

Abstract
Inflammation is a major risk factor for carcinogenesis in patients affected by chronic colitis, yet the molecular mechanisms underlying the progression from chronic inflammation to cancer are not completely understood. Activation of the Toll-like receptor 4 (TLR4)-NFκB signaling axis is associated with inflammation. Thus, we hypothesized that inhibition of TLR4-NFκB signaling might help in limiting inflammatory responses and inflammation-induced oncogenesis. In this work, we studied the effects of a TLR4-interacting surfactant protein A-derived (SPA4) peptide on lipopolysaccharide (LPS)-induced TLR4-NFκB signaling and cancer progression. We first characterized this peptide for its ability to bind the TLR4 ligand-LPS and for physico-chemical characteristics. Inflammation was induced by challenging the colon cancer SW480 cells with Escherichia coli LPS. Cells were then treated with varying amounts of the SPA4 peptide. Changes in the expression of TLR4, interleukin (IL)-1β and IL-6, in intracellular NFκB-related signal transducers (IKBα, p65, phosphorylated IKBα, phosphorylated p65, RelB, COX-2) as well as in the transcriptional activity of NFκB were studied by immunocytochemistry, immunoblotting and NFκB reporter assay, respectively. Simultaneously, the effects on LPS-induced cell migration and invasion were determined. We found that the SPA4 peptide does not bind to LPS. Rather, its binding to TLR4 inhibits the LPS-induced phosphorylation of p65, production of IL-1β and IL-6, activity of NFκB, migration and invasion of SW480 cells. In conclusion, our results suggest that the inhibition of TLR4-NFκB signaling by a TLR4-binding peptide may help for the treatment of chronic inflammation and prevention of inflammation-induced cancer in patients with colitis.
Keywords: NFκB, TLR4, cancer, inflammation, surfactant protein A
Introduction
Colorectal cancer is the third most common cancer and leading cause of cancer-related mortality in the United States. As per the recent annual report, there were 141,210 new cases of colorectal cancer and 49,380-associated deaths in the US (National Cancer Institute at the National Institute of Health, Washington DC). An exaggerated inflammatory response has been reported to increase the risk of colorectal cancer in patients with inflammatory bowel disease (IBD), ulcerative colitis (UC) or Crohn’s disease.
This inflammation-induced progression of cancer at least theoretically can be suppressed by anti-inflammatory agents. Common anti-inflammatory medications include aminosalicylates, corticosteroids and immunosuppressive drugs (e.g., methotrexate, cyclosporin A). While it remains to be established whether conventional anti-inflammatory agents can have chemopreventive effects against cancer, recent advances in our understanding of the mucosal immune system has led to the development of targeted medications against IBD such as anti-tumor necrosis factor α (TNFα) products.1 A number of anti-TNFα products (antibodies and receptor antagonists) are approved by the Food and Drug Administration for reducing intestinal inflammation in patients with colitis.2 These biologics provide only transient relief because of their binding to secreted TNFα. Thus, a large and continued dose of these agents is usually needed to achieve persistent therapeutic effects, which leads to significant toxicity, including an increased risk of infections and perturbed physiological functioning.3 Presumably, a therapy that inhibits intracellular inflammatory signaling can exert sustained anti-inflammatory effects by preventing the production of inflammatory mediators and hence blocking the progression from chronic inflammation to cancer.
Key molecules involved in inflammatory pathways include Toll-like receptors (TLRs), nuclear factor (NF)κB, cytokines, growth factors, kinases, cyclooxygenases and nitric oxide synthases. TLRs are unique because they sense “danger signals” in the form of infectious agents or stress-related ligands and—by the virtue of their intracellular Toll/Interleukin-1 receptor (TIR) domain—are associated with a complex intracellular signaling network that involves NFκB. Thus, new therapies targeting TLRs may be beneficial for suppressing inflammation in a sustained fashion. Among a number of TLRs, TLR4 was first discovered in 1996 as an innate immune receptor for Gram-negative bacterial lipopolysaccharide (LPS). TLR4 is now recognized as pattern recognition receptor against a diverse array of ligands including endogenous stress ligands or damage-associated molecular patterns (DAMPs) such as heat-shock proteins and fibronectin. A number of recent studies have reported the involvement of TLR4 in colitis and cancer progression.4-6 Constitutive activation of TLR4 augments inflammation in colitis patients and contributes to colitis-induced tumorigenesis.4 The colon cancer cell lines: SW480 and SW620 have been shown to constitutively express TLR4.7 It has been suggested that TLR4 signaling might provide cancer cells with the capacity to evade the immune system and metastatize.8,9 Accordingly, LPS has been reported to stimulate metastasis in SW480 cells by increasing migration, adhesion and invasion in a TLR4-dependent fashion.10-12 Thus, we postulate that the suppression of LPS-stimulated TLR4-signaling will not only help to control inflammation, but also mitigate inflammation-induced cancer progression.
We recently identified a TLR4-interacting peptide that is derived from a natural endogenous defense protein surfactant protein A (SP-A).13 Although SP-A is mainly found in the lung, epithelial cells at other mucosal sites, including the intestine, have also been shown to express SP-A.14 The expression of SP-A is important for the maintenance of immune homeostasis because of its anti-microbial and anti-inflammatory properties.15-17 Full-length SP-A has been shown to exert pro-inflammatory effects through its N-terminal collagen domain, and anti-inflammatory effects through its C-terminal region.18 Furthermore, we and others have recently reported that the anti-inflammatory effects of SP-A are partly mediated by the interaction between C-terminal region of SP-A and TLR4.13,19,20 Thus, the TLR4-interacting C-terminal region of SP-A may be of therapeutic value. Using cutting edge computer-modeling and immunobiochemical approaches, we discovered that a 20 amino acids long peptide derived from the C-terminal region of SP-A, which we named SPA4, interacts with TLR4 and reduces the secretion of TNFα by dendritic cells treated with LPS.13
In this study, we investigated whether SPA4 peptide inhibits LPS-induced TLR4-NFκB signaling and inflammatory responses in colon cancer SW480 cells. Simultaneously, we investigated the effects of SPA4 peptide on LPS-induced migration and invasion of SW480 cells.
Results
Characteristics of the SPA4 peptide
All the batch preparations of the SPA4 peptide (amino acids: GDFRYSDGTPVNYTNWYRGE) were tested for purity by HPLC chromatography and mass spectrometry as well as for any endotoxin contamination by the Limulus Amoebocyte Lysate (LAL) assay. The endotoxin level was undetectable (below the lower limit of 0.001 ng/mL) in reconstituted SPA4 peptide suspensions of all the batches.
