of NHL, HB22.7-SAP significantly inhibited the growth of established lesions and completely prevented tumor development when treatment was initiated within 24 h from tumor-cell inoculation. HB22.7-SAP had no significant in vivo toxicity. In conclusion, HB22.7 constitutes a potential platform for CD22-targeted ADCs.
Keywords: CD22, HB22.7, antibody drug conjugate, non-Hodgkin’s lymphoma, saporin
Introduction
Monoclonal antibodies (mAbs) have demonstrated utility in cancer research and treatment because of their exquisite specificity and relatively manageable side effects. However, antibody-based therapeutics have limited efficacy as single agents and thus are frequently used as an adjunct to chemotherapy. In such combination regimens, efficacy is limited by the toxicity associated to chemotherapy. The next generation of antibody-based therapy will involve the use of mAbs armed with potent cytotoxic drugs, radioisotopes and toxins for tumor-specific intracellular delivery. Based on their specificity and potency, antibody drug conjugates (ADCs) may enhance currently available chemotherapeutic approaches and at some point even obviate the need for systemic chemotherapy.
Among B-cell-specific antigens, CD22 is an ideal target for ADC therapy because: (1) it is broadly expressed by B-cell malignancies, (2) it undergoes rapid internalization following antibody binding and (3) it is not expressed by stem cell precursors, allowing for the regeneration of normal B cells following ADC-based therapy. In line with this, at least 3 different CD22-targeted ADCs are under clinical investigation.1-3 CD22 is a 140 kDa sialo-adhesion protein specifically expressed by normal and malignant B cells, and appears to be involved in the regulation of B-cell function and survival.4 CD22 expression has been observed in 60–80% of B-cell malignancies and in more than 90% of the most common types of NHL, namely, follicular and diffuse large B-cell lymphoma (DLBCL).5 One of the most appealing features of CD22 as a target for ADC therapy is that the binding of mAb facilitates its rapid internalization, in turn promoting the efficient delivery of conjugated drugs into target cells. In vitro studies with CD22+ human B-cell NHL have demonstrated that within 15 min from the binding of an anti-CD22 mAb, approximately 80% CD22 molecules are internalized.6
HB22.7 is an anti-CD22 mAb developed (and humanized) by our group for the treatment of NHL. HB22.7 is classified as a “ligand blocking” antibody because it binds to the same epitopes of CD22 as its natural ligands and effectively blocks binding.7 While in primary B cells HB22.7 induces a proliferative response, it activates apoptotic responses in neoplastic B cells, primarily through the stress activated protein kinase (SAPK) pathway.8 Using xenograft models of NHL, we have previously shown the lymphomacidal properties of HB22.7.9-13 In vivo studies demonstrated that HB22.7 is significantly more efficient than non-ligand blocking anti-CD22 mAbs.11 For the development of a CD22-targeted ADC, we hypothesized that it would be advantageous to use HB22.7 because of its cytotoxic effect, which are not observed with other anti-CD22 mAbs. In the present study, we provide the “proof of principle” that HB22.7 is highly efficient when used as a vehicle for the specific delivery of toxins, such as the plant-derived molecule saporin (SAP), to CD22+ NHL cell lines.
Results
HB22.7-SAP in vitro cytotoxicity
To ensure that SAP conjugation to HB22.7 did not affect CD22 binding, the CD22+ NHL cell lines Raji and Ramos were used to compare the binding of unmodified HB22.7 to that of HB22.7-SAP. SAP conjugation to HB22.7 had indeed no effect on its binding to CD22 (Fig. 1).

Figure 1. HB22.7-SAP binding and assessment of CD22 expression by flow cytometry. The NHL cell lines Ramos, Raji, DOHH-2, Granta 519 and SU-DHL-4 were either left untreated (red), treated with anti-mouse IgG FITC (black) or HB22.7 plus anti-mouse IgG FITC (green). In some cases, cells were incubated with HB22.7-SAP plus anti-mouse IgG FITC (blue). The T cell leukemia cell line Jurkat was used as a CD22- control.
