

Abstract
Myelin basic protein (MBP, 18.5 kDa isoform) is a peripheral membrane protein that is essential for maintaining the structural integrity of the multilamellar myelin sheath of the central nervous system. Reconstitution of the most abundant 18.5 kDa MBP isoform with lipid vesicles yields an aggregated assembly mimicking the protein’s natural environment, but which is not amenable to standard solution NMR spectroscopy. On the other hand, the mobility of MBP in such a system is variable, depends on the local strength of the protein–lipid interaction, and in general is of such a time scale that the dipolar interactions are averaged out. Here, we used a combination of solution and solid-state NMR (ssNMR) approaches: J-coupling-driven polarization transfers were combined with magic angle spinning and high-power decoupling to yield high-resolution spectra of the mobile fragments of 18.5 kDa murine MBP in membrane-associated form. To partially circumvent the problem of short transverse relaxation, we implemented three-dimensional constant-time correlation experiments (NCOCX, NCACX, CONCACX, and CAN(CO) CX) that were able to provide interresidue and intraresidue backbone correlations. These experiments resulted in partial spectral assignments for mobile fragments of the protein. Additional nuclear Overhauser effect spectroscopy (NOESY)-based experiments revealed that the mobile fragments were exposed to solvent and were likely located outside the lipid bilayer, or in its hydrophilic portion. Chemical shift index analysis showed that the fragments were largely disordered under these conditions. These combined approaches are applicable to ssNMR investigations of other peripheral membrane proteins reconstituted with lipids.
Keywords: Myelin basic protein (MBP), Solid-state NMR, Magic angle spinning, J-couplings, INEPT, TOBSY, Multidimensional NMR
1. Introduction
The “classic” 18.5 kDa isoform of myelin basic protein (MBP) is a peripheral membrane protein that plays diverse functional roles in maintenance of mature myelin in the central nervous system [1,2]. The most important function of this protein is to adhere the cytoplasmic faces of the oligodendrocyte membrane in close apposition to form the multilamellar myelin sheath. Various biophysical approaches have been applied to study MBP’s interactions with the myelin membrane [1], including site-directed spin labelling (SDSL) and electron paramagnetic resonance (EPR) spectroscopy [3–5]. These latter studies have shown that portions of the protein are relatively deeply embedded in the lipid bilayer, whereas other segments are exposed to the cytoplasm. Recent solid-state NMR spectroscopic studies have shown that MBP does not significantly disrupt the integrity of the lipid bilayer [6].
All known forms of MBP (splice and post-translationally modified variants) are “intrinsically disordered”, a large class of proteins which are generally extended and conformationally adaptable, only adopting a definite tertiary fold in the presence of some interacting partner [1,7]. The most suitable direct structural probe for this class of proteins is NMR spectroscopy [1,8,9]. Previously, we have described solution NMR studies of 18.5 kDa rmMBP (or rmC1, a completely unmodified form, 176 residues including a C-terminal LEH6 tag) in 30% TFE-d2 (deuterated trifluoroethanol), a membrane-mimetic solvent [10]. More recently, these studies have been extended to rmMBP in KCl solution, a more physiological condition [11]. A peptide fragment encompassing an immunodominant epitope of MBP has also been characterised by solution NMR spectroscopy in aqueous buffer, in 30% TFE-d2, and in dodecylphosphocholine micelles [12]. However, solution NMR methodology is difficult to apply to studies of MBP:lipid complexes formed with the full-length protein, conditions where MBP is known to have an increased proportion of ordered secondary structure (reviewed in [1]). Such reconstituted systems can be adapted, in principle, for solid-state NMR studies.
Solid-state NMR (ssNMR) spectroscopy is proving to be a powerful, emerging technique in structural biology, in its ability to yield biomolecular information at the atomic level. Spectral assignments in uniformly 13C,15N-labelled microcrystalline proteins, and more recently in amyloid fibrils and membrane proteins, appear with increasing regularity [13–23]. In particular, several notable studies include the determination of high-resolution structures of the tripeptide MLF [24], of the alpha-spectrin SH3 domain [17], and of a short peptide fragment of transthyretin [25]. Recent progress in magic angle spinning (MAS) ssNMR spectroscopy [26] has also enabled studies of large, macroscopically unoriented membrane proteins [27–30].
Peripherally membrane-associated MBP represents a completely different system from most globular, amyloid-forming and integral membrane proteins. In contrast to other systems where a significant degree of mobility for relatively small fragments has been reported [21,23,29,31], much of the MBP is surface-accessible when it interacts with lipids, and is therefore highly conformationally flexible. These observations stem from pioneering NMR studies of this protein [32,33], as well as from more recent SDSL-EPR investigations [3]. Fast molecular motions result in averaging of dipolar interactions in the protein, thus necessitating the development of new NMR spectroscopic strategies. The motions can be stopped by going to low temperatures, but the resulting sample exhibits a very high degree of disorder, rendering standard ssNMR approaches inapplicable. At higher temperatures, on the other hand, the results of previous NMR spectroscopic investigations of bovine 18.5 kDa MBP [32,33], and our SDSL-EPR studies on 18.5 kDa rmMBP [3], suggest that fragments of the protein would be sufficiently mobile to remove most of the dipolar interactions and to facilitate through-bond polarization transfers. Here, we combined solution NMR INEPT [34] (insensitive nuclear enhancement of polarization transfer)-based methodology with magic angle spinning and high-power decoupling to study the mobile portion of the membrane-associated protein.
In order to progress in resonance assignment in such a semi-mobile system, constant-time NCACX/NCOCX experiments were performed that combined NC INEPT transfer steps and indirect chemical shift evolutions to minimise the relaxation losses. We have also devised complementary three-dimensional constant-time experiments, CONCACX and CAN(CO)CX, to facilitate the sequential assignment process. Using these combined approaches, we have achieved a partial assignment of the mobile fragments of membrane-associated 18.5 kDa MBP.