As per the Solvent AccesiBiLitiEs (SABLE), version 2 program, SPA4 peptide is predicted to have coiled and β strand structures. Relative solvent accessibility (RSA) measurements suggest that SPA4 peptide is easily soluble in water (Fig. 1). Furthermore, we found the Ko/w partition coefficient of the SPA4 peptide to be 0.56. These results confirm that the SPA4 peptide is hydrophilic.
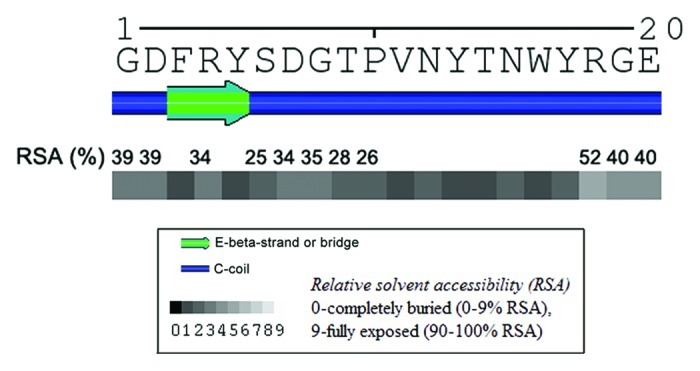
Figure 1. Secondary structure and relative solvent accessibility (RSA) of the 20mer SPA4 peptide. Values were predicted by the SABLE program, version 2. The values indicate the percent RSA real values of amino acids exhibiting > 25% RSA (predicted with wApproximator setting).
SPA4 peptide does not bind LPS
We have previously shown that the SPA4 peptide binds to recombinant TLR4-MD2 protein.13 In order to further validate that the anti-inflammatory effects of SPA4 peptide are not mediated by the binding of SPA4 peptide to the TLR4 ligand-LPS, we studied the association of SPA4 peptide to LPS in vitro. Our results demonstrate no binding of SPA4 peptide to LPS (Fig. 2A).

Figure 2. Binding of SPA4 peptide to LPS-TLR4-MD2. (A) SPA4 peptide does not bind to lipopolysaccharide (LPS) as measured by the LAL assay. The assay reaction was read after 6 and 12 min of substrate-addition. An equivalent amount of polymyxin B was included as positive control. (B) Computer model of LPS-TLR4-MD2 showing the binding sites of LPS (within red shadowed area) and SPA4 peptide (shown within gray shadowed area). LPS, TLR4 and MD2 structures are shown in red, blue/green and gray colors, respectively. This figure is reprinted and adapted by permission from MacMillan Publisher Ltd: Nature.22
Additional evidence is provided by superimposing the predicted SPA4 peptide binding site on a computer model highlighting the LPS-binding site within the TLR4-MD2 complex:21,22 the binding site of SPA4 peptide to TLR4 is far away from the LPS-binding site (Fig. 2B). These results support the notion that SPA4 peptide most likely does not compete with LPS for binding to the TLR4-MD2 complex. Most of the experiments in this study were performed with cells that had been challenged with LPS 4h prior to the administration of SPA4 peptide. In these conditions, the LPS-TLR4-MD2 complex was likely to have already formed prior to the addition of the SPA4 peptide.
SPA4 peptide reduces the expression of TLR4
We investigated the effects of the SPA4 peptide on the expression of TLR4 by confocal microscopy. SW480 cells were found to constitutively express TLR4, and TLR4 expression was further increased in response to LPS from Gram-negative bacteria. Immunostaining of TLR4 was uniform throughout the cytoplasm and periphery in both untreated and LPS-challenged cells. TLR4 staining intensity decreased in SW480 cells treated with the SPA4 peptide (Fig. 3).

Figure 3. Effect of SPA4 peptide on TLR4 expression. (A, B) SW480 cells were challenged with 1 µg/mL lipopolysaccharide (LPS) for 4 h and treated with SPA4 peptide (1, 10 and 100 µM) for 1 h. Cells were subsequently stained with Alexa fluor 488-labeled antibody specific for TLR4 (green), nuclear stain Hoechst 33342 (blue) and cytoplasmic phalloidin stain (red). (A) Confocal images of representative fields are shown for untreated cells, cells treated with 1, 10 or 100 µM SPA4 peptide, and cells challenged with 1 µg/mL LPS alone, and LPS-challenged cells treated with 1, 10 or 100 μM SPA4 peptide. (B) Confocal images of different fields were acquired for the cells in which both the nucleus and the cytoplasm were visible in the same plane. The fluorescent staining for TLR4 was quantified by densitometry. Mean (± SEM) densitometric units are shown as bars. Results are from one out of three independent experiment. *p < 0.05 vs. LPS-challenged cells (ANOVA).
The SPA4 peptide inhibits LPS-induced MYD88-dependent NFκB activity
Myeloid differentiation primary response gene 88 (MYD88) is a known adaptor molecule of TLR4 that operates downstream of the LPS-TLR4-MD2 complex but upstream of the transcription factors AP-1 and NFκB. MYD88 engages IL-1 receptor-associated kinase (IRAK) through its death domain and hence transduces TLR4-derived signals.21 In this study, we transfected SW480 cells with a construct coding for a dominant negative variant of MYD88 (MYD88DN).23,24 We observed a transfection efficiency of 63–68%.NFκB activity was then measured using a reporter plasmid (pGL4.32) that contains five copies of the NFκB-responsive element driving the expression of a luciferase reporter. Treatment with the SPA4 peptide (at 10 and 50 µM concentrations) inhibited LPS-induced NFκB activity after 5 h. After 9 h of treatment, the LPS-induced activity of NFκB was no more significantly inhibited by the SPA4 peptide (employed at 1, 10 and and 50 µM concentrations). MYD88DN-transfected cells exhibited reduced NFκB activity following LPS stimulation as compared with cells transfected with a control plasmid. SPA4 did not further reduce the NFκB activity of MYD88DN-transfected cells. These results suggest that the SPA4 peptide does not affect MYD88-independent NFκB signaling (Fig. 4).

Figure 4. SPA4 peptide inhibits lipopolysaccharide (LPS)-induced NFκB transcriptional activity. (A-C) Luciferase activity was measured in lysates of LPS-challenged SW480 cells harvested (A) after 1 h and 5 h of treatment with SPA4 peptide. SW480 cells were co-transfected with either (B) pcDNA3.0 and NFκB luciferase reporter plasmids or (C) MYD88DN and NFκB luciferase reporter plasmids. The bars indicate mean (± SEM) luminescence values normalized to total protein content. The results are from one out of four independent experiments performed in triplicate. *p < 0.05 and ns: not significant, vs. cells challenged with LPS for 5 h or 9 h (ANOVA).
Expression and activation status of NFκB-related signaling molecules is affected by the SPA4 peptide
The expression and activation status of NFκB-related signaling molecules was measured in cell lysates by immunoblotting. The SPA4 peptide led only to subtle changes in the expression levels of NFκB-related signaling molecules yet it caused a considerable decrease in the phosphorylation of p65. Conversely, LPS stimulated p65 phosphorylation (Fig. 5).