Using flow cytometry with HB22.7 as a probe, CD22 expression was assessed in cell lines representing the following types of NHL: Burkitt’s lymphoma (Ramos and Raji), transformed follicular lymphoma (DOHH-2), mantle cell lymphoma (Granta 519) and DLBCL (SU-DHL-4). All cell lines were CD22+, with Ramos and Raji exhibiting the highest levels of CD22 expression followed by Granta 519, DOHH-2 and SU-DHL-4 (Fig. 1). Jurkat T cells were used as a CD22- control. Accordingly, staining with HB22.7 did not result in a fluorescence signal higher than that obtained with FITC-conjugated anti-mouse control IgGs.
Next, the cytotoxicity of HB22.7-SAP against NHL cell lines in vitro was evaluated using an MTS assay. HB22.7-SAP exerted a potent cytotoxic effect against all CD22+ NHL cell lines with IC50 values ranging from 1 to 8.4 ng/mL (Fig. 2). Conversely, HB22.7-SAP exhibited no cytotoxicity against CD22- Jurkat cells. Further, an isotype-matched control ADC, mouse-IgG-SAP, was not cytotoxic against any of the cell lines (data not shown). These data suggest that the in vitro cytotoxicity of HB22.7-SAP is dependent upon CD22 expression. As a control, the NHL cell lines were also treated with free HB22.7 plus free SAP at molar concentrations equivalent to those of each component in the HB22.7-SAP conjugate. At all tested concentrations, free HB22.7 plus free SAP exerted no cytotoxicity (Fig. 2 and data not shown). It should be noted that free HB22.7 does have independent cytotoxic effects in vitro, but only at concentrations higher than those used in this assay.8

Figure 2. In vitro cytotoxicity of HB22.7-SAP against NHL cell lines. NHL cell lines were incubated with titrations of HB22.7-SAP continuously for 72 h. The concentration of HB22.7-SAP (ng/mL) corresponds to the final antibody concentration. Cell viability was measured using an MTS assay and the mean of 3 separate experiments performed in triplicate is shown. Jurkat T cells were used as a CD22- control cell line. Additionally, Ramos cells were treated with free HB22.7 plus free SAP at concentrations equivalent to those of each component of the HB22.7-SAP conjugate.
To examine the impact of incubation time on HB22.7-SAP-induced in vitro cytotoxicity, Raji cells were incubated with dilutions of HB22.7-SAP for 2 h, washed to remove unbound antibody, resuspended in fresh media and incubated for additional 70 h. Using these conditions, the IC50 of HB22.7-SAP increased approximately 5-fold to 22.2 ng/mL (data not shown).
In vivo efficacy and toxicity of HB22.7-SAP in an established NHL xenograft model
The antitumor activity of HB22.7-SAP was investigated with a xenograft model of NHL, in which tumors were allowed to reach a volume of 100–200 mm3 before treatment initiation (designated as day 0). During the first two weeks of treatment, HB22.7-SAP (1 mg/kg) significantly inhibited (p < 0.01) tumor growth as compared with PBS and free HB22.7 (0.6 mg/kg) plus free SAP (0.36 mg/kg) control groups (Fig. 3A). As measured on days 5 and 8, the average tumor volume of mice that received HB22.7-SAP was more than 2-fold smaller than that of mice that received PBS or free HB22.7 plus free SAP. Despite an initial control of tumor growth, however, by day 19 the average tumor volume of mice treated with HB22.7-SAP was no different than that of control animals. It should be noted that the dose of HB22.7 used in “free HB22.7 plus free SAP” group was well below the dose that we previously used to inhibit tumor growth in a xenograft model of NHL.11 Thus, it was expected that free HB22.7 plus free SAP had no antitumor activity.