2. Materials and methods
2.1. Materials
Electrophoresis grade chemicals such as acrylamide were purchased from ICN Biomedicals (Costa Mesa, CA). Most other chemicals were reagent grade and acquired from either Fisher Scientific (Unionville, ON) or Sigma-Aldrich (Oakville, ON). Electrophoresis grade sodium dodecyl sulphate (SDS) was obtained from Bio-Rad Laboratories (Mississauga, ON). The Ni2+-NTA (nitrilotriacetic acid) agarose beads were purchased from Qiagen (Mississauga, ON). For uniform labelling of protein for NMR spectroscopy, the stable isotopic compounds 15NH4Cl, and 13C6-glucose were obtained from Cambridge Isotope Laboratories (C.I.L., Andover, MA). The lipids DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) and DMPG (1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]) were purchased from Avanti Polar Lipids (Alabaster, AL). The phosphorus assay standard [35,36] and the Peterson assay kit were purchased from Sigma-Aldrich (Oakville, ON), and the latter assay was performed according to the manufacturer’s instructions. The Micro Bicinchoninic Acid assay kit was purchased from Pierce (Rockford, IL), and the assay was performed according to the manufacturer’s instructions [37].
2.2. Purification and characterisation of uniformly 13C,15N-labelled rmMBP
The unmodified “classic” 18.5 kDa recombinant murine MBP (rmMBP, or component rmC1, with a C-terminal LEH6 tag) was expressed in Escherichia coli and purified as previously described by nickel-affinity chromatography [38,39]. An additional step of ion exchange chromatography served to remove minor contaminating material [40]. Protein eluate from the column was dialysed (using tubing with Mr cutoff 6000–8000 Da) twice against 2 L of buffer (50 mM Tris–HCl, pH 7.4, 250 mM NaCl), twice against 2 L of 100 mM NaCl, and finally four times against 2 L ddH2O. Uniformly 13C,15N-labelled protein was cultured in cells grown in M9 minimal media [10]. Protein concentrations were determined by measuring the absorbance at 280 nm, using the extinction coefficient ε=12950 M−1 cm−1 for rmMBP (as calculated by SwissProt for protein in 6.0 M guanidine hydrochloride, 0.02 M phosphate buffer, pH 6.5). Purity of the protein preparation was assayed by SDS-polyacrylamide gel electrophoresis, with results (a major band representing undegraded, unmodified rmMBP) consistent with our previous investigations [38,39].
2.3. Preparation of large unilamellar lipid vesicles (LUVs)
The only “functional” assay of MBP is its ability to aggregate lipid vesicles [41,42], and it was decided to use this phenomenon as the basis for preparing reconstituted protein–lipid samples for ssNMR spectroscopy. To prepare large unilamellar vesicles (LUVs), the DMPC and DMPG lipids were combined in a 1:1 mass ratio, placed at the bottom of 15-mL glass tubes, and dissolved in methanol:chloroform:ddH2O=2:1:1. The organic solvent was dried under a stream of nitrogen gas, followed by vacuum pump drying overnight. The LUVs were prepared by hydrating the lipids in physiological buffer (2 mM HEPES–NaOH, pH=7.8, 100 mM NaCl, 1 mM EDTA) overnight with intermittent vortexing in the first hour. Water bath heating was used to hasten the hydration of the lipids. An Avanti mini-extruder was used to extrude the mixture of lipids through the 100-nm polycarbonate filter 35 times. The concentration of phospholipids was determined by a standard phosphorus assay [35,36]. Vesicle size distribution was characterised by dynamic light scattering using a Zetasizer Nano-S model ZEN1600 (633-nm “red” laser —Malvern Instruments) apparatus, which indicated that the initial (prior to addition of MBP) particle size distribution was unimodal, with a mean of 100 nm.
2.4. Aggregation of LUVs by rmMBP
The standard assay for the ability of MBP to adhere lipid monolayers together is the measurement of the optical density (due to scattering) at 450 nm [41]. The aggregation of DMPC:DMPG LUVs after the addition of our rmMBP preparations was, therefore, monitored in this way [42]. The LUVs were prepared as described above. The rmMBP preparation was diluted in the same buffer as the LUVs to a final concentration of 1 mg/mL. The lipid vesicle aggregation assay was performed by adding the rmMBP to a fixed amount of LUVs (1 mg lipids in 1 mL buffer) with an initial 1:1 protein-to-lipid mass ratio in 2-mL microcentrifuge tubes, and mixing by inversion of the tube several times. Samples were incubated for 2 h at room temperature, and optical density at 450 nm measured in a spectrophotometer.
After measurement of the optical density, the sample was spun down at 14,000 rpm for 10 min in a microcentrifuge for measurement of lipid and protein composition. The pellet was dissolved in 200 μL 1% SDS, and 800 μL MilliQ water was added to make the final volume 1 mL. The amount of lipid present was determined by the standard phosphorus assay [35,36]. The amount of protein in the reconstituted samples was determined by a Micro Bicinchoninic Acid assay [37].
2.5. Reconstitution of rmMBP into lipid vesicles for NMR experiments
Reconstituted protein–lipid samples were prepared for NMR spectroscopy by adding 1 mg rmMBP (at 1 mg/mL) in buffer to 1 mg of lipids (in LUV form, also at 1 mg/mL), mixing by inversion of the sample tube, and incubating for 2 h at room temperature. In general, ten vials (a total of ~7–8 mg of uniformly 13C,15N-labelled protein) were prepared in this way, spun down at 14,000 rpm in a benchtop microcentrifuge, and packed into a 3.2-mm rotor. We used 31P NMR spectroscopy to confirm that all spectra were recorded in the liquid crystalline phase [43]. The 13C spectra recorded for different protein:lipid mass ratios up to 0.7 were essentially identical. Carbon and proton linewidths were found to be independent of the spinning frequency in the range of 6 kHz to 22 kHz.