Figure 5. Effects of the SPA4 peptide on NFκB signal transduction. (A, B) Ten µg total proteins were run on 4–20% Tris-glycine SDS-PAGE gels in partially-reducing condition (heating at 95°C for 5 min, no reducing agent). Separated proteins were immunoblotted with specific antibodies. Images of immunoreactive bands were acquired and analyzed densitometrically. (A) Acquired images of immunoreactive bands of IKBα, phosphorylated IKBα, p65, phosphorylated p65, RelB and COX-2 in SW480 cells treated with lipopolysaccharide (LPS) ± SPA4 peptide for a total period of 5 h and 9 h (see Figure 4). (B) Densitometric readings of the indicated immunoreactive bands normalized to those of β-actin, which was employed as a loading control. Results are from one out of two independent experiments.
When LPS-challenged SW480 cells were treated with SPA4 peptide for 1 h, a significant decrease was observed in the phosphorylation state of p65. This decrease was also evident when the treatment with SPA4 peptide was extended to 5 h. SPA4 peptide led to a modest initial increase in the expression of IKBα, which later reached to control level. However, SPA4 peptide did not alter, or did so only slightly, the LPS-stimulated changes in the expression or activation of other NFκB-related signaling molecules (Fig. 5).
SPA4 peptide inhibits the expression of interleukin-1β and interleukin-6
Immunoblotting of SW480 cell lysates with an anti-human interleukin (IL)-1β antibody identified three major immunoreactive bands (identified as 1, 2 and 3 in Figure 6). IL-1β is produced as an inactive 31 kDa precursor (also known as pro-IL-1β) that undergoes enzymatic cleavage to a biologically active form (~17.5 kDa)25 (identified as 2 and 3, respectively, in Figure 6). The uppermost immunoreactive band most likely represents the IL-1β/binding protein complex.26,27 On comparison of β actin-normalized densitometric units, the SPA4 peptide was found to inhibit the generation of the active (17.5 kDa) IL-1β form in a dose-dependent manner (Fig. 6A).

Figure 6. Inhibition of lipopolysaccharide (LPS)-induced expression of interleukin-1β and interleukin-6 by SPA4 peptide. (A, B) Colon carcinoma SW480 cells were challenged with 1 µg/mL LPS for 4 h and then with the SPA4 peptide (1, 10, 50 µM) for additional 1 h and 5 h. Total proteins were probed with antibodies specific for (A) interleukin (IL)-1β and (B) IL-6. Densitometric readings of immunoreactive bands were normalized to those of β-actin, which served as loading control. Results are from one out of two independent experiments.
We also studied the expression of IL-6 in SW480 cell lysates treated with LPS alone or in the presence of the SPA4 peptide. Three major immunoreactive bands of IL-6 were detected. IL-6 has been recognized to be secreted as a heterogeneous set of proteins with molecular weights ranging from 19 to 70 kDa. Immunoreactive bands 2 and 3 represent the IL-6 dimer and monomer, respectively. The heaviest immunoreactive protein band represents the multimeric form of IL-6.28 Only subtle changes were observed in IL-6 expression by SW480 cells treated with LPS and the SPA4 peptide for 1 h or 5 h (Fig. 6B).
SPA4 inhibits LPS-induced migration and invasion of SW480 cells
Increased cell migration and invasion are known features of metastatic tumor cells.12 As expected, LPS was found to induce metastatic properties, i.e., cell migration (Fig. 7) and invasion (Fig. 8) in SW480 cells. Treatment with SPA4 inhibited the LPS-induced migration of SW480 cells (Fig. 7). The experiments were designed so that the SPA4 peptide was added to the cells 4 h after the challenge with LPS, implying that LPS and SPA4 remained in the culture medium for the total duration of the assay (Figs. 3–6). The inhibitory effect of SPA4 on LPS-induced cell migration was apparent as early as 24 h after treatment (data not shown) and remained consistent till 72 h. Thus, it is likely that the inhibition of migration of SW480 cells is initiated early and is maintained on the long-term. Finally, we found that the LPS-stimulated invasion of SW480 cells through a Matrigel matrix is significantly inhibited by SPA4 over a period of 96 h (p < 0.001; Figure 8). Treatment with the SPA4 peptide alone did not affect the invasive properties of SW480 cells.
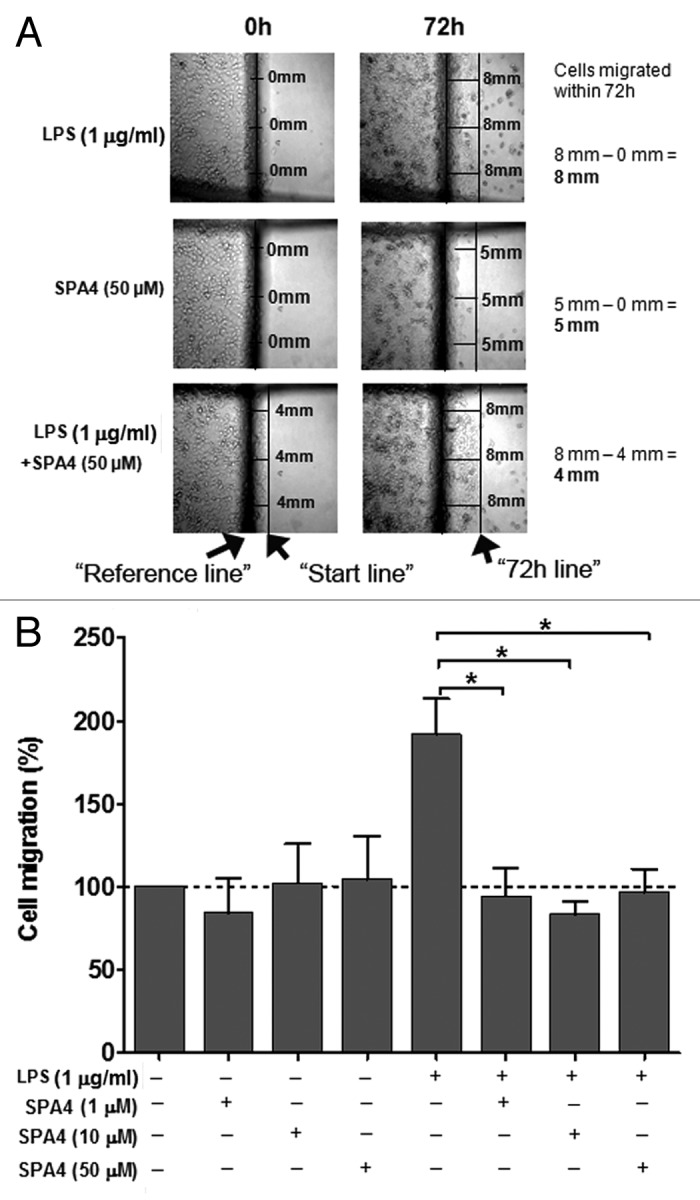
Figure 7. Inhibition of lipopolysaccharide (LPS)-stimulated migration of SW480 cells by the SPA4 peptide. (A) Photomicrographs are shown from one out of four independent experiments. At the beginning of the experiment, a “reference line” (central dark line) and markers were drawn at the bottom of the plate. After scraping, the cells were challenged with 1 µg/mL LPS for 4 h and then treated with SPA4 (1, 10 and 50 µM). On 0 h images, a “start line” was drawn to represent the starting points for cells. On 72 h images, a second line was drawn along the edge of cells to represent the migration. (B) Percent cell migration was calculated for LPS ± SPA4-treated cells as compared with untreated control cells for each experiment. The bar chart represents means (+ SEM) of four independent experiments. *p < 0.05 vs. LPS-challenged cells (ANOVA).

