

Abstract
In spite of the fact that they occur at high rates, the clinical responses of BRAFV600 mutant metastatic melanoma to BRAF inhibitors are usually short-lasting, with most cases progressing within less than 8 mo. Immunomodulatory strategies initiated after progression have recently been reported to be poorly efficient. By characterizing the immunological interactions between T cells and cancer cells in clinical material as well as the influence of the FDA-approved BRAF inhibitor vemurafenib on the immune system, we aimed at unraveling new strategies to expand the efficacy of adoptive T-cell transfer, which represents one of the most promising approaches currently in clinical development for the treatment of metastatic melanoma. Here we show that blocking the BRAF-MAPK pathway in BRAF signaling-addicted melanoma cells significantly increases the ability of T cells contained in clinical grade tumor-infiltrating lymphocytes to recognize autologous BRAFV600 mutant melanoma cell lines in vitro. Antitumor reactivity was improved regardless of the class of antigen recognized by tumor-specific CD8+ T cells. Microarray data suggests that improved tumor recognition is associated with modified expression of MHC Class I-associated proteins as well as of heat-shock proteins. In conclusion, our preclinical data suggest that an appropriately timed sequential treatment of BRAFV600 mutant melanoma with vemurafenib and adoptive T-cell transfer might result in synergistic antineoplastic effects owing to an increased immunogenicity of cancer cells.
Keywords: Adoptive T-cell therapy, BRAF inhibitors, combination therapies, melanoma, Vemurafenib, tumor-infiltrating lymphocytes
Introduction
Treatment of BRAFV600E mutant melanoma with vemurafenib (Vem), a selective BRAF inhibitor, has been shown to result in impressive rates (up to 50%) of objective responses and improved progression-free and overall survival, as compared with standard chemotherapy.1 However, clinical responses to Vem are usually short-lasting and most patients progress within less than 8 mo.2 Upon progression, patients should be offered alternative treatments. Unfortunately, recent data indicate that FDA-approved immune-activating anti-CTLA4 antibodies may have limited efficacy if administered after post-Vem progression.3 Thus, other kinds of immunotherapy should be designed for (appropriately selected) melanoma patients that progress upon therapy with BRAF inhibitors.
Adoptive T-cell transfer (ACT) with tumor-infiltrating lymphocytes (TILs) is another immunotherapeutic strategy in clinical development for patients with metastatic melanoma. It has been associated with a response rate of approximately 50% in Phase II clinical trials and potentially induces durable complete responses in a large fraction (up to 20%) of treated patients, regardless of BRAF mutational status.4 ACT may therefore obtain regulatory approval as a standard treatment within the next few years.5
Recent studies have shown that BRAF-targeting agents do not affect the viability and functionality of T lymphocytes when used at therapeutic doses,6-8 suggesting that a combination of BRAF inhibitors and immunomodulatory therapies is feasible. In addition, melanoma cells treated with the selective BRAF inhibitor PLX4720 may be more visible to the immune system as this drug directly stimulates the expression and presentation of melanoma-differentiation antigens (MDAs),8 recognized by a subset of antitumor T cells that we and others have shown to be commonly found in TIL products for ACT.9,10 Moreover, Koya et al. have very recently demonstrated that a combination regimen involving Vem plus ACT with lymphocytes genetically modified to express a T-cell receptor (TCR) that recognizes chicken ovalbumin expressed by SM1-OVA tumors or gp100 (an MDA) endogenously expressed by murine melanoma SM1 cells, exerts superior antitumor effects in vivo as compared with either agent alone.11 However, current ACT protocols are based on the recognition of multiple classes of tumor-associated antigens (TAAs), including differentiation, cancer-testis, mutated and overexpressed antigens as well as a large array of uncharacterized antigens that may account for up to 50% of the whole cell population in clinical grade TIL products.9,10,12
In order to verify whether a combination of BRAF-blocking agents and ACT may have direct synergistic effects against melanoma, we have studied in vitro tumor recognition by clinically relevant TIL preparations after the blockade of BRAF signaling in BRAFV600 mutant autologous melanoma cells. Subsequently, we characterized tumor-specific immune responses against selected differentiation, cancer-testis and overexpressed antigens.