2.6. Solid-state NMR experiments—instrumentation and processing
One-dimensional, two-dimensional 1H–13C, 1H–15N, and three-dimensional 1H–13C–13C correlation experiments were performed on a Bruker (Bruker BioSpin GmbH, Rheinstetten, Germany) Avance III spectrometer operating at a proton Larmor frequency of 600.13 MHz. Triple resonance 1H–15N–13C experiments for spectroscopic assignments were all performed on a Bruker Avance III spectrometer operating at a proton Larmor frequency of 800.13 MHz. In all experiments, we used 3.2-mm triple-channel HCN solid-state NMR magic angle spinning probes. The external magnetic field stability was controlled using an external D2O-based lock. One-dimensional control experiments were always collected after each multidimensional experiment to monitor the stability of the lock. Unless stated otherwise, the experiments described below were performed at a temperature of 32 °C. In the course of our studies, we found that the line intensities recorded at high magic angle spinning frequencies in the range of 15–22 kHz decayed over the course of a few days. This decay occurred reversibly, i.e., the original intensities could be restored when the spinning was stopped for a few hours, probably owing to a reversible water:lipid phase separation occurring in the sample. Thus, the magic angle spinning frequency of 10 kHz was used in all experiments presented in this work. Further experimental details are discussed in the following sections.
All spectra were processed with NMRPipe [44]. Additional acquisition and processing parameters for each spectrum are included in the figure captions. Chemical shifts were referenced to adamantane, with the downfield 13C resonance assigned to 40.48 ppm (parts per million) [45]. Spectra were analysed with the programs Sparky, Version 3.1 (T.D. Goddard and D.G. Kneller, University of California, San Francisco, CA), and CARA (computer-aided resonance assignment) [46].
Static 31P NMR spectroscopy was performed on the Bruker Avance 500-MHz spectrometer, using an echo [47] experiment with 83 kHz two-pulse phase modulation (TPPM) decoupling [48] during the echo delay and acquisition.
2.7. Multidimensional ssNMR spectroscopy—overview
This work focuses entirely on the identification and characterisation of the solvent-exposed portions of lipid bilayer-associated MBP. Dipolar interactions are largely averaged out in these mobile regions of the protein, thus necessitating the use of through-bond coherence transfers for establishing two-dimensional and three-dimensional correlations. The pulse sequences used in this study are shown in Fig. 1. In all experiments, heteronuclear transfers were established through a refocused INEPT [34,49] experiment. We found that rotor-synchronised TOBSY (total through-bond correlation spectroscopy) [50,51] mixing was the most efficient way to establish homonuclear 13C–13C connectivities. We used P913 TOBSY mixing with 60 kHz 13C radio frequency field strength in all experiments.
Fig. 1.

Experimental pulse sequences. In all pulse sequences, narrow rectangles represent 90° pulses, while wide rectangles are 180° pulses. (A) The HCC three-dimensional experiment. Phase cycling in the experiment was as follows: ϕ1 = ϕ2 = ϕ4 = x, ϕ3 =(y, −y), ϕ5 = ϕ6 = y, ϕ7 =(8(x), 8(−x)), ϕ8 =(2(x), 2(y), 2(−x), 2(−y)), ϕrec =(x, −x, y, −y, −x, x, −y, y, −x, x, −y, y, x, −x, y, −y). The INEPT periods were as follows: τ1 =1.4 ms, τ2 =0.9 ms. The 13C carrier frequency was placed at 45 ppm for mixing between aliphatic carbons, and at 75 ppm for mixing between aliphatic and aromatic carbons. (B) The HHCC NOE experiment used to probe through-space 1H–1H interactions. Phase cycling was the same as in panel A, with two additional pulses phase-cycled to eliminate T1 relaxation effects during NOE mixing: ϕ10 =(−x, x), ϕ11 =(x, −x). (C) Pulse sequence for the three-dimensional constant-time NCACX/NCOCX experiments. The INEPT periods τ3 and τ4 were 1.7 ms. Selective pulses were implemented as a Gaussian cascade [69] of 200 μs for Cα, and as a single Gaussian pulse of 300 μs for carbonyl atoms. The value of ε1 was equal to the length of the selective pulse ϕ7 = ϕ17. Time constants T1 and T2 were set to 21 ms and 25 ms, respectively, in the NCACX experiment, and 24 ms (both) in the NCOCX experiment. Phase cycling in this experiment was as follows: ϕ1 = ϕ2 = ϕ4 = ϕ5 = ϕ6 = ϕ7 = ϕ8 = ϕ9 = ϕ10 = ϕ11 = ϕ12 = ϕ17 = x, ϕ3 = (y, −y), ϕ13 = ϕ18 = y, ϕ19 = (2(y), 2(−x), 2(−y), 2(x)), ϕrec =(x, −x, y, −y, −x, x, −y, y). The pulse sequence and the basic principles in the NCACX experiment were very similar to those of the NCOCX experiment, except that the selective pulses applied to the 13C′ and 13Cα spins were switched. In addition, the 13Cα chemical shift evolution dimension was kept less than 10 ms ( ), to minimise the effect of J-coupling between 13Cα spins and other aliphatic spins. The selective pulses used on 13Cα and 13C′ were implemented as Gaussian cascades [69] and Gaussian pulses, respectively. (D) Pulse sequence for the three-dimensional constant-time CONCACX/CAN(CO)CX experiments. Time constants in CONCACX were T1 =24 ms, T2 =26 ms, T3 =30 ms, τ3 =10 ms, and in CAN(CO)CX they were T1 =20 ms, T2 =24 ms, T3 =26 ms, τ3 =15 ms. Here, ε1 is the length of the selective pulse with phase ϕ29, while ε2 is the length of selective pulse with phase ϕ7. The phase cycling in the pulse sequence was as follows: ϕ1 = ϕ2 = ϕ4 = ϕ6 = ϕ7 = ϕ8 = ϕ9 = ϕ10 = ϕ12 = ϕ17 = ϕ23 = ϕ28 = ϕ29 = x, ϕ3 = ϕ18 = y, ϕ5 =(x, −x), ϕ11 =(2(x), 2(y), 2(−x), 2(−y)), ϕ13 =(2(y), 2(−x), 2(−y), 2(x)), ϕ25 = (−x), ϕrec =(x, −x, y, −y, −x, x, −y, y). GARP (globally optimised alternating phase rectangular pulses) [70] decoupling was used to remove NC J-couplings.