Figure 8. Inhibition of lipopolysaccharide (LPS)-stimulated invasion of SW480 cells by the SPA4 peptide. (A, B) SW480 cells were challenged with 1 µg/mL LPS for 4 h and then with SPA4 (1 and 10 µM). After incubation for 96 h, the matrix was scrubbed off from the top of the insert, and the bottom of the insert was stained with Diff-Quick Wright-Giemsa stain. The photomicrographs of invading cells were taken using a 20X objective. (A) Representative photomicrographs of Wright-Giemsa stained-, untreated control, SPA4 peptide-treated and LPS ± SPA4 peptide-treated SW480 cells. (B) The numbers of invading cells were counted in at least 15 representative microscopic fields, and percentages of cell that invaded the Matrigel under various conditions were calculated relative to the ones obtained with LPS only. The dotted line indicates 100% invasion, as set for LPS-challenged cells. The bar chart represents means (± SEM) from one out of three independent experiments. **p < 0.001 vs. LPS-challenged cells (ANOVA).
Discussion
TLR4 is a well-characterized molecule expressed on immune and non immune cells and plays a critical role in sensing pathogen-derived signals and DAMPs as well as in regulating immune responses. Although its involvement in cancer has not yet been fully established, an increased expression of TLR4 is associated with inflammation-induced carcinogenesis.29-33 Correspondingly, reduced TLR4 activity has been shown to inhibit the secretion of inflammatory cytokines, cancer cell proliferation and cancer-associated pain.34-36
Based on these premises, it has been proposed that novel therapeutics designed to block TLR4 should help reducing inflammation and inflammation-associated oncogenesis. We utilized colorectal cancer SW480 cells as a model system for inflammation-induced cancer progression.11 This work was focused on the effects of a TLR4-interacting peptide, SPA4 (which has recently been discovered in our lab), on LPS-induced TLR4-signaling, inflammatory responses, cell migration and invasion.
The SPA4 peptide is derived from an endogenous host defense protein: SP-A.13 SP-A is mainly expressed as a component of the lung surfactant, yet its expression has also been noted at other mucosal surfaces, such as the intestine, skin, eye and urogenitary systems.37 Accordingly, we detected a constitutive expression of SP-A by SW480 cells (results not shown). Although the expression levels of SP-A in inflammatory or tumor microenvironments in the intestine are not known, we and others have shown that the SP-A levels in lung alveoli are reduced significantly under infectious and inflammatory conditions.15,16,38 SP-A is known to maintain immune homeostasis under normal conditions. Indeed, reduced levels of SP-A in infectious/inflammatory environments can hypothetically affect the immune equilibrium and promote tumor-progression. Using computer-modeling and functional screening of a small peptide library, we identified a short peptide out of the TLR4-interacting region of SP-A, which we called SPA4.13 The SPA4 peptide was found to inhibit LPS-induced inflammatory responses in JAWS II dendritic cells.13
TLR4 signaling is induced in response to a number of endogenous and exogenous ligands released during infectious or noninfectious injuries. LPS from Gram-negative bacteria is a well-defined TLR4 ligand, and has been widely studied. LPS binds to the TLR4-MD2 complex and induces an inflammatory response by activating a complex intracellular signaling network. SW480 cells constitutively express TLR4 and LPS further increases TLR4 expression and induces an inflammatory response, cell adhesion and migration.11 Thus, we utilized this system to investigate the effects of the SPA4 peptide on LPS-induced TLR4-NFκB signaling, inflammation and malignant properties of SW480 cells.
In order to understand the mechanisms of action of the SPA4 peptide, we first confirmed that SPA4 peptide does not directly bind to the TLR4 ligand LPS, and that the binding site of LPS on the TLR4-MD2 complex is physically distant from the binding site of SPA4 peptide. These observations indicate that SPA4 peptide neither binds LPS nor interferes with the binding of LPS to the TLR4-MD2 complex. Furthermore, since the SPA4 peptide is hydrophilic (Ko/w = 0.56), it is expected not to directly affect intracellular inflammatory signaling by itself. Direct binding of SPA4 peptide to TLR4 is further confirmed by our recent findings that the SP-A-TLR4 interaction is inhibited in absence of SPA4 peptide region in a two-hybrid mammalian system (data not shown). Most of the experiments were designed so that LPS-TLR4-signaling was initiated 4 h prior to the treatment with SPA4 peptide. This experimental design rules out the possibility that SPA4 peptide would interfere with CD14 and LBP, LPS-TLR4-MD2 complex formation or the initiation of LPS-TLR4 signaling. Overall, these results support the notion that the anti-inflammatory effects of SPA4 peptide are due to its interaction with TLR4 and not to an interference with the LPS-TLR4-MD2 complex formation.
LPS from Gram-negative bacteria is known to induce inflammation by activating the TLR4-NFκB signaling pathway in MYD88-dependent as well as MYD88-independent manners. Here, we show that the SPA4 peptide inhibits LPS-triggered MYD88-dependent NFκB activity and inflammatory responses. Full-length SP-A has been shown to inhibit LPS-induced NFκB activity,39-41 but upstream molecular events occurring upon the SP-A-TLR4 interaction remain unknown. The N-terminal region of SP-A has been shown to exert pro-inflammatory effects through its binding to calreticulin/CD91.18 In the same study, the C-terminal region of SP-A was shown to block the production of pro-inflammatory mediators by binding to SIRPα. No information is available in the literature about the SPA4 peptide in particular.