Figure 3. In vivo efficacy and toxicity of HB22.7-SAP. (A-C) To establish tumors, nude mice were subcutaneously injected with 5 x 106 Raji NHLcells. Tumors were allowed to grow until they reached a volume of 100–200 mm3 and this was designated as day 0. Mice were then randomly separated into three treatment groups (n = 8–10 per group) consisting of PBS, free HB22.7 (0.6 mg/kg) plus free SAP (0.36 mg/kg) and HB22.7-SAP (1 mg/kg). The dose of free HB22.7 and free SAP was equivalent to each component in the HB22.7-SAP conjugate. All treatments were administered by intreperitoneal injection weekly for a period of three weeks (indicated by arrows). (A) Tumor volume was calculated using the formula (L × W2)/2. (B) Body weight was measured twice a week and the percent of starting weight was calculated. (C) On days 6, 13 and 20, blood was collected from animals in each group (n = 3) for complete blood counts. *p < 0.01 compared with PBS and free HB22.7 plus free SAP groups.
In the evaluation of HB22.7-SAP, body weight and complete blood counts (CBCs) were also assessed as indicators of treatment-induced toxicity. Notably, HB22.7-SAP induced a slight weight loss, which could be completely recovered prior to the next injection (Fig. 3B). As a comparison, mice treated with PBS or free HB22.7 plus free SAP gained weight over the same period of time. Additionally, the blood of mice from each treatment group was collected on days 6, 13 and 20 for CBCs. Mice treated with HB22.7-SAP had blood counts within the normal range and the counts did not fluctuate significantly over time (Fig. 3C).
In vivo efficacy and toxicity of preemptive HB22.7-SAP treatment in a NHL xenograft model
To determine if the degree of tumor burden would affect HB22.7-SAP-mediated efficacy, a second NHL xenograft model was used. Twenty-four hours after subcutaneous implantation of Raji cells (designated as day 0), mice were started on the same treatment regimen mentioned above. Using the preemptive treatment model, HB22.7-SAP was highly effective, as no mice in this treatment group developed measureable tumors during the 50 d study period (Fig. 4A). On the contrary, free HB22.7 plus free SAP had no effect on tumor growth and mice from this group exhibited tumor volumes similar to mice treated with PBS.
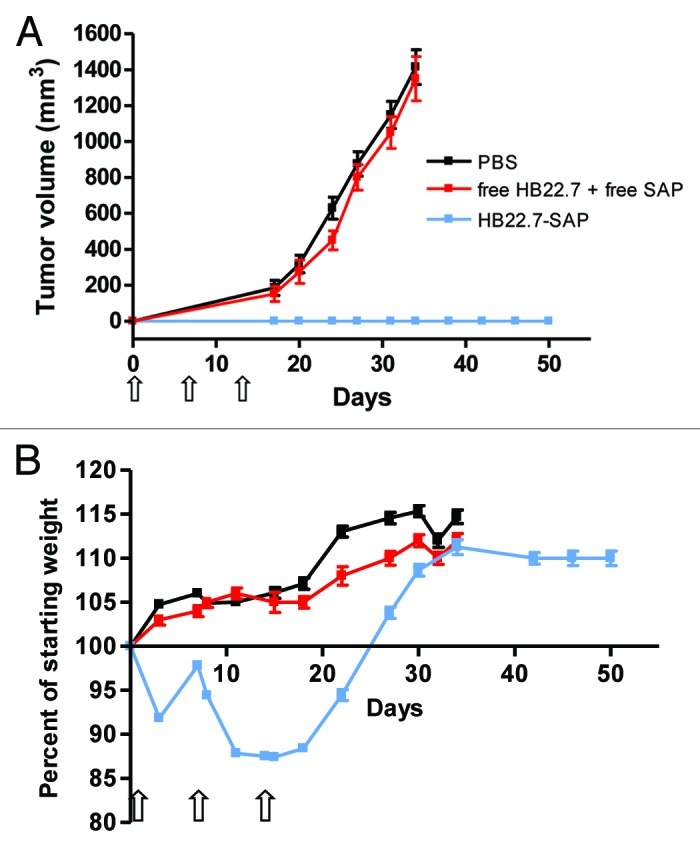
Figure 4. In vivo efficacy of preemptive HB22.7-SAP treatment. (A, B) Nude mice were subcutaneously injected with 5 x 106 Raji cells. Twenty-four h later, designated as day 0, mice were randomly separated into three treatment groups (n = 8–10 per group) consisting of PBS, free HB22.7 (0.6 mg/kg) plus free SAP (0.36 mg/kg) and HB22.7-SAP (1 mg/kg). The dose of free HB22.7 and free SAP was equivalent to those of each component in the HB22.7-SAP conjugate. Starting on day 0, treatments were administered by intraperitoneal injection weekly for a period of three weeks (indicated by arrows). (A) Tumor volume was calculated using the formula (L × W2)/2. (B) Body weight was measured twice a week and the percent of starting weight was calculated.