In the experiment depicted in Fig. 1A, intraresidue HCC correlations are recorded. Polarization is first transferred from 1H to directly bonded 13C through the HC INEPT step, and then further to directly bonded carbon spins. Frequency discrimination in the indirect t1 and t2 dimensions was achieved through time-proportional phase incrementation (TPPI) [52]. In the following, all experimental parameters, such as INEPT delays, TOBSY mixing times, etc., were directly optimised to result in maximum signal intensities. The optimal τ1 and τ2 INEPT delay times were 1.4 ms and 0.9 ms, respectively. To remove the effect of residual 1H–13C interactions during indirect and direct 13C detection, different decoupling schemes were tested including WALTZ-16 (wideband, alternating phase, low-power technique for zero-residual splitting) [53,54] and TPPM [48]. We found that TPPM decoupling generally led to narrower carbon lines, which was especially important in the triple resonance experiments, as explained below.
An HHCC experiment is shown in Fig. 1B. It is, in general, very similar to HCC correlation but involves one additional polarization transfer step between protons. In this work, we used through-space nuclear Overhauser effect (NOE) mixing [55,56].
Sequential backbone assignments of the backbone fragments of the protein were obtained using three-dimensional chemical shift correlation NCOCX/NCACX and CONCACX/CAN(CO)CX experiments (Fig. 1C and D). The CONCACX experiment correlates the indirectly recorded chemical shifts of 13C′[i−1] and 15N[i] with the directly detected shifts of Cα[i]/CX[i]. The latter CAN(CO)CX experiment is capable of establishing correlations between Cα[i], N[i], and Cα[i−1]. Because of the restricted and possibly anisotropic mobility in membrane-associated rmMBP, the transverse relaxation was about an order of magnitude shorter than would be the case for isotropically tumbling molecules in solution, and the NC INEPT polarization transfer steps were associated with significant signal losses. Additional signal losses occur during indirect t1 and t2 chemical shift evolutions. To reduce these losses, the three-dimensional NCACX, NCOCX, CONCACX, and CAN(CO)CX experiments were implemented in a constant-time manner as shown in Fig. 1. In these experiments, the t1/t2 15N/13C chemical shift evolutions and J-driven INEPT steps are performed simultaneously, thus minimising detrimental effects of the transverse relaxation [57,58].
2.8. Multidimensional ssNMR spectroscopy—details of constant-time experiments
Here, we briefly describe how the NCOCX experiment works. The other constant-time experiments (NCACX, CONCACX, and CAN(CO)CX) described in Fig. 1C and D are based on similar principles. The NCOCX experiment starts with the excitation of 15N coherence, which is accomplished by the HN INEPT step. As with HC INEPT excitation, the optimal τ4 and τ3 INEPT periods were shorter than 1/(4JNH) and were equal to 1.7 ms. We will assume that at point “a” in the pulse sequence (Fig. 1C), the density matrix for an individual nitrogen spin is in the state Nx, where N denotes the 15N spin operator. In the NCOCX experiment, J-coupling results in the formation of NxCz coherence after an overall period T1 made up of t1/2, T1/2, and T1/2 − t1/2 periods in Fig. 1C. The 15N chemical shift encoding is achieved by the incrementation of t1.
For the purpose of the following discussion, it is convenient to break down the T1 time as follows. First, starting after the pulse with phase ϕ6, there are two t1/2 periods with a phase ϕ7 pulse in the middle. During this total t1 time, the 15N chemical shift and NCO J-coupling evolve, while unwanted NCA J-interaction is refocused by the selective 180° pulse with phase ϕ7, applied to the aliphatic spins. Second, the following two periods are of (T1/2 − t1/2) duration each, with a hard 180° pulse with phase ϕ8 and a selective pulse with phase ϕ9 in the middle; these pulses refocus the NCA J-interactions and 15N chemical shifts, while the NCO J-coupling evolves. The hard pulse with phase ϕ6 refocuses the 15N chemical shift evolution that occurs during the selective pulse with phase ϕ7. The net results are that the NCO J-coupling evolves for a total time of T1, while the 15N chemical shift evolves for a time t1. Likewise, the conversion of the 13C antiphase coherence created by the 90° pulses with phases ϕ10 and ϕ11, starting at point “b” in Fig. 1C, is accomplished in a very similar constant-time manner.
An advantage of such an implementation of NCO INEPT transfer is twofold. First, the J-couplings and 15N chemical shifts evolve simultaneously, without any additional signal losses. To further minimise signal losses due to relaxation, a high-power TPPM decoupling (usually 50 kHz) is applied to protons. Second, because the total t1 evolution time is kept constant, the linewidth in the indirect dimension is entirely determined by the inhomogeneous contribution.
Following the conversion period (point “c” in Fig. 1C), 13C′[i−1] coherences are created, which indirectly encode the 15N[i] and 13C′[i−1] chemical shifts. Polarization is further relayed to other 13CX[i−1] nuclei during the subsequent TOBSY mixing step, where (X=α, β, γ), resulting in a three-dimensional data set that correlates the 15N[i], 13C′[i−1], and 13CX[i−1] spins. Depending on the length of the TOBSY mixing time, 13C′ polarization could be transferred to carbons within the same amino acid up to three bonds away, and this phenomenon could be used to identify the amino acid type.
The pulse sequence and the basic principles in the NCACX experiment are very similar to those of the NCOCX experiment, with differences noted in the caption to Fig. 1C. In general, the NCOCX and NCACX experiments provide complementary pieces of information: NCOCX establishes interresidue N[i]–Cα[i−1]–CX[i−1] correlations, whereas NCACX is capable of giving both intraresidue N[i]–Cα[i]–CX[i] and interresidue N[i]–Cα[i−1]–CX[i−1] correlations. However, there were several factors that complicated the spectral assignments in MBP. First, the solvent-exposed residues observed in INEPT experiments were located in disordered segments of the protein. Thus, high spectral degeneracy, especially in the Cα region, resulted in overlap in cross-peaks in the NCACX spectrum. Second, we often did not see two-bond interresidue transfers in the NCACX experiment. To circumvent these problems, we have designed two additional experiments, CONCACX and CAN(CO)CX, that provided connectivity between three backbone atoms: C′[i−1]–N[i]–Cα[i]/CX[i] in the former, and Cα[i]–N[i]–C′[i−1]/CX[i−1] in the latter. The experimental pulse sequences are shown in Fig. 1D. Both experiments started with selective excitation of the C′/Cα regions. To minimise transverse relaxation losses, the NC J-coupling and C/N chemical shift evolutions were encoded together in a constant-time fashion, similar to the NCOCX experiment.