Our data suggest that SPA4 peptide upregulates IKBα, an inhibitor of NFκB. This increased expression of IKBα is in accordance with the published data on full-length SP-A.40 The SPA4 peptide was also found to significantly inhibit the LPS-induced phosphorylation of p65 (Fig. 5) and p65 DNA-binding activity (data not shown). Since NFκB is composed of five different subunits: c-Rel, RelB, p65, p50 and p52, reduced phosphorylation of p65 by SPA4 peptide translates into the inhibition of NFκB activity. A number of positive and negative regulators of inflammations besides TLR4 are known to modulate the inflammatory response. It is still possible that interaction between SPA4 peptide and TLR4 affects molecular pathways other than those mediated by NFκB. A detailed study is warranted to fully elucidate the mechanisms of action of SPA4 peptide and its functions in immune and non-immune cell systems.
Concurrent to the inhibition of NFκB signaling by the SPA4 peptide, we observed a decrease in the LPS-induced levels of IL-1β and IL-6. This inhibition of inflammatory signaling and cytokine production correlated well with the inhibition of LPS-induced cell migration and invasion, two hallmarks of cancer metastasis. Although the underlying mechanisms remain to be established, TLR4 signaling has been shown to promote tumor survival and metastasis.42,43 We have investigated a direct effect of SPA4 peptide on the inflammation-induced metastatic properties of SW480 cells; a composite effect is unknown in the in vivo cancer models where multiple cell types and receptors are present. In the ongoing studies employing a mouse model of LPS-induced inflammation, we have found that SPA4 peptide inhibits inflammation and alleviates clinical symptoms (data not shown). These results support our overall hypothesis that the SPA4 peptide inhibits inflammation. Further studies in appropriate in vivo models will have to address whether SPA4 peptide can prevent inflammation-induced cancer progression. The work presented here is a preliminary step in this direction: we have tested our hypothesis in vitro, in cell culture models, which sets the foundation for future in vivo testing. Further validation of these results will promote the development of a novel chemopreventive immunotherapy to control inflammation-induced cancer progression in patients with colitis. In conclusion, our results suggest that the SPA4 peptide has potential to be further developed as an immunotherapeutic strategy to limit inflammation-induced cancer. It is expected that SPA4 peptide may be of clinical utility in other types of cancer where the activation of TLR4 has been shown to exert oncogenic effects.32,33,44,45
Materials and Methods
Cell cultures
Human colorectal adenocarcinoma SW480 cells (original stock from ATCC), were obtained from the laboratory of Dr. Shrikant Anant (University of Kansas Medical Center). Cells were maintained in Dulbecco's minimum essential medium (D-MEM, Invitrogen), supplemented with 4.5 g/L glucose, 1 mM sodium pyruvate, 4 mM L-glutamine, 10% fetal bovine serum and 1% antibiomyco (Invitrogen) at 37°C in a humidified 5% CO2 incubator.
SPA4 peptide
The 20-mer SPA4 peptide (amino acid sequence: GDFRYSDGTPVNYTNWYRGE, derived from the C-terminal region of SP-A), was synthesized by Genscript, NJ. The mass spectroscopy and high-performance liquid chromatography were performed on all the batch-preparations of the SPA4 peptide to confirm its purity. The peptide was suspended in endotoxin-free Hyclone Cell culture grade water, and endotoxin content was measured by Limulus Amoebocyte Lysate (LAL) assay (Charlesriver Lab).
Measurement of n-octanol/water partition coefficient (Ko/w) of SPA4 peptide
The n-octanol/water partition coefficient (Ko/w) is a measure of hydrophobicity/hydrophilicity.46 It is calculated as the ratio of the concentration of a chemical in n-octanol to that in water in a two-phase system at equilibrium. Equal volumes of MiliQ water and n-Octanol were mixed in a microcentrifuge tube, and shaken for 4 h at 25°C. A weighed amount of the SPA4 peptide was then added to this n-Octanol-water mixture and shaken overnight at 25°C. The SPA4 peptide-n-Octanol-water mixture was allowed to settle for 2 h. The aqueous phase was separated by centrifugation at 16,000 × g for 10 min. The concentration of SPA4 peptide in aqueous phase was measured by spectrophotometric absorbance readings at 280 nm. The concentration of the SPA4 peptide in n-Octanol phase was obtained after subtracting the amount of peptide in water phase from that of the total SPA4 peptide added. Finally, the Ko/w of the SPA4 peptide was determined using following formula: Ko/w = concentration of SPA4 in octanol phase / concentration of SPA4 peptide in aqueous phase.
Binding of SPA4 peptide to LPS
The binding of SPA4 peptide to LPS was studied by the LAL assay as per the method described by Giacometti et al.47 Briefly, 0–20 µM SPA4 peptide or polymyxin B (positive control) solutions were added to 0.01 ng/mL Escherichia coli O111:B4 LPS (supplied with the kit, Charles River, MA) in the wells of a 96-well plate and incubated at 37°C for 40 min. Fifty µL of LAL substrate solution were then added to each well, and the plate was incubated for another 10 min. Finally, substrate-buffer solution was added, and optical density (OD) readings were obtained at 405 nm 0, 6 and 12 min after the addition of substrate. ∆OD values for SPA4 peptide or polymyxin B incubated with LPS (∆ODtreatment), LPS alone (∆ODLPS) and blank (∆ODBlank) were calculated by subtracting the OD values obtained at 6 or 12 min from those obtained at 0 min. Percent binding of SPA4 peptide and polymyxin B was calculated at 6 and 12 min of reaction using following formula: Percent binding = 100 × 1-(∆ODtreatment-∆ODblank) / ∆ODLPS-∆ODBlank
Expression of TLR4
We investigated the effect of SPA4 peptide on the expression of TLR4 protein in SW480 cells by immunocytochemistry and laser confocal microscopy. Briefly, about 2.5 × 104 cells were seeded in an 8-well chamber slide (Thermo Fisher, NY) in complete medium. The cells were challenged with 1 µg/mL E. coli O111:B4-derived, highly-purified, low-protein LPS (Calbiochem) for 4 h and then treated with the SPA4 peptide (1, 10 and 100 µM) for additional 1 h. Cells were fixed in 3.5% paraformaldehyde in Dulbecco’s PBS (DPBS) and permeabilized with 0.05% saponin solution.48 Wells were washed with DPBS supplemented with 1% FBS and 0.05% saponin, and stained with TLR4-specific antibody (1:50 dilution, Abcam, MA) and 10 µg/mL Alexa-fluor 488-labeled secondary anti-rabbit IgG antibody (Invitrogen). After washing, cells were stained with 100 nM rhodamin-phallodin (Cytoskeleton Inc.) and 1 µg/mL Hoechst 33342 (Invitrogen). Confocal microscopic images were acquired at the Imaging core facility of the Oklahoma Medical Research Foundation or University of Oklahoma Health Sciences Center, using a Zeiss LSM-510 META or a Leica SP2 confocal microscope.