Results
BRAF inhibition promotes the recognition of autologous melanoma cells by clinical grade TILs
Twenty-two independent clinical grade TIL cultures generated either with the standard or with the “young TIL” method were obtained from five patients bearing BRAFV600E mutant metastatic melanoma. These TILs were tested in co-culture assays with autologous, short-term cultured melanoma cell lines. Target cells were pre-treated with Vem at low (approximately corresponding to the 50% growth inhibitory, GI50, concentration) or high doses (close to the maximal drug effect). Figure S1 depicts the sensitivity of the melanoma cell lines used in this study to Vem.
Treatment with Vem significantly increased the frequency of TILs recognizing autologous melanoma cells and responding to them by producing Type 1 helper cytokines (Fig. 1A) or by mobilizing cytotoxic granules (Fig. 1B), confirming the polyfunctionality of newly-induced responses.
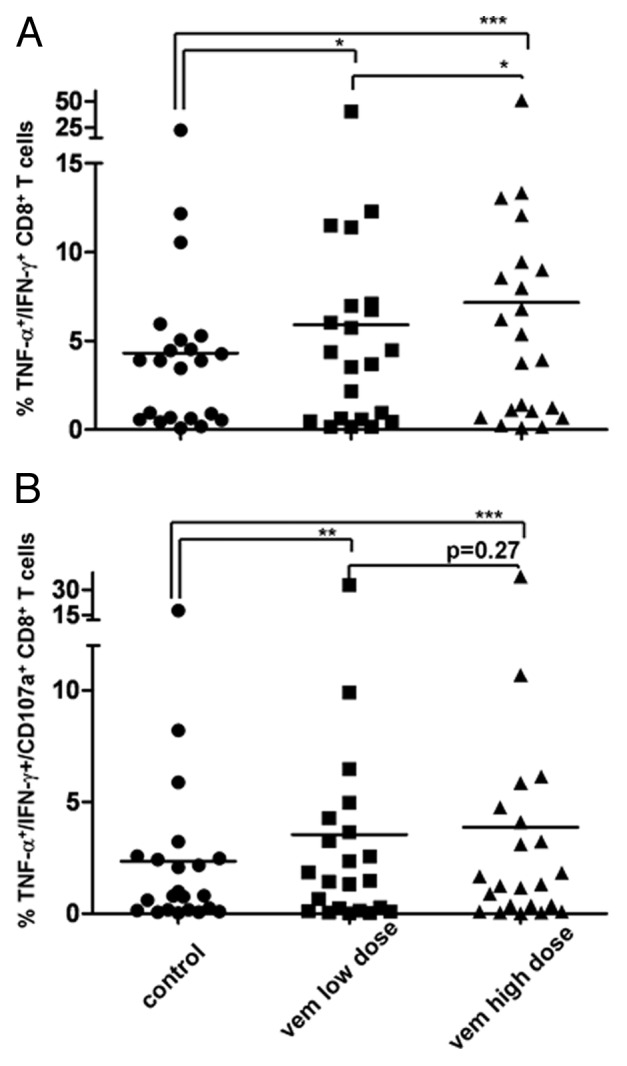
Figure 1. BRAF inhibition improves autologous tumor recognition by CD8+ tumor-infiltrating lymphocytes. (A, B) Tumor-infiltrating lymphocytes (TILs) were co-cultured with autologous BRAFV600E mutant melanoma target cell lines treated with vemurafenib (Vem) at low or high dose, or left untreated. (A) Frequency of tumor necrosis factor α (TNFα)− and interferon γ (IFNγ)-producing CD8+ TILs. (B) Frequency of CD8+ TILs producing TNFα and IFNγ and simultaneously mobilizing CD107a upon co-culture with autologous tumor cells. *p < 0.05 ; ** p < 0.01; ***p < 0.001
Effects of BRAF inhibition on TILs enriched in tumor-specific cells
Unselected clinical-grade TILs apparently contain only a small fraction of tumor-specific T cells and can include CD8+ T cells specific for non-tumor associated viral antigens.9 Therefore, the antitumor effects resulting from an improved recognition of cancer cells by T cells (Fig. 1) may be diluted by large amounts of non-tumor specific T cells.