Fig. 2 summarises the information that can be obtained from all four experiments. The primary task of assigning carbon spin systems and linking them to the nitrogen spin system of the next residue was done using the NCOCX experiment. Then, the CONCACX and CAN(CO)CX experiments helped to link the spin systems, and NCACX data were primarily used to reduce ambiguity by establishing long-range intraresidue nitrogen–sidechain correlations and identifying spin systems not assigned to a particular residue type. Generally, correlations up to three bonds were established in 13C–13C mixing in NCOCX and NCACX experiments. The CONCACX and CAN(CO)CX spectra had lower signal-to-noise ratios because of the two NC INEPT transfer steps, and only CONCA and CAN(CO)CA correlations were established.
Fig. 2.

Schematic representation of the four triple-resonance experiments used to obtain sequential assignments. Correlated residues are indicated by light gray boxes. Solid arrows show dominating one-bond polarization transfers, while dashed arrows show two-bond N[i]–Cα[i−1] transfers, which could not be observed in our spectra.
3. Results and discussion
3.1. Preparation of membrane-associated rmMBP for ssNMR
The 18.5 kDa MBP isoform is peripheral membrane protein located on the cytoplasmic side of the oligodendrocyte membrane in the spiral-wrapped, multilamellar myelin sheath [1,2], and thus represents a highly mobile system compared with integral membrane proteins. Uniformly 13C,15N-labelled rmMBP was prepared with high purity and no or negligible degradation [10,38,39], and interacted with LUVs in an aggregation assay as previously described [38,39,41,42]. These aggregates were spun down to yield a semi-solid sample that could be packed into an MAS rotor for ssNMR. Since the aggregates formed after the addition of MBP are large and relatively amorphous, the results of dynamic light scattering studies of these protein–lipid assemblies are more difficult to interpret [59] and were not necessary for our purposes.
Ideally, the lipid composition of the LUVs would mimic that of central nervous system myelin and would include zwitterionic lipids such as phosphatidylcholine, negatively charged lipids such as phosphatidylserine, and a significant amount (>40%) of cholesterol. However, our primary initial concern was sample stability during spectroscopy, anticipating that a significant amount of ssNMR methodology development would be required. Natural lipids such as phosphatidylcholine, phosphatidylserine, and cholesterol would oxidise over extended periods of time, such as those required for ssNMR data collection. Thus, to initiate this project, we chose to use synthetic DMPC and DMPG lipids, which have no unsaturated carbons in their acyl chains, and which are also more homogeneous than lipid preparations derived from natural sources.
We found that a typical protein-to-lipid mass ratio in the pellet was approximately 0.7, indicating that roughly 70% of the added protein was incorporated into the protein–lipid assembly. This value is about 10 times greater than that in myelin, and what was used in our SDSL/EPR studies [3], but it was essential to maximise the amount of protein to achieve adequate signal-to-noise ratios in the ssNMR experiments.
3.2. One-dimensional ssNMR of rmMBP indicates differential mobility of the protein
One-dimensional ssNMR spectroscopy was first used to evaluate sample quality. The cross-polarization [60], magic angle spinning [61] (CPMAS), and INEPT [34,49] experiments exploit different polarization transfer mechanisms, and can be used to select regions with different dynamic properties in the protein—relatively immobile vs. mobile fragments, respectively. The results of CPMAS and INEPT experiments obtained from lipid-reconstituted rmMBP at 32 °C are shown in Fig. 3. Based on the total number of repetitions required to obtain comparable signal intensities, INEPT was much more efficient at this temperature, indicating that much of the protein was sufficiently mobile to yield INEPT 13C spectra with high sensitivity. Since this mobility resulted in averaging of dipolar interactions, cross-polarization excitation was rendered inefficient. Nonetheless, the CPMAS experiment could still give a signal, probably indicating that a small portion of rmMBP strongly interacted with lipids under these conditions. In addition to the protein signal, sharp lipid lines were observed in both the CPMAS and INEPT spectra, most notably at ~16–17 ppm.
Fig. 3.
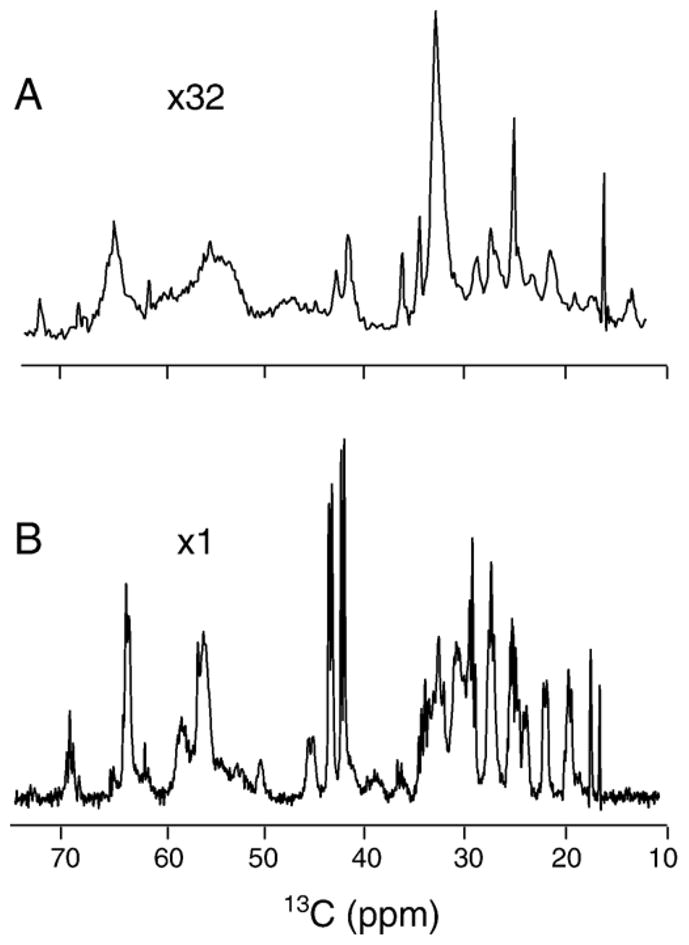
The CPMAS carbon spectrum (A) and INEPT carbon spectrum (B) of fully 13C,15N-labelled 18.5 kDa rmMBP samples reconstituted with lipids. TPPM decoupling of 71.4 kHz was applied during acquisition. Both spectra were taken at 600 MHz, and at a temperature of 32 °C. The CPMAS spectrum shown in the image was collected with a contact time of 3 ms, and with 4180 scans, and at a spinning frequency of 20 kHz. Experiments with shorter cross-polarization mixing times and different cross-polarization power levels resulted in spectra of similar intensities. The INEPT spectrum was collected at 32 °C with 32 scans, at a spinning frequency of 10 kHz. The acquisition lengths were 21 ms and 40 ms in the CPMAS and INEPT experiments, respectively. All spectra were processed with exponential function apodisation of 10 Hz.