NFκB reporter activity assay
Since binding of LPS to TLR4 activates NFκB through MYD88-dependent and independent pathways, we investigated the effects of SPA4 peptide on basal and LPS-induced NFκB activity in SW480 cells transfected with a construct coding for a dominant negative variant of MYD88 (MYD88DN) and an NFκB firefly-luciferase reporter plasmid. We studied the short- (1 h) and long-term (5 h) effects of the SPA4 peptide on NFκB activity.
SW480 cells were transiently transfected with the NFκB firefly-luciferase reporter plasmid pGL4.32 (luc2P/NFκB-RE/Hygro, Promega; provided by Dr. Kelly Standifer, Department of Pharmaceutical Sciences, University of Oklahoma Health Sciences Center) and MYD88DN plasmid construct (provided by Dr. Ruslan Medzhitov, Yale University, CT). As a note, MYD88DN lacks the death domain and intermediate domain.24 Briefly, NFκB firefly-luciferase reporter and MYD88DN plasmids (1 µg each) were mixed with 6 µL of Fugene HD reagent (Roche) in 92 µL of serum-free low-glucose DMEM medium (Invitrogen), and incubated for 20 min at room temperature. The transfection-mix was then added to SW480 cells. SW480 cells transfected with a plasmid DNA construct expressing enhanced green fluorescent protein (pHYG-EGFP; Clontech) were observed under Leica DM4000B fluorescent microscope. The transfection efficiency was calculated as percent of cells expressing EGFP over total number of cells in the brightfield channel. An empty vector plasmid DNA (pcDNA 3.0; obtained from Dr. Brian Ceresa, Department of Cell Biology, University of Oklahoma Health Sciences Center) was used as negative control. Cells were incubated for 18–20 h at 37°C in humidified 5% CO2 chamber. After the incubation period, fresh complete medium was added. Cells were then challenged with LPS (1 µg/mL) for 4 h following and then with the SPA4 peptide (1, 10, 50 µM) for 1 h (total period of 5h) or 5 h (total period of 9 h). LPS remained in the medium throughout the incubation period.
After the completion of incubation period, supernatants were removed and cells were washed with DPBS. Cell extracts were prepared using the reporter assay cell-lysis buffer (Promega,) and stored at -80°C for further analysis. The firefly-luciferase activity (measure of NFκB activity) was measured using the luciferase reporter assay system (Promega). Briefly, 50 µL of luciferase assay reagent was added to the 20 µL cell-lysate by automated dispenser of Synergy HT multi-mode microplate reader (Biotek), and luminescence was read within 10 sec. Total protein content in cell lysates was estimated using BCA protein assay kit (Pierce). The luciferase activity units were finally normalized with the total protein content of lysates.
Expression and activation of NFκB pathway-related molecules by immunoblotting
For immunoblotting, 10 µg of total cell lysate proteins were separated on Novex 4–20% Tris–glycine gradient SDS-PAGE gel (Invitrogen, CA) by electrophoresis. Separated proteins were electro-transferred onto a nitrocellulose membrane using iBlot gel transfer device (Invitrogen). Non-specific binding sites were blocked by incubating the membrane with 7% skimmed milk in Tris-buffered saline with 0.4% Tween-20 (TBST) for 1 h at room temperature. Membranes were incubated overnight at 4°C with 1:1000 diluted antibodies against NFκB canonical pathway molecules: phosphorylated IKBα, total IKBα, p65, RelB (Cell Signaling Technology), phosphorylated p65 (Santacruz Biotech) and cyclooxygenase-2 (COX-2; Santacruz Biotech). Membranes were washed with TBST and incubated at room temperature for 45 min with 1:3500 diluted horseradish peroxidase (HRP)-conjugated-secondary antibodies (Sigma-Aldrich). Immunoreactive bands were detected by Super Signal West Femto detection reagent (Thermo Fisher Scientific). In order to ensure equal protein loading in the wells, the membranes were stripped of probing antibodies at 60°C for 45 min using a stripping solution containing 10% SDS, 0.5 M Tris and β-mercaptoethanol (35 μL/mL), and re-probed with an anti-β-actin antibody (Sigma-Aldrich; 1:1000 in TBST). Immunoblots were imaged using the Ultraquant image acquisition program (Ultralum Inc.). Densitometric readings were obtained for immunoreactive bands with Image J 1.42q program (NIH). Finally, arbitrary densitometric values for the proteins of interest were normalized to those for β-actin.
Expression of IL-1β and IL-6
We utilized a post-LPS treatment model for assessing the effects of SPA4 peptide on LPS-induced cytokines IL-1β and IL-6. The models are described above. Cell lysates were prepared in commercially available cell-culture lysis reagent (Promega). Ten µg of total proteins were separated on 4–20% Novex Tris–glycine gradient SDS-PAGE gel (Invitrogen, CA) or 10% separating-5% stacking acrylamide gel. The expression levels of cytokines were measured by immunoblotting as described above using 1:1000 diluted antibodies against IL-1β and IL-6. Both antibodies were purchased from Santa Cruz Biotech.
Cell migration
SW480 cells were plated in 30 mm tissue culture-treated dishes at a density of 1 × 106 cells per plate. At 80–90% confluence, a “reference line” was drawn at the bottom of the plate. The cells were scratched off from one side of the reference line using a rubber policeman. A picture was taken at 0 h that helped in marking the “start line.” Cells were then washed with complete medium and incubated with 1 µg/mL LPS and/or SPA4 peptide (1, 10 and 50 µM). Photomicrographs of cells migrated across the “start line” were taken in different fields after each treatment at 24, 48 and 72 h (± 2 h) following traceable inscriptions made under the plate at three different points, with a Canon digital camera. On 24, 48 and 72 h images, a second line was drawn along the edge of cells to represent the migration of cells. Cell migration was calculated by measuring the distance cells migrated from the “start line.” Only the continuous migration of cells was considered for measurement. The islets of cells were disregarded. The cell migration was calculated using the following formula: (distance between “start line” at 0h – “reference line”) - (distance between “72h line” – “reference line”).
Cell invasion
The effects of SPA4 peptide on LPS-induced invasiveness of SW480 cells were studied by a modified Boyden chamber Matrigel method using 8 µm transwell chambers.49 The insert wells were prepared by rehydrating the Matrigel matrix with DMEM medium for 2 h at 37°C. The rehydration solution was carefully removed and complete DMEM medium was added to the bottom of the insert. The cells (125,000 cells per well) suspended in complete DMEM medium were added onto the top of the insert with 8 µm filters (BD Biosciences). One µg/mL LPS was added onto the top of the insert at the time of seeding the cells. After 4 h of incubation with LPS, cells were treated with SPA4 peptide (1 µM and 10 µM). After 96 h of incubation, the medium was removed from the inserts, and the top layer of Matrigel was scrubbed. The inserts were removed and cells at the bottom of the inserts were stained with Diff-Quik Wright-Giemsa stain as per manufacturer’s instructions (Dade Behring). Stained cells at the bottom of the insert were observed under the microscope using 20X objective. Multiple representative photomicrographs were taken for each well and the numbers of cells invaded through the matrix were counted.