To further scrutinize the effects of BRAF blockade on tumor recognition, we performed additional experiments using TIL preparations that were highly enriched in tumor-specific T cells. These TILs were produced either with conventional methods (standard TILs) and cultures selected based on a high frequency of in vitro tumor-reactive T cells or upon flow cytometry based sorting of tumor-specific T cells from TIL bulk cultures (based on CD107a expression upon co-culture with autologous tumor cells), followed by one cycle of rapid expansion protocol (REP), as required for clinical purposes. As expected, improved tumor recognition was more evident in this setting than with unselected TILs (Fig. 2A and B). Of note, in some cases, a dose-dependent increased recognition could be evidenced (Figs. 1A and 2A).

Figure 2. Effects of BRAF inhibition on tumor recognition by tumor-infiltrating lymphocytes highly enriched in antitumor T cells. (A, B) Two representative tumor-infiltrating lymphocyte (TIL) products containing a high frequency of tumor-specific cells were co-cultured with autologous BRAFV600E mutant melanoma target cell lines as described in Materials and Methods. Plots show the frequency of tumor necrosis factor α (TNFα)- and interferon γ (IFNγ)-producing CD8+ TILs upon co-culture with autologous tumor cells, which was largely increased by BRAF inhibition. (A) TILs produced with conventional methods with a high frequency of in vitro reactive T cells. (B) TILs produced with FACS-sorting of tumor-specific T cells from TIL bulk cultures (based on CD107a expression upon co-culture with autologous tumor cells), followed by one cycle of rapid expansion protocol (REP).
BRAF inhibition increases T-cell recognition of different classes of tumor antigens
We have previously reported that clinical grade TILs may contain T cells specific for different classes of TAAs.9,13 Therefore, to further dissect the antitumor immune responses elicited by BRAF inhibition, we assessed the ability of Vem to increase tumor recognition by T cells specific for defined TAAs, using combined fluorochrome-conjugated peptide-HLA multimers and intracellular cytokine staining.
MDAs
TILs enriched in T cells specific for the HLA-A2-restricted MART-1/Melan-A-derived peptide ELAGIGILTV (ELA) and for the gp100-derived peptide YLEPGPVTA generated from Patient 5 were available for analysis. In line with previous reports by Boni A et al.,8 our data indicate that BRAF inhibition increases the recognition of one BRAFV600E mutant melanoma cell line (FM92) by allogeneic T cells specific for MDAs, although apparently at a lower extent than what observed earlier (Fig. 3A).

Figure 3. Effects of BRAF inhibition on tumor recognition by tumor-infiltrating lymphocytes specific for defined tumor-associated antigens. (A, B) Tumor-infiltrating lymphocytes (TILs) enriched in CD8+ T cells specific for defined melanoma differentation antigens (MART-1 and gp100), overexpressed (AIM-2) or cancer-testis (TAG) antigens were co-cultured with BRAFV600E (FM92, cell line Pt.2 and Pt. 4) or BRAFV600K (FM57 and FM88) mutant melanoma cell lines as indicated in Materials and Methods. Simultaneous tumor necrosis factor α (TNFα) and interferon γ (IFNγ) production of antigen-specific CD8+ T cells was analyzed with combined fluorochrome conjugated peptide-HLA multimers and intracellular cytokine staining. (A) Frequency of antigen-specific T cells co-cultured with vemurafenib (Vem)-treated or untreated cell lines. (B) TILs specific for the cancer-testis antigen TAG co-cultured with BRAFV600E mutant autologous melanoma cells.
In addition, we tested the effects of BRAF inhibition on BRAFV600K mutant cell lines. In this setting, T-cell recognition was increased to a very high extent for the cell line FM88, indicating that improved antigen-presentation may occur also in cancer cells harboring this mutation. However, no increase was observed for the cell line FM57. This latter cell line was constitutively recognized by a very high frequency (over 60%) of both MART-1- and gp100-specific T cells (Fig. 3A).
When both MART-1- and gp100-specific T cells were tested in co-culture assays with autologous short-term cultured BRAFV600E mutant tumor cells, no recognition was detected both in the absence and in the presence of Vem. PCR analyses revealed that the tumor cell line expressed both MART-1 and gp-100 (data not shown). However, this finding is not surprising in view of the fact that our group has previously shown that the wild-type immunodominant MART-1 epitope EAAGIGILTV is not presented on the surface of many melanoma cell lines despite antigen expression.14 Probably, this situation reflects a process of immunoediting that has lead to BRAF inhibition-insensitive defects in antigen presentation.