Similar to the results obtained with the xenograft model described above (Fig. 3B), HB22.7-SAP induced a slight weight loss (8%) that could be recovered by the second injection (Fig. 4B). However, following the second and third injections, a moderate weight loss (13%) and a prolonged recovery period were observed.
Discussion
The goal of the present study was to demonstrate that the anti-CD22 mAb HB22.7 can serve as a vehicle for the targeted delivery of a conjugated cytotoxic agent. To this end, as a proof-of-principle, we conjugated the plant-derived toxin, SAP to HB22.7 and showed that the resulting HB22.7-SAP conjugate has potent and specific antitumor activity in vitro and in vivo.
Using a panel of cell lines representing the major types of NHL, we demonstrated the potent in vitro cytotoxicity of HB22.7-SAP (Fig. 2). Interestingly, the expression of CD22 by the cell lines does not correlate well with HB22.7-SAP-induced cytotoxicity. For instance, Raji cells expressed 4 to 5-fold more CD22 molecules than DOHH-2 cells, yet DOHH-2 cells were almost 5-fold more sensitive to HB22.7-SAP. This phenomenon was previously documented by Polson et al. who showed that, among 18 NHL cell lines, CD22 receptor density do not correlate well with cytotoxicity as induced by an anti-CD22-MC-DM1 conjugate.14 Similarly, it has been previously shown that, in cases of low CD22 expression levels, such as by chronic lymphocytic leukemia (CLL) cells, CD22-targeted ADCs retain activity.15 The lack of correlation between CD22 expression and cytotoxicity suggests that factors such as the kinetics of receptor internalization, release of free drug inside the target cell and sensitivity of the target cell to the drug may play a role in the overall sensitivity of a particular cell type to ADCs.