Statistics
The results were analyzed by one-way analysis of variance (ANOVA) using a statistical analysis program (Graphpad Prism). A p value < 0.05 was noted, unless otherwise indicated.
Acknowledgments
Authors acknowledge Kaustuv Sahoo, graduate student in Department of Pharmaceutical Sciences for his help in the determination of Ko/w partition co-efficient of SPA4 peptide. We also acknowledge Dr. Dharmalingam Subramaniam, University of Kansas Medical Center, Kansas City, KS, for his help in maintaining the SW480 cells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/22089
References
- 1.Bosani M, Ardizzone S, Porro GB. Biologic targeting in the treatment of inflammatory bowel diseases. Biologics. 2009;3:77–97. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Ho RJ, Chien JY. Drug delivery trends in clinical trials and translational medicine: growth in biologic molecule development and impact on rheumatoid arthritis, Crohn’s disease, and colitis. J Pharm Sci. 2012;101:2668–74. doi: 10.1002/jps.23154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ngo B, Farrell CP, Barr M, Wolov K, Bailey R, Mullin JM, et al. Tumor necrosis factor blockade for treatment of inflammatory bowel disease: efficacy and safety. Curr Mol Pharmacol. 2010;3:145–52. doi: 10.2174/1874467211003030145. [DOI] [PubMed] [Google Scholar]
- 4.Fukata M, Shang L, Santaolalla R, Sotolongo J, Pastorini C, España C, et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm Bowel Dis. 2011;17:1464–73. doi: 10.1002/ibd.21527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rahman FZ, Smith AM, Hayee B, Marks DJ, Bloom SL, Segal AW. Delayed resolution of acute inflammation in ulcerative colitis is associated with elevated cytokine release downstream of TLR4. PLoS One. 2010;5:e9891. doi: 10.1371/journal.pone.0009891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Török HP, Glas J, Tonenchi L, Mussack T, Folwaczny C. Polymorphisms of the lipopolysaccharide-signaling complex in inflammatory bowel disease: association of a mutation in the Toll-like receptor 4 gene with ulcerative colitis. Clin Immunol. 2004;112:85–91. doi: 10.1016/j.clim.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Tang XY, Zhu YQ, Wei B, Wang H. Expression and functional research of TLR4 in human colon carcinoma. Am J Med Sci. 2010;339:319–26. doi: 10.1097/MAJ.0b013e3181cef1b7. [DOI] [PubMed] [Google Scholar]
- 8.He W, Liu Q, Wang L, Chen W, Li N, Cao X. TLR4 signaling promotes immune escape of human lung cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Mol Immunol. 2007;44:2850–9. doi: 10.1016/j.molimm.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Szajnik M, Szczepanski MJ, Czystowska M, Elishaev E, Mandapathil M, Nowak-Markwitz E, et al. TLR4 signaling induced by lipopolysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene. 2009;28:4353–63. doi: 10.1038/onc.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Killeen SD, Wang JH, Andrews EJ, Redmond HP. Bacterial endotoxin enhances colorectal cancer cell adhesion and invasion through TLR-4 and NFkappaB-dependent activation of the urokinase plasminogen activator system. Br J Cancer. 2009;100:1589–602. doi: 10.1038/sj.bjc.6604942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu RY, Chan CH, Spicer JD, Rousseau MC, Giannias B, Rousseau S, et al. LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res. 2011;71:1989–98. doi: 10.1158/0008-5472.CAN-10-2833. [DOI] [PubMed] [Google Scholar]
- 12.Wang JH, Manning BJ, Wu QD, Blankson S, Bouchier-Hayes D, Redmond HP. Endotoxin/lipopolysaccharide activates NFkappa B and enhances tumor cell adhesion and invasion through a beta 1 integrin-dependent mechanism. J Immunol. 2003;170:795–804. doi: 10.4049/jimmunol.170.2.795. [DOI] [PubMed] [Google Scholar]
- 13.Awasthi S, Brown K, King C, Awasthi V, Bondugula R. A toll-like receptor-4-interacting surfactant protein-A-derived peptide suppresses tumor necrosis factor-α release from mouse JAWS II dendritic cells. J Pharmacol Exp Ther. 2011;336:672–81. doi: 10.1124/jpet.110.173765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rubio S, Lacaze-Masmonteil T, Chailley-Heu B, Kahn A, Bourbon JR, Ducroc R. Pulmonary surfactant protein A (SP-A) is expressed by epithelial cells of small and large intestine. J Biol Chem. 1995;270:12162–9. doi: 10.1074/jbc.270.20.12162. [DOI] [PubMed] [Google Scholar]
- 15.Awasthi S, Coalson JJ, Crouch E, Yang F, King RJ. Surfactant proteins A and D in premature baboons with chronic lung injury (Bronchopulmonary dysplasia). Evidence for an inhibition of secretion. Am J Respir Crit Care Med. 1999;160:942–9. doi: 10.1164/ajrccm.160.3.9806061. [DOI] [PubMed] [Google Scholar]
- 16.Awasthi S, Coalson JJ, Yoder BA, Crouch E, King RJ. Deficiencies in lung surfactant proteins A and D are associated with lung infection in very premature neonatal baboons. Am J Respir Crit Care Med. 2001;163:389–97. doi: 10.1164/ajrccm.163.2.2004168. [DOI] [PubMed] [Google Scholar]
- 17.Awasthi S, Magee DM, Coalson JJ. Coccidioides posadasii infection alters the expression of pulmonary surfactant proteins (SP)-A and SP-D. Respir Res. 2004;5:28. doi: 10.1186/1465-9921-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. 2003;115:13–23. doi: 10.1016/S0092-8674(03)00758-X. [DOI] [PubMed] [Google Scholar]
- 19.Awasthi S, Madhusoodhanan R, Wolf R. Surfactant protein-A and toll-like receptor-4 modulate immune functions of preterm baboon lung dendritic cell precursor cells. Cell Immunol. 2011;268:87–96. doi: 10.1016/j.cellimm.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamada C, Sano H, Shimizu T, Mitsuzawa H, Nishitani C, Himi T, et al. Surfactant protein A directly interacts with TLR4 and MD-2 and regulates inflammatory cellular response. Importance of supratrimeric oligomerization. J Biol Chem. 2006;281:21771–80. doi: 10.1074/jbc.M513041200. [DOI] [PubMed] [Google Scholar]
- 21.Carpenter S, O’Neill LA. Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signalling proteins. Biochem J. 2009;422:1–10. doi: 10.1042/BJ20090616. [DOI] [PubMed] [Google Scholar]