The CPMAS and INEPT experiments were all repeated over a wide range of temperatures down to −30 °C. The INEPT efficiency was found to drop as the temperature decreased, especially around and below 0 °C, while the cross-polarization efficiency increased. A sharp increase in CPMAS transfer efficiency was observed in the range from −5 °C to −15 °C. This increase may have been associated with water freezing outside the lipid bilayer and in its hydrophilic portion, thereby inhibiting the motions of the protein fragments. The CPMAS 13C spectra below −15 °C were inhomogeneously broadened in appearance, reflecting the presence of multiple protein conformations.
Fig. 4 demonstrates the effect of decoupling on the INEPT 13C spectra. Due to the mobile nature of the observed residues, relatively high resolution is achieved even at low-power WALTZ-16 decoupling. The resolution improves even further as the decoupling power increases, as demonstrated in the inserts in Fig. 4. In addition, new lines appear in the spectra, most notably the glycine resonances at around 45 ppm. This observation is consistent with our estimation of T2 relaxation times, which show that the carbon (especially Cα) spins relax more slowly at higher power decoupling.
Fig. 4.

A comparison of the effect of the decoupling on the resolution of 13C INEPT spectra. High resolution is seen even at low-power WALTZ-16 [53,54] decoupling, indicating the mobile nature of the residues contributing to the INEPT spectra. Even higher resolution is observed at moderate- and high-power TPPM decoupling. All experiments were obtained on a 600-MHz Bruker spectrometer with 10 kHz MAS. The acquisition and spectral processing parameters are given in the caption to Fig. 1.
3.3. Two- and three-dimensional 1H–13C correlation spectroscopy
Two-dimensional and three-dimensional 1H–13C experiments were performed to identify spin systems by amino acid type, to characterise the proton and carbon chemical shift dispersion and secondary structure distribution in the mobile fragments. The 1H–13C two-dimensional HSQC (13C-detected, heteronuclear single quantum coherence) spectrum is shown in Fig. 5A. One notable feature of this correlation spectrum is that the Hα and Cα dimensions are poorly dispersed, which is considered to be a characteristic feature of unstructured proteins [62,63]. The proton linewidths are of the order of 0.15 ppm to 0.2 ppm at 600-MHz field strength. Such fairly narrow lines for solid-state NMR spectra are indicative of high mobility in the observed protein fragments.
Fig. 5.

(A) The two-dimensional 1H–13C HSQC 13C-detected spectrum, obtained using the pulse sequence shown in Fig. 1A, with t2 evolution and TOBSY mixing set to zero. The total 1H evolution time was 16 ms with 200 points. The spectrum was collected with 16 scans per point and with the recycling delay of 2 s. The spectrum was processed in NMRPipe [44], employing exponential function apodisation of 20 Hz in the direct dimension and cosine square apodisation in the indirect dimension. Data were zero filled up to 4096×2048 points in the direct and indirect dimensions, respectively. (B, C) Representative two-dimensional planes of the three-dimensional HCC chemical shift correlation experiment. The total 1H evolution time was 8.2 ms with 88 points, while the total indirect carbon evolution time was 10 ms with 240 points. The spectrum was collected with 8 scans per point. The recycling delay was set to 1.7 s. The carrier frequency was set to 45 ppm. Both spectra were processed in NMRPipe [44], employing exponential function apodisation of 10 Hz in the direct dimension, and cosine square apodisation in the two indirect dimensions. Data were zero filled up to 4096×2048×2048 points in the direct and two indirect dimensions, respectively.
To assign spin systems to amino acid types, we performed the three-dimensional HCC correlation experiment shown in Fig. 1A. In principle, this experiment can distinguish different residue types, based on distinct correlation patterns between sidechain carbons in different amino acids. Fig. 5B and C show two two-dimensional planes of the full three-dimensional HCC spectrum. A number of residues can be identified in these spectra. For instance, serine Cα–Cβ and proline Cα–Cβ–Cγ–Cδ correlation patterns are clearly distinguishable in Fig. 5B. In general, P913 TOBSY mixing of 10.5 ms was sufficient to relay polarization over three bonds, as evident from the example of proline in Fig. 5B, and two more examples of lysyl and arginyl residues shown in Fig. 5C. A total of 30 spin systems could be assigned to a particular residue type in this HCC experiment.
3.4. Sequential resonance assignments
In order to obtain site-specific assignments, we performed three-dimensional constant-time NCOCX, NCACX, CONCACX, and CAN(CO)CX experiments. Carbon spin systems were first identified from NCOCX experiments. The TOBSY mixing of 9.9 ms allowed up to three bonds transfer, thus establishing intraresidue correlations of C′–Cα–Cβ and sometimes C′–Cα–Cβ–Cγ type along the sidechain. Typical two-dimensional planes of three-dimensional NCOCX and NCACX spectra are shown in Fig. 6. In these particular examples, polarization can be transferred up to two bonds, allowing one to establish G121-P120 connectivity in the NCOCX experiment (upper panel), and to identify Ala134 in the NCACX experiment (lower panel). Amino acid types were identified as follows. Spin systems with Cα shifts around 45 ppm, and with N shifts around 110 ppm or less, were assigned to glycines. Spin systems with Cα shifts around 52 ppm, Cβ shifts around 19 ppm, and N shifts around 125 ppm were assigned to alanines. Systems with Cα shifts around 62 ppm, Cβ shifts around 69 ppm, and N shifts around 115 ppm were assigned to threonines, and systems with Cα shifts around 58 ppm, Cβ shifts around 64 ppm, and N shifts around 116 ppm were assigned to serines. A total of 80 spin systems were identified, of which eight were assigned to Ala, nine to Gly, eight to Ser, four to Thr, three to Asp, and one to Pro. Many other spin systems could be identified in the spectrum (Table S2 in the Supplementary data), but could not be assigned unambiguously to a single amino acid type.