Altogether, these data suggest that the recognition of MDAs expressed on melanoma cells can be enhanced by BRAF inhibition only when tumor recognition is constitutively present to some extent. This finding is particularly striking in view of the close association between antigen-specific T-cell reactivity in TILs and antigen expression in fresh tumor samples,10 because it highlights the relative importance of functional assays to detect tumor-specific responses.
Overexpressed antigens
Antigen isolated from immunoselected-melanoma (AIM-2) is a widely expressed tumor antigen, which can be found in melanoma as well as tumors of other histological origin.15 Therefore, we have generated HLA-A1-restricted T cells specific for the non-spliced AIM-2-derived peptide RSDSGQQARY, starting from pre-REP TILs as obtained from one patient with metastatic melanoma (the autologous melanoma cell line from this patient was not available) and tested in co-culture assays with short-term cultured allogeneic BRAFV600E mutant cell lines from Patients 1, 2, 3 and 4 (all of which were HLA-A1+). AIM-2-specific T cells recognized a single cell line (from Patient 2), and tumor recognition was increased by the pre-treatment of melanoma cells with Vem (Fig. 3A).
Cancer-testis antigens
TAG genes encoding the shared cancer-testis antigens TAG-1 and TAG-2 are expressed in an unusually high percentage of melanoma patients (over 80%).16 TILs specific for the HLA-A3-restricted TAG-derived peptide RLSNRLLLR were generated from Patient 4, and tested in co-culture with short-term cultured autologous BRAFV600E mutant cancer cells. Importantly, recognition by antigen-specific T cells was augmented in an autologous setting (Fig. 3A and B).
Changes in gene expression upon increased antigen recognition as induced by BRAF inhibition
In an attempt to decipher the mechanisms responsible for the enhanced immunogenicity of melanoma cells subjected to BRAF inhibition, we have analyzed two publicly available cDNA microarray data sets from several established malignant melanoma cell lines treated with DMSO or with either 0.25 or 1 μM of Vem. Among the genes screened, we have selected two gene sets that are likely to be involved in tumor recognition.
We have recently compiled a list of all described TAAs.9 The first gene set was derived from 68 antigens (among the indicated 230 TAAs) whose immunogenicity has previously been described in melanoma (Table S1). The second gene set was derived from the “Antigen Processing and Presentation” pathway of the KEGG database, and included genes involved in the MHC Class I-restricted antigen presentation process (Table S2).
Of the 68 TAAs, 37 and 48 were annotated in the GSE20051 and GSE24862 data sets, respectively. Upregulated TAA-encoding genes included MLANA, RAB38 and PRAME in GSE20051 and MUM1, PRAME, BIRC5 and RAB38 in GSE24862. Downregulated genes included EPHA2, SNRPD1, WDR46 and MC1R in GSE200051, BIRC7 and EPHA in GSE24462, respectively (Fig. 4A and B). With regard to the MHC Class I antigen presentation pathway, among 32 genes selected 23 and 14 were annotated in the GSE20051 and GSE24462 data sets, respectively. Upregulated genes included HLA-G, PDIA3, HLA-F, PSME1, HLA-E, CREB1, LGMN and TAPBP in GSE20051 and HSP90AA1, HSP90AA2, CALR, CANX, HSPA2, B2M, CREB1 in GSE24862. Downregulated genes encompassed PSME3, HSPA8, NFYA, HSP90AB1, HSPA6, NFYB, HSP90AA1 and HSPA5 in GSE20051 while PSM3 only was observed in GSE24862 (Fig. 4C and D).

Figure 4. Expression analysis of genes associated with the immune response upon BRAF inhibition. (A-D) The two independent data sets GSE20051 and GSE24862 containing gene expression data from malignant melanoma cell lines treated with vemurafenib (Vem) were analyzed as described in Materials and Methods. Hierarchical clustering of significantly modulated genes in the TAAs (A, B) and in the MHC class I antigen processing and presentation pathway gene sets (C, D), as found in the GSE20051 (A, C) and GSE24862 (B,D) data sets.
Discussion
Nowadays, patients bearing BRAFV600E mutant melanoma are in most cases offered first-line treatment with Vem. However, slowly progressing and asymptomatic patients could also be good candidate for immunotherapy. In particular, in the future ACT might become an alternative treatment for patients with a surgically resectable lesion and a good performance status.5
Constitutive activation of the BRAF-MAPK signaling pathway has previously been identified as one major mechanism of immune evasion by human melanoma,17 and BRAF-targeted therapy has been linked to increased infiltration of tumors by T cells,18 pointing to this pathway as to a potential target for overcoming cancer immune escape.