- 22.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 23.Medzhitov R, Janeway CA., Jr. Innate immune recognition and control of adaptive immune responses. Semin Immunol. 1998;10:351–3. doi: 10.1006/smim.1998.0136. [DOI] [PubMed] [Google Scholar]
- 24.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–8. doi: 10.1016/S1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 25.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–74. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 26.Symons JA, Eastgate JA, Duff GW. A soluble binding protein specific for interleukin 1 beta is produced by activated mononuclear cells. Cytokine. 1990;2:190–8. doi: 10.1016/1043-4666(90)90015-L. [DOI] [PubMed] [Google Scholar]
- 27.Eastgate JA, Symons JA, Duff GW. Identification of an interleukin-1 beta binding protein in human plasma. FEBS Lett. 1990;260:213–6. doi: 10.1016/0014-5793(90)80106-S. [DOI] [PubMed] [Google Scholar]
- 28.May LT, Viguet H, Kenney JS, Ida N, Allison AC, Sehgal PB. High levels of “complexed” interleukin-6 in human blood. J Biol Chem. 1992;267:19698–704. [PubMed] [Google Scholar]
- 29.Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66:3859–68. doi: 10.1158/0008-5472.CAN-05-3948. [DOI] [PubMed] [Google Scholar]
- 30.Kutikhin AG. Association of polymorphisms in TLR genes and in genes of the Toll-like receptor signaling pathway with cancer risk. Hum Immunol. 2011;72:1095–116. doi: 10.1016/j.humimm.2011.07.307. [DOI] [PubMed] [Google Scholar]
- 31.Slattery ML, Herrick JS, Bondurant KL, Wolff RK. Toll-like receptor genes and their association with colon and rectal cancer development and prognosis. Int J Cancer. 2012;130:2974–80. doi: 10.1002/ijc.26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petricevic B, Vrbanec D, Jakic-Razumovic J, Brcic I, Rabic D, Badovinac T, et al. Expression of Toll-like receptor 4 and beta 1 integrin in breast cancer. Med Oncol. 2012;29:486–94. doi: 10.1007/s12032-011-9885-0. [DOI] [PubMed] [Google Scholar]
- 33.Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, et al. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer. 2010;102:908–15. doi: 10.1038/sj.bjc.6605558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang H, Zhou H, Feng P, Zhou X, Wen H, Xie X, et al. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J Exp Clin Cancer Res. 2010;29:92. doi: 10.1186/1756-9966-29-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan LS, Ping YJ, Na WL, Miao J, Cheng QQ, Ni MZ, et al. Down-regulation of Toll-like receptor 4 gene expression by short interfering RNA attenuates bone cancer pain in a rat model. Mol Pain. 2010;6:2. doi: 10.1186/1744-8069-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Earl TM, Nicoud IB, Pierce JM, Wright JP, Majoras NE, Rubin JE, et al. Silencing of TLR4 decreases liver tumor burden in a murine model of colorectal metastasis and hepatic steatosis. Ann Surg Oncol. 2009;16:1043–50. doi: 10.1245/s10434-009-0325-8. [DOI] [PubMed] [Google Scholar]
- 37.van Rozendaal BA, van Golde LM, Haagsman HP. Localization and functions of SP-A and SP-D at mucosal surfaces. Pediatr Pathol Mol Med. 2001;20:319–39. doi: 10.1080/152279501750412243. [DOI] [PubMed] [Google Scholar]
- 38.Awasthi S. Surfactant protein (SP)-A and SP-D as antimicrobial and immunotherapeutic agents. Recent Pat Antiinfect Drug Discov. 2010;5:115–23. doi: 10.2174/157489110791233559. [DOI] [PubMed] [Google Scholar]
- 39.Moulakakis C, Adam S, Seitzer U, Schromm AB, Leitges M, Stamme C. Surfactant protein A activation of atypical protein kinase C zeta in IkappaB-alpha-dependent anti-inflammatory immune regulation. J Immunol. 2007;179:4480–91. doi: 10.4049/jimmunol.179.7.4480. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, Adam S, Hamann L, Heine H, Ulmer AJ, Buwitt-Beckmann U, et al. Accumulation of inhibitory kappaB-alpha as a mechanism contributing to the anti-inflammatory effects of surfactant protein-A. Am J Respir Cell Mol Biol. 2004;31:587–94. doi: 10.1165/rcmb.2004-0003OC. [DOI] [PubMed] [Google Scholar]
- 41.Alcorn JF, Wright JR. Surfactant protein A inhibits alveolar macrophage cytokine production by CD14-independent pathway. Am J Physiol Lung Cell Mol Physiol. 2004;286:L129–36. doi: 10.1152/ajplung.00427.2002. [DOI] [PubMed] [Google Scholar]
- 42.Lee CH, Wu CL, Shiau AL. Toll-like receptor 4 signaling promotes tumor growth. J Immunother. 2010;33:73–82. doi: 10.1097/CJI.0b013e3181b7a0a4. [DOI] [PubMed] [Google Scholar]
- 43.Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, et al. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009;69:3105–13. doi: 10.1158/0008-5472.CAN-08-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gatti G, Quintar AA, Andreani V, Nicola JP, Maldonado CA, Masini-Repiso AM, et al. Expression of Toll-like receptor 4 in the prostate gland and its association with the severity of prostate cancer. Prostate. 2009;69:1387–97. doi: 10.1002/pros.20984. [DOI] [PubMed] [Google Scholar]
- 45.Ehrchen JM, Sunderkötter C, Foell D, Vogl T, Roth J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol. 2009;86:557–66. doi: 10.1189/jlb.1008647. [DOI] [PubMed] [Google Scholar]
- 46.Ghose AK, Crippen GM. Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships. I. Partition coefficients as a measure of hydrophobicity. J Comput Chem. 1986;7:565–77. doi: 10.1002/jcc.540070419. [DOI] [PubMed] [Google Scholar]
- 47.Giacometti A, Cirioni O, Ghiselli R, Mocchegiani F, D’Amato G, Circo R, et al. Cathelicidin peptide sheep myeloid antimicrobial peptide-29 prevents endotoxin-induced mortality in rat models of septic shock. Am J Respir Crit Care Med. 2004;169:187–94. doi: 10.1164/rccm.200307-971OC. [DOI] [PubMed] [Google Scholar]
- 48.Inaba K, Turley S, Yamaide F, Iyoda T, Mahnke K, Inaba M, et al. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J Exp Med. 1998;188:2163–73. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim HR, Wheeler MA, Wilson CM, Iida J, Eng D, Simpson MA, et al. Hyaluronan facilitates invasion of colon carcinoma cells in vitro via interaction with CD44. Cancer Res. 2004;64:4569–76. doi: 10.1158/0008-5472.CAN-04-0202. [DOI] [PubMed] [Google Scholar]