To study the efficacy of HB22.7-SAP in vivo, we employed two xenograft models of NHL. In the first model, treatment was initiated when tumors reached a volume of 100–200 mm3. Using this model, mice treated with HB22.7-SAP experienced an inhibition of tumor growth for the first two weeks of treatment. However, mice eventually developed large, unresponsive tumors (Fig. 3A). In the second model, treatment was initiated 24 h after tumor cell implantation. The difference in treatment initiation had a significant impact on efficacy, as none of the mice treated with HB22.7-SAP in this second model developed tumors for the entire duration of the study (Fig. 4A). In previous studies with naked HB22.7, we reported similar findings that are consistent with the hypothesis that mAb-based therapeutics are more effective for the treatment of small tumors. Mice with tumors < 200 mm3 at the time of HB22.7 treatment had a higher response rate and greater inhibition of tumor growth as compared with mice whose tumors were > 200 mm3 at the start of treatment.11
Unlike most other mAbs used in the construction of ADCs, HB22.7 has unique binding and functional characteristics that we surmise make it a superior platform for ADC-based therapeutics. Specifically, HB22.7 binds to the two NH2-terminal Ig domains of CD22 and blocks CD22 ligand binding. In neoplastic B cells, this induces an apoptotic response and we have previously reported on the lymphomacidal properties of HB22.7 in xenograft models of NHL. On the contrary, anti-CD22 mAbs that do not block ligand binding have only modest functional effects. It remains unknown whether the efficacy of HB22.7-SAP demonstrated in the current study is due in part to the direct cytotoxic effects of HB22.7 or solely due to the intracellular delivery of the toxic payload. Certainly, using HB22.7 as the platform for the development of an ADC has theoretical advantages over other anti-CD22 mAbs that are not capable of independent cytotoxicity. It should be noted that among CD22-targeted ADCs that are currently in preclinical and clinical development (inotuzumab ozogamicin, moxetumomab pasudotox and RG7593), none are based on an anti-CD22 mAb with documented independent cytotoxicity.14,16,17
Several groups have conjugated SAP to mAbs for targeted delivery to a variety of tumor cells and demonstrated significant preclinical antitumor activity.18-23 In this study, SAP was chosen because of its potency and ease of chemical conjugation.24 However, clinical development of mAb-SAP conjugates may be limited by issues stemming from the toxin itself. SAP is a plant-derived toxin and when injected into humans induces an immunogenic response in the form of antitoxin antibodies.25 This leads to a rapid clearance of mAb-SAP conjugates and hence a reduction in the level of conjugate available for uptake by target cells. Additionally, the use of mAb-SAP conjugates in humans is limited by the possible development of a vascular leak syndrome and hepatotoxicity.26 Stirpe et al. showed that the non-specific uptake of a mAb-SAP conjugate by hepatocytes was most likely due to the SAP moiety of the conjugate rather than the antigen-binding sites, carbohydrate moieties or Fc region of the mAb.27 As an alternative, potent small molecule drugs such as those from the auristatin (MMAE) and maytansine (DM1) families have shown promising results as cytotoxic components for ADCs. These drugs have similar potency as SAP but have the consistent advantage of not producing any of the above mentioned side-effects.
ADCs represent novel targeted therapeutics capable of simultanesouly improving the efficacy and reducing the toxicity of drugs that are too potent to be administered systemically. HB22.7 is an ideal candidate as the platform for ADC because it targets an internalizing antigen expressed on neoplastic B cells and because of its unique functional characteristics not observed with other anti-CD22 mAbs.
Materials and Methods
HB22.7 and saponin
The anti-CD22 mAb HB22.7 was prepared and characterized as described previously.8 SAP, mouse-IgG-SAP and the custom conjugate, HB22.7-SAP, were produced by Advanced Targeting Systems.
Cell lines
Ramos, Raji, Granta 519 NHL and Jurkat T cells were purchased from the American Type Culture Collection. SU-DHL-4 and DOHH-2 NHL cells were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). All cell lines were maintained with RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and incubated at 37°C, 5% CO2 and 90% humidity.
Flow cytometry
To assess CD22 expression, 0.5 × 106 cells per sample were resuspended in 100 μL FACS buffer (PBS + 0.5% FBS) and chilled on ice. HB22.7 (10 μg/mL) was incubated with cells for 30 min on ice, followed by 3 washes with ice cold FACS buffer. Cells were then incubated with a 1/50 dilution of goat anti-mouse IgG-FITC (Invitrogen) for 30 min on ice and in the dark. Cells were washed 3 times and 10,000 events were analyzed on a FACScan (BD Biosciences).