The data presented in this report suggest that an appropriately timed sequential treatment with Vem and ACT might result in synergistic antitumor effects. Moreover, the use of ACT may (at least theoretically) clear all the possible occult sites of disease including early foci of resistant (or partially resistant) cells that would be responsible for relapse after an initial response to the Vem-based monotherapy. In this regard, ACT could help consolidating the beneficial anticancer effects of Vem and significantly improve the long-tem prognosis of melanoma patients.
Indeed, apart from the well-known effects of BRAF inhibitors, leading to cell cycle arrest and apoptosis,19 our data prove that Vem additionally exert potent effects on BRAFV600E mutant cancer cells, leading to improved antigen presentation and eventually recognition by T cells that do not appear to be restricted for any particular class of TAAs. Of note, clinical grade ACT products contain CD8+ T cells specific for several classes of TAAs.9
The BRAFV600K mutation accounts for about 10% of all BRAFV600 mutations, and recent data indicate that Vem can induce objective responses in patients bearing this mutation.1,2,20 Here, we additionally show that Vem may exert immunosensitizing effect also in BRAFV600K mutant melanoma cells.
By using a meta-analysis approach, we have studied the factors that may be responsible for the observed increased recognition of melanoma cells treated with Vem. To this aim, we have analyzed two publicly available cDNA microarray data sets on Vem-treated melanoma cell lines. Although in the platforms used to perform the analysis cell lines and drug concentrations were different as compared with those used in our study, we were able to discriminate possible factors involved in the enhanced immune recognition of melanoma cells after treatment with Vem.
The analysis of the expression of melanoma-associated antigens revealed a significant upregulation of RAB38 and PRAME, in both data sets. However, we were not able to detect specific responses to any of these two antigens in TIL cultures, neither in this study or previously.9 Limitations of currently available libraries allowed us to include only one RAB-38 and four PRAME-derived peptides in our screening for melanoma associated epitopes, invariably restricted for HLA-A2,. However, many patients screened in this study or previously were HLA-A2-. Therefore, the contribution of T cells specific for these antigens to the observed increased autologous tumor recognition could not be established in this study. Of note, our results suggest not only that BRAF inhibition may synergize with ACT but also that novel vaccination approaches specifically targeting RAB38 or PRAME may additionally benefit from a combination therapy. Interestingly, immunization strategies with RAB38 (ISRCTN trial identifier: ISRCTN21224989) or PRAME21 are already being tested as monotherapy in clinical trials.
Conversely, in both data sets a significant downregulation of the EPHA2 tumor antigen was observed, but—again—the specific contribution of EPHA2-specific T cells could not be established. On the other hand, the analysis of the genes associated to the MHC Class I-restricted antigen processing and presentation pathway revealed differences between the two data sets, although one major problem was represented by a great number of annotated genes missing in GSE24862. In both data sets, a significant downregulation of PSME3 was observed.
In contrast, a significant upregulation of MHC Class I-associated as well as HSP-coding genes was observed in both data sets. Notably, HSPs are involved in chaperoning proteins to degradation and in funneling the relevant degradation products to the MHC class I presentation pathway.22,23 Theoretically, their increased expression may be linked to the situation of stress induced by BRAF inhibitors in cells that otherwise exhibit a constitutive activation of this signaling pathway.
Although MHC class I upregulation has not been recognized previously as a feature of BRAFV600 mutant melanoma cells responding to BRAF inhibitors,8 this modulated expression of significant genes associated with MHC Class I antigen processing and presentation pathways may explain the observed (apparent) non-antigen class-restricted improved tumor recognition.
In conclusion, this report further supports the concept of combining BRAF-targeted agents and immunotherapies, especially ACT. Recently,two trials assessing the clinical efficacy of this combination were initiated (clinicaltrials.gov identifier: NCT01585415 and NCT01659151). We expect that this study will provide clinical evidence on the feasibility of this approach.
Materials and Methods
Tumor specimens
All the materials were obtained after approval by the Scientific Ethics Committee for the Capital Region of Denmark. Written informed consent was obtained from patients before any procedure, according to the Declaration of Helsinki.