Fig. 6.

The two-dimensional 13C–13C planes of (A) the three-dimensional NCOCX, and (B) NCACX experiments. In the NCOCX experiment, a total of 120 t1 points were collected, with a maximum evolution time of 12.3 ms. A total of 100 points were taken in t2, resulting in the total evolution time of 11 ms. In the NCACX experiment, the t1 evolution time was 9.6 ms with 96 points, and the t2 evolution time was 8.3 ms with 100 points. TOBSY mixing times of 9.9 ms and 7.5 ms were used in the NCOCX and NCACX experiments, respectively. The number of scans per free induction decay (FID) was 16 in the NCOCX and 24 in the NCACX experiments. The carrier frequency was set at 175 ppm for carbon in NCOCX, at 50 ppm for carbon in NCACX, and at 116 ppm for nitrogen. The recycling delay was set to 2 s. A 50 kHz TPPM decoupling was applied on the 1H channel during 13C′–15N/13Cα–15N polarization transfers, and during direct and indirect detections.
As was explained previously, the NCACX experiment often fails to provide interresidue connectivity. Moreover, it only correlates two backbone atoms and suffers from a higher degree of degeneracy, compared to the NCOCX experiment. In contrast, the CONCACX experiment correlates three backbone atoms, C′ [i−1], N[i], and Cα[i]. Two of these nuclei overlap with those correlated in NCOCX. Thus, this experiment was used in conjunction with NCOCX to link spin systems through establishing connectivity between Cα[i] and Cα[i−1] atoms. A total of 62 spin systems could be linked in non-contiguous fragments (Table S2 in the Supplementary data), and 49 of them could be assigned to particular fragments of the protein. The remaining amino acids could not be assigned to the protein sequence because of the low signal-to-noise ratio for the side-chain signals, and the lack of information on the amino acid type. Additional information was derived from the NCACX experiment, where intraresidue correlations were used to establish amino acid types for the remaining residues. The CAN(CO)CX experiment did not provide any additional information and could only be used to verify some of the assignments.
The spectral analysis was performed using the CARA software package [46]. The spin systems identified from each spectrum were linked to each other based on at least two matching chemical shifts. The connectivities were established using the Autolink program, which is built into the CARA software package. One example of strip plots showing connectivities between spin systems is shown in Fig. 7. The final assignments are given in Table S1 in the Supplementary data.
Fig. 7.

Strip plots from the three-dimensional NCOCX, NCACX, and CONCACX experiments, showing the D143-T147 amino acid stretch. The NCOCX experiment establishes N[i+1]–C′ [i]–Cα[i]/Cβ[i] correlations shown in green, whereas the CONCACX experiment gives correlations between C′ [i], N[i+1], and Cα[i+1] spins, indicated in red. Shared C′[i] and N[i+1] shifts allow one to establish interresidue Cα[i]–Cα[i+1] correlations, and “walk” sequentially along the backbone, as shown by the horizontal and vertical dashed lines. Carbonyl shifts for each strip are shown on the top. The NCACX slice for T147 is shown in blue. The NCOCX and NCACX spectra were acquired as described in the caption of Fig. 6. The CONCACX spectrum was acquired with total t1 and t2 acquisition lengths of 11.4 ms and 10 ms, respectively, with 92 and 96 points in the carbonyl and nitrogen indirect dimensions, respectively. TPPM decoupling of 50 kHz was applied during NC INEPT transfers and direct acquisition. The number of scans per FID was 16, with a recycling delay of 2 s. The CAN(CO)CX experiment was collected with similar parameters, except for TOBSY mixing, which was 6 ms.
To improve sequential assignments, and potentially to collect structural restraints, we performed a set of NOESY (nuclear Overhauser effect spectroscopy) [55,56] experiments. The NOE mixing was incorporated into the three-dimensional HHCC experiment, as shown in Fig. 1B. NOE mixing times of 50 ms to 300 ms were used. Unfortunately, we did not observe any interresidue correlations. However, a series of cross-peaks were observed in the spectra, at the proton resonance frequency of H2O (~4.6 ppm) as shown in Fig. 8. These peaks indicated that the sidechain and backbone protons strongly interacted with water molecules. Further examination of the two-dimensional 13C–13C plane taken at 4.6 ppm showed that most of the carbon nuclei show up at that frequency (Figure S1 in the Supplementary data). This hydration of many amino acids seen in the NMR spectra suggests that they are either located outside the membrane, or in the hydrophilic region of the phospholipid headgroups. Similar effects have been observed for unstructured fragments of globular and membrane proteins [64,65].
Fig. 8.

A comparison of the two-dimensional (ω1-1H, ω3-13C) projections of the (A) HCC, and (B) HHCC experiments. A series of extra peaks appearing at 4.6 ppm (the 1H frequency of H2O) is indicated by the arrows in panel B. The NOE mixing time was 50 ms.
3.5. Chemical shift index analysis
The chemical shift values determined for mobile residues (Table S1, Supplementary data) can be used to analyse the residual secondary structure. We used shifts of Cα resonances for chemical shift index (CSI) analysis [66], using sequence-corrected random coil values for all amino acids [67]. The CSI analysis shown in Fig. 9A indicates that most residues exhibit chemical shifts typical of unstructured regions, although some fragments appear to show some propensity towards α-helicity. For example, residues H21-A22-R23 have Cα shifts slightly above the random coil “threshold” [68]. Residues D143, Q145, L148, and L154 show some propensity towards α-helix, consistent with solution NMR studies of the protein in 30% TFE-d2 [10,11]. The relatively high degree of randomness of the mobile fragments is consistent with them being exposed to solvent.