MTS assays
The in vitro cytotoxicity of HB22.7-SAP was evaluated using an MTS assay.28 Cells were seeded in 96-well plates at a density of 1 × 104 cells/well in 90 μL media. HB22.7-SAP was serially diluted with media and 10 μL of each dilution was added to the appropriate well and incubated continuously for 72 h. As a control, another group of cells was treated with free HB22.7 plus free SAP at molar concentrations equivalent to each component in the HB22.7-SAP conjugate. In a separate experiment, Raji cells were incubated with HB22.7-SAP for 2 h, washed 3 times, resuspended in fresh media and cultured for an additional 70 h. After a 72 h incubation, the cell viability of all treatment groups was assessed using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay according to the manufacturer's instructions. MTS solution (20 μL) was added to each well and allowed to incubate for 1 h at 37°C. Cell viability as a percent of the untreated control was calculated as follows: [(OD490 treated - OD490 background) / (OD490 control - OD490 background)*100]. The mean ± standard deviation (SD) of 3 separate experiments performed in triplicate is shown.
In vivo efficacy and toxicity
Female athymic nude mice (6–8 weeks old) were obtained from Harlan Sprague–Dawley and maintained in micro-isolation cages under pathogen free conditions. All animal experiments were performed in compliance with institutional guidelines and according to protocol No.16322 approved by the Animal Use and Care Administrative Advisory Committee at the University of California, Davis. Mice were allowed to acclimatize for at least 4 d prior to the start of any experiment. Three days prior to tumor cell implantation, mice received 400 rads of whole body radiation. To establish tumors, 5 × 106 Raji cells were resuspended in PBS and subcutaneously injected into the flank of each mouse. Tumors were allowed to grow until they reached a volume of 100–200 mm3 and this was designated as day 0. Mice were then randomly separated into 3 treatment groups (n = 8–10 per group) consisting of PBS, free HB22.7 (0.6 mg/kg) plus free SAP (0.36 mg/kg) and HB22.7-SAP (1 mg/kg). The doses of free HB22.7 and free SAP were equivalent to each component in the HB22.7-SAP conjugate. All treatments were administered weekly for three weeks by intraperitoneal injection. Tumor volume was measured using digital calipers and calculated by the formula (L × W2)/2, where L is the longest and W is the shortest in tumor diameters (mm).
To assess the significance of starting tumor volume on HB22.7-SAP-mediated tumor growth inhibition, we used a second xenograft model in which treatment was initiated 24 h following tumor cell implantation (designated as day 0). Mice (n = 8–10 per group) were separated into treatment groups as mentioned above and treated using the same doses and dosing regimen.
Body weight and other symptoms and signs of toxicity (unkempt fur, ataxia, piloerection) were observed daily. Mice were sacrificed when tumor volume exceeded 1500 mm3 or 20 mm in diameter. On days 6, 13 and 20, blood was drawn from the lateral saphenous vein of animals in each group (n = 3) and collected into EDTA-lined tubes for complete blood counts (CBCs).
Statistical analysis
Statistical analysis was performed by Student’s t-tests for two groups, and one-way ANOVA for multiple groups. All results were expressed as the mean ± standard error (SEM) unless otherwise noted. p values < 0.05 were considered statistically significant. Survival analysis was performed using GraphPad Prism 4 software.