TIL generation and melanoma cell lines
Clinical grade TILs were generated as previously described with a two-step method.24 Several TILs were produced from each patient, either with the standard or the “young TIL” procedure. Each TIL culture was considered “independent” from the others, because they were generated by starting from different tumor fragments which were kept separated during the entire process of clinical-grade TIL generation.
Autologous short-term melanoma cell lines were established from additional tumor fragments originating from the same lesions used for TIL generation by serial passage of adherent cells, as previously described.24 BRAF mutational status testing was performed at the Department of Pathology, Copenhagen University Hospital at Herlev by Pyrosequencing technology using primers as described by Richman et al.25 We used a Pyromark Q24 with a sensitivity of 5% to detect mutations. Vem was obtained from Selleckchem (S1267). Sensitivity to Vem was tested in a flow-cytometry based cell proliferation assay after 72 h of exposure to various drug concentrations, as previously described.12 The sensitivity of the melanoma cell lines used in this study to Vem is shown in Figure S1.
We generated short-term melanoma cell lines bearing the BRAFV600 mutation and relevant clinical grade TIL culture from five patients with AJCC stage III or IV melanoma. A total of 22 independent clinical grade TIL cultures were produced: four from Patient 1; four from Patient 2; three from Patient 3; seven from Patient 4; four from Patient 5. HLA-A typing (performed at Herlev Hospital by PCR on selected HLA-A alleles) revealed the following HLA-A distribution: Patient A1+, A24+; Patient 2 A1+, A3+; Patient 3 A1+; Patient 4 A1+; A3+; Patient 5 A2+.
Cell lines FM57 (BRAFV600K), FM88 (BRAFV600K) and FM92 (BRAFV600E) (from the ESTDAB, see http://www.ebi.ac.uk/ipd/estdab/ for further information) were a gift of Prof. Per Thor Straten (Copenhagen, Denmark). All three cell lines were HLA-A2+ and expressing high levels of both MART-1 and gp100 (refer to ESTDAB database). Information on the BRAF mutational status of these cell lines was obtained from Jonsson et al.26
TILs from the five patients included in this study, or obtained previously both from patients bearing BRAFV600 mutant or wild-type melanoma, were screened for T-cell responses against known TAAs by combinatorial encoding of fluorochrome conjugated peptide-MHC multimers, as previously described.27 Subsequently, TIL cultures enriched in defined antigen-specific CD8+ populations were produced by FACS-sorting (on a FACSAria instrument, BD) of specific T cells from pre-REP cultures, followed by one cycle of REP, performed as previously described.24 By doing so, these “enriched” TILs were comparable in number of in vitro proliferation cycles and differentiation state to regular clinical grade non-selected TILs which are used for patient treatment.
Antitumor activity of TILs
The following fluorochrome-conjugated antibodies were used for flow cytometry: anti CD8-PerCP (345774), CD4-FITC (345768), CD107a-PE (555801), IFNγ-PeCy7 (557643); TNFα-APC (554514).; TNFα-PE (340512) (all from BD). Fixation/Permeabilization Buffer (00–5223–56 and 00–5123–43), Permeabilization Buffer (00–8333–56) and Fixable Viability Dye eFluor® 450 (65–0863–14) were from Ebiosciences, GolgiPlug was from BD (555029). Fluorochrome conjugated (PE or APC) peptide-HLA multimers were produced in house, as previously described.28
Analysis of antitumor activity was performed as previously described.24 Autologous or HLA-A-matched cancer cells used as targets were pretreated with the BRAF selective inhibitor Vem at a dose around the predicted GI50 (between 100 and 250 nM) or which induced an effect close to the maximum (1 to 2 μM). These doses were chosen on the basis of cell sensitivity to the drug (Fig. S1). Allogeneic cell lines were considered HLA-A-matched when they had at least one HLA-A allele in common with the relevant TILs. Antitumor responses of CD8+ TILs specific for defined tumor antigens were evaluated with combined intracellular cytokine and fluorochrome conjugated peptide-MHC multimer staining, as described.29 In selected experiments (all those involving the cell line from Patient 3), cancer cells were concurrently treated with 100 IU/mL IFNγ (Imukin, from Boehringer-Ingelheim) to increase in vitro antigen processing and presentation, as previously described.12
Fluorescence data were acquired using a BD FACSCanto II flow cytofluorometer. At least 100 000 live TILs were acquired. Analysis was performed with BD FACSDiva Software.