Acknowledgments
This work was supported by the Schwedler Family Foundation and the deLeuze Non-toxic Cure for Lymphoma Fund, and by Department of Defense grant W81XWH-10-1-0716.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/21815
References
- 1.Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, et al. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J Clin Oncol. 2012;30:1822–8. doi: 10.1200/JCO.2011.38.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogura M, Hatake K, Ando K, Tobinai K, Tokushige K, Ono C, et al. Phase I study of anti-CD22 immunoconjugate inotuzumab ozogamicin plus rituximab in relapsed/refractory B-cell non-Hodgkin lymphoma. Cancer Sci. 2012;103:933–8. doi: 10.1111/j.1349-7006.2012.02241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, et al. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: preclinical studies and phase I clinical trial. Clin Cancer Res. 2010;16:1894–903. doi: 10.1158/1078-0432.CCR-09-2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tedder TF, Poe JC, Haas KM. CD22: a multifunctional receptor that regulates B lymphocyte survival and signal transduction. Adv Immunol. 2005;88:1–50. doi: 10.1016/S0065-2776(05)88001-0. [DOI] [PubMed] [Google Scholar]
- 5.Tu X, LaVallee T, Lechleider R. CD22 as a target for cancer therapy. J Exp Ther Oncol. 2011;9:241–8. [PubMed] [Google Scholar]
- 6.Carnahan J, Wang P, Kendall R, Chen C, Hu S, Boone T, et al. Epratuzumab, a humanized monoclonal antibody targeting CD22: characterization of in vitro properties. Clin Cancer Res. 2003;9:3982S–90S. [PubMed] [Google Scholar]
- 7.Tuscano J, Engel P, Tedder TF, Kehrl JH. Engagement of the adhesion receptor CD22 triggers a potent stimulatory signal for B cells and blocking CD22/CD22L interactions impairs T-cell proliferation. Blood. 1996;87:4723–30. [PubMed] [Google Scholar]
- 8.Tuscano JM, Riva A, Toscano SN, Tedder TF, Kehrl JH. CD22 cross-linking generates B-cell antigen receptor-independent signals that activate the JNK/SAPK signaling cascade. Blood. 1999;94:1382–92. [PubMed] [Google Scholar]
- 9.Tuscano JM, O’Donnell RT, Miers LA, Kroger LA, Kukis DL, Lamborn KR, et al. Anti-CD22 ligand-blocking antibody HB22.7 has independent lymphomacidal properties and augments the efficacy of 90Y-DOTA-peptide-Lym-1 in lymphoma xenografts. Blood. 2003;101:3641–7. doi: 10.1182/blood-2002-08-2629. [DOI] [PubMed] [Google Scholar]
- 10.O’Donnell RT, Pearson D, McKnight HC, Ma YP, Tuscano JM. Treatment of non-Hodgkin’s lymphoma xenografts with the HB22.7 anti-CD22 monoclonal antibody and phosphatase inhibitors improves efficacy. Cancer Immunol Immunother. 2009;58:1715–22. doi: 10.1007/s00262-009-0688-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Donnell RT, Ma Y, McKnight HC, Pearson D, Tuscano JM. Dose, timing, schedule, and the choice of targeted epitope alter the efficacy of anti-CD22 immunotherapy in mice bearing human lymphoma xenografts. Cancer Immunol Immunother. 2009;58:2051–8. doi: 10.1007/s00262-009-0713-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Donnell RT, Pearson D, McKnight HC, Ma YP, Tuscano JM. Phosphatase inhibition augments anti-CD22-mediated signaling and cytotoxicity in non-hodgkin’s lymphoma cells. Leuk Res. 2009;33:964–9. doi: 10.1016/j.leukres.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 13.Martin SM, Churchill E, McKnight H, Mahaffey CM, Ma Y, O’Donnell RT, et al. The HB22.7 Anti-CD22 monoclonal antibody enhances bortezomib-mediated lymphomacidal activity in a sequence dependent manner. J Hematol Oncol. 2011;4:49. doi: 10.1186/1756-8722-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polson AG, Williams M, Gray AM, Fuji RN, Poon KA, McBride J, et al. Anti-CD22-MCC-DM1: an antibody-drug conjugate with a stable linker for the treatment of non-Hodgkin’s lymphoma. Leukemia. 2010;24:1566–73. doi: 10.1038/leu.2010.141. [DOI] [PubMed] [Google Scholar]