The D'Agostino-Pearson normality test was performed to check for normal distribution of the values. Differences between different groups were compared with two-tailed Stundet’s t-tests for paired data and Wilcoxon matched pairs tests (comparison with Vem-treated or untreated) for normally or non-normally distributed sets of data, respectively. Statistical analyses were performed with GraphPad Prism 5 (GraphPad Software).
Microarray data processing and analysis
Two independent data sets (GSE20051 and GSE24862) containing gene expression data from malignant melanoma cell lines treated with Vem were identified in the Gene Expression Omnibus (GEO) databank. Raw data files (.CEL files) were normalized using the RMA algorithm followed by gene-wise mean centering. Normalized data were annotated using the corresponding platform annotation files available from GEO and multiple probes collapsed by averaging the expression values across the probes.
Two manually curated gene signatures were used for the analysis. The first gene signature included all the melanoma genes known to be immunogenic. The second gene signature belonged to the MHC Class I antigen processing and presentation pathway and was derived from the “Antigen Processing and Presentation” pathway of the KEGG database (please visit http://www.genome.jp/kegg/ for further information).
Statistical analyses on the available genes were performed using two class-paired. Significance analyses of microarrays and unsupervised hierarchical clustering of significant genes were executed using the Pearson’s correlation as similarity measurement.
Supplementary Material
Acknowledgments
Tina Juanita Seremet and Kirsten Nikolajsen are acknowledged for technical assistance. The studies were supported by grants from Roche A/S Denmark, Aase and Ejnar Danielsens Foundation, The Danish Cancer Society, the Lundbeck Foundation and the Capital Region of Denmark Research Foundation.
Glossary
Abbreviations:
- ACT
adoptive T-cell transfer
- AJCC
American Joint Committee on Cancer
- ESTDAB
European Searchable Tumour Line Database
- GI50
50% growth inhibitory
- HSP
heat-shock protein
- MDA
melanoma differentiation antigen
- REP
rapid expansion protocol
- TAA
tumor-associated antigen
- TCR
T-cell receptor
- TIL
tumor-infiltrating lymphocyte
- Vem
vemurafenib (PLX4032)
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/21940
References
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–14. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ackerman A, McDermott DF, Lawrence DP, Gunturi A, Flaherty KT, Giobbie-Hurder A, et al. Outcomes of patients with malignant melanoma treated with immunotherapy prior to or after vemurafenib. J Clin Oncol. 2012 [Google Scholar]
- 4.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber J, Atkins M, Hwu P, Radvanyi L, Sznol M, Yee C, Immunotherapy Task Force of the NCI Investigational Drug Steering Committee White paper on adoptive cell therapy for cancer with tumor-infiltrating lymphocytes: a report of the CTEP subcommittee on adoptive cell therapy. Clin Cancer Res. 2011;17:1664–73. doi: 10.1158/1078-0432.CCR-10-2272. [DOI] [PubMed] [Google Scholar]
- 6.Hong DS, Vence L, Falchook G, Radvanyi LG, Liu C, Goodman V, et al. BRAF(V600) inhibitor GSK2118436 targeted inhibition of mutant BRAF in cancer patients does not impair overall immune competency. Clin Cancer Res. 2012;18:2326–35. doi: 10.1158/1078-0432.CCR-11-2515. [DOI] [PubMed] [Google Scholar]
- 7.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16:6040–8. doi: 10.1158/1078-0432.CCR-10-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 9.Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebæk E, et al. Dissection of T-cell antigen specificity in human melanoma. Cancer Res. 2012;72:1642–50. doi: 10.1158/0008-5472.CAN-11-2614. [DOI] [PubMed] [Google Scholar]
- 10.Kvistborg P, Shu CJ, Heemskerk B, Fankhauser M, Thrue CA, Toebes M, et al. TIL therapy broadens the tumor-reactive CD8(+) T cell compartment in melanoma patients. Oncoimmunology. 2012;1:409–18. doi: 10.4161/onci.18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:1642–50. doi: 10.1158/0008-5472.CAN-11-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donia M, Hansen M, Sendrup SL, Zeeberg Iversen T, Ellebæk E, Andersen MH, et al. Methods to improve adoptive T-cell therapy for melanoma: IFN-γ enhances anticancer responses of cell products for infusion. J Invest Dermatol. doi: 10.1038/jid.2012.336. In press. [DOI] [PubMed] [Google Scholar]