- 15.Kreitman RJ, Margulies I, Stetler-Stevenson M, Wang QC, FitzGerald DJ, Pastan I. Cytotoxic activity of disulfide-stabilized recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) toward fresh malignant cells from patients with B-cell leukemias. Clin Cancer Res. 2000;6:1476–87. [PubMed] [Google Scholar]
- 16.Salvatore G, Beers R, Margulies I, Kreitman RJ, Pastan I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin Cancer Res. 2002;8:995–1002. [PubMed] [Google Scholar]
- 17.DiJoseph JF, Armellino DC, Boghaert ER, Khandke K, Dougher MM, Sridharan L, et al. Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood. 2004;103:1807–14. doi: 10.1182/blood-2003-07-2466. [DOI] [PubMed] [Google Scholar]
- 18.Bolognesi A, Tazzari PL, Olivieri F, Polito L, Lemoli R, Terenzi A, et al. Evaluation of immunotoxins containing single-chain ribosome-inactivating proteins and an anti-CD22 monoclonal antibody (OM124): in vitro and in vivo studies. Br J Haematol. 1998;101:179–88. doi: 10.1046/j.1365-2141.1998.00665.x. [DOI] [PubMed] [Google Scholar]
- 19.Flavell DJ, Boehm DA, Noss A, Warnes SL, Flavell SU. Therapy of human T-cell acute lymphoblastic leukaemia with a combination of anti-CD7 and anti-CD38-SAPORIN immunotoxins is significantly better than therapy with each individual immunotoxin. Br J Cancer. 2001;84:571–8. doi: 10.1054/bjoc.2000.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foehr ED, Lorente G, Kuo J, Ram R, Nikolich K, Urfer R. Targeting of the receptor protein tyrosine phosphatase beta with a monoclonal antibody delays tumor growth in a glioblastoma model. Cancer Res. 2006;66:2271–8. doi: 10.1158/0008-5472.CAN-05-1221. [DOI] [PubMed] [Google Scholar]
- 21.Kuroda K, Liu H, Kim S, Guo M, Navarro V, Bander NH. Saporin toxin-conjugated monoclonal antibody targeting prostate-specific membrane antigen has potent anticancer activity. Prostate. 2010;70:1286–94. doi: 10.1002/pros.21164. [DOI] [PubMed] [Google Scholar]
- 22.Pasqualucci L, Wasik M, Teicher BA, Flenghi L, Bolognesi A, Stirpe F, et al. Antitumor activity of anti-CD30 immunotoxin (Ber-H2/saporin) in vitro and in severe combined immunodeficiency disease mice xenografted with human CD30+ anaplastic large-cell lymphoma. Blood. 1995;85:2139–46. [PubMed] [Google Scholar]
- 23.Tazzari PL, Polito L, Bolognesi A, Pistillo MP, Capanni P, Palmisano GL, et al. Immunotoxins containing recombinant anti-CTLA-4 single-chain fragment variable antibodies and saporin: in vitro results and in vivo effects in an acute rejection model. J Immunol. 2001;167:4222–9. doi: 10.4049/jimmunol.167.8.4222. [DOI] [PubMed] [Google Scholar]
- 24.Polito L, Bortolotti M, Pedrazzi M, Bolognesi A. Immunotoxins and other conjugates containing saporin-s6 for cancer therapy. Toxins (Basel) 2011;3:697–720. doi: 10.3390/toxins3060697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.French RR, Bell AJ, Hamblin TJ, Tutt AL, Glennie MJ. Response of B-cell lymphoma to a combination of bispecific antibodies and saporin. Leuk Res. 1996;20:607–17. doi: 10.1016/0145-2126(96)00007-0. [DOI] [PubMed] [Google Scholar]
- 26.Pasqualucci L, Flenghi L, Terenzi A, Bolognesi A, Stirpe F, Bigerna B, et al. Immunotoxin therapy of hematological malignancies. Haematologica. 1995;80:546–56. [PubMed] [Google Scholar]
- 27.Stirpe F, Derenzini M, Barbieri L, Farabegoli F, Brown ANF, Knowles PP, et al. Hepatotoxicity of immunotoxins made with saporin, a ribosome-inactivating protein from Saponaria officinalis. Virchows Arch B Cell Pathol Incl Mol Pathol. 1987;53:259–71. doi: 10.1007/BF02890252. [DOI] [PubMed] [Google Scholar]
- 28.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–12. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]