- 13.Junker N, Kvistborg P, Køllgaard T, Straten Pt, Andersen MH, Svane IM. Tumor associated antigen specific T-cell populations identified in ex vivo expanded TIL cultures. Cell Immunol. 2012;273:1–9. doi: 10.1016/j.cellimm.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Sørensen RB, Junker N, Kirkin A, Voigt H, Svane IM, Becker JC, et al. The immunodominant HLA-A2-restricted MART-1 epitope is not presented on the surface of many melanoma cell lines. Cancer Immunol Immunother. 2009;58:665–75. doi: 10.1007/s00262-008-0588-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harada M, Li YF, El-Gamil M, Ohnmacht GA, Rosenberg SA, Robbins PF. Melanoma-Reactive CD8+ T cells recognize a novel tumor antigen expressed in a wide variety of tumor types. J Immunother. 2001;24:323–33. doi: 10.1097/00002371-200107000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Hogan KT, Coppola MA, Gatlin CL, Thompson LW, Shabanowitz J, Hunt DF, et al. Identification of novel and widely expressed cancer/testis gene isoforms that elicit spontaneous cytotoxic T-lymphocyte reactivity to melanoma. Cancer Res. 2004;64:1157–63. doi: 10.1158/0008-5472.CAN-03-2209. [DOI] [PubMed] [Google Scholar]
- 17.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–6. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–94. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 19.Søndergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, et al. Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032. J Transl Med. 2010;8:39. doi: 10.1186/1479-5876-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubinstein JC, Sznol M, Pavlick AC, Ariyan S, Cheng E, Bacchiocchi A, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med. 2010;8:67. doi: 10.1186/1479-5876-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weber JS, Vogelzang NJ, Ernstoff MS, Goodman OB, Cranmer LD, Marshall JL, et al. A phase 1 study of a vaccine targeting preferentially expressed antigen in melanoma and prostate-specific membrane antigen in patients with advanced solid tumors. J Immunother. 2011;34:556–67. doi: 10.1097/CJI.0b013e3182280db1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Callahan MK, Garg M, Srivastava PK. Heat-shock protein 90 associates with N-terminal extended peptides and is required for direct and indirect antigen presentation. Proc Natl Acad Sci U S A. 2008;105:1662–7. doi: 10.1073/pnas.0711365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Menoret A, Srivastava P. Roles of heat-shock proteins in antigen presentation and cross-presentation. Curr Opin Immunol. 2002;14:45–51. doi: 10.1016/S0952-7915(01)00297-7. [DOI] [PubMed] [Google Scholar]
- 24.Donia M, Junker N, Ellebaek E, Andersen MH, Straten PT, Svane IM. Characterization and comparison of “Standard” and “Young” tumor infiltrating lymphocytes for adoptive cell therapy at a Danish Translational Research Institution. Scand J Immunol. 2011 doi: 10.1111/j.1365-3083.2011.02640.x. In press. [DOI] [PubMed] [Google Scholar]
- 25.Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–7. doi: 10.1200/JCO.2009.22.4295. [DOI] [PubMed] [Google Scholar]
- 26.Jönsson G, Dahl C, Staaf J, Sandberg T, Bendahl PO, Ringnér M, et al. Genomic profiling of malignant melanoma using tiling-resolution arrayCGH. Oncogene. 2007;26:4738–48. doi: 10.1038/sj.onc.1210252. [DOI] [PubMed] [Google Scholar]
- 27.Andersen RS, Kvistborg P, Frøsig TM, Pedersen NW, Lyngaa R, Bakker AH, et al. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat Protoc. 2012;7:891–902. doi: 10.1038/nprot.2012.037. [DOI] [PubMed] [Google Scholar]
- 28.Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. 2006;1:1120–32. doi: 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]
- 29.Dimopoulos N, Jackson HM, Ebert L, Guillaume P, Luescher IF, Ritter G, et al. Combining MHC tetramer and intracellular cytokine staining for CD8(+) T cells to reveal antigenic epitopes naturally presented on tumor cells. J Immunol Methods. 2009;340:90–4. doi: 10.1016/j.jim.2008.09.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.