Fig. 9.
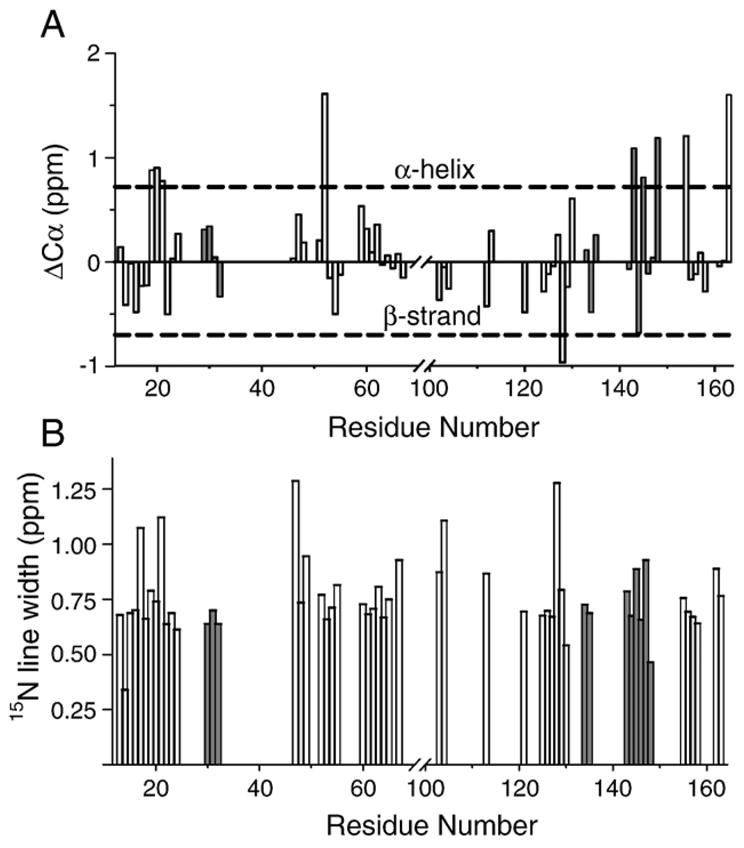
(A) Chemical shift index analysis of Cα of the backbone assignments. All of the random coil chemical shifts are sequence-corrected based on [67]. Secondary chemical shifts for tentatively assigned residues are shown in gray. (B) The 15N linewidths for assigned residues. Linewidths for tentatively assigned residues are shown in gray.
Further information on the type of motion can be derived from the analysis of the linewidths for observed residues. For this purpose, the three-dimensional NCOCX spectrum, which has the highest signal-to-noise ratio, was reprocessed with linear prediction and without line broadening. The 15N linewidth was chosen as the one which was least affected by the spin–spin interactions. The corresponding plot is shown in Fig. 9B. Most mobile residues observed in the spectra have linewidths in the range of 0.5 to 0.8 ppm, dominated by inhomogeneous broadening because of the constant time nature of the experiment. Contributions associated with instrumental imperfections such as instability of the external lock were estimated to be small compared to the observed line width. Magnetic susceptibility effects can also be ruled out. Indeed, the observed proton linewidths, typically 0.15–0.2 ppm (120–160 Hz at 800 MHz), suggest that the susceptibility effects would not result in 15N linewidths larger than 0.15–0.2 ppm, which is much smaller than the observed values. Thus, the most likely explanation is conformational heterogeneity. The heterogeneous linewidths of 0.5 ppm to 0.8 ppm (40–60 Hz at 800 MHz proton Larmor frequency) suggest that the local conformation-averaging backbone motions are at least slower than the inverse linewidth, i.e., their time scale is of the order of tens to hundreds of milliseconds. These slow motions in this experimental system are not sufficiently fast to average large proton–proton or proton–carbon dipolar interactions. Thus, other motions, fast and non-local, must be present. The 31P chemical shift anisotropy (CSA) values determined from static spectra (Figure S2, Supplementary data) indicate that the overall tumbling motions of the lipid vesicles are not sufficiently fast to eliminate large proton–proton and proton–carbon dipolar couplings. Thus, the most likely motion responsible for the averaging of dipolar couplings would be a large amplitude motion of the entire solvent-exposed fragment. The mobile residues comprise almost contiguous protein fragments: T15-R23, D46-S67, P120-L148, and L154-S163. Overall undulating motions of these large fragments can average dipolar interactions, but will not affect local structure, and therefore will not contribute to the conformational averaging.
4. Conclusions
In this study, we have developed and implemented a set of three-dimensional heteronuclear ssNMR experiments suitable for resonance assignments in proteins in a semi-solid state with a significant degree of internal motion. The experiments were implemented in a constant-time manner, which minimised signal losses due to strong relaxation effects. The type of excitation used in the experiments filtered out the contributions from the immobilised part of the molecule, and guaranteed that only mobile fragments of the membrane-associated 18.5 kDa rmMBP were observed. Since the problems of low sensitivity and short transverse relaxation times would likely arise in ssNMR studies of any peripheral membrane-associated protein at physiological temperatures, the strategies developed here for 18.5 kDa rmMBP are of wider applicability.
Using this methodology, we could assign many of the mobile amino acid fragments of 18.5 kDa rmMBP. These fragments interacted strongly with water and were likely located either outside the lipid bilayer, or associated with its hydrophilic portion. Furthermore, amino acid type-specific chemical shift indexing indicated that the observed fragments were largely disordered under these conditions. On the other hand, the protein fragments containing highly hydrophobic pairs Phe42/Phe43, Phe86/Phe87 were not seen in our spectra. Other hydrophobic residues such as Ile and Val were also not seen. Presumably, these residues were at least partially buried and immobilised in the membrane, as suggested by previously reported SDSL-EPR measurements [3], and thus could not be observed in the INEPT type of experiment. Based on the linewidth analysis, the mobile fragments likely undergo large amplitude undulating motions outside the membrane, which results in averaging of dipolar interactions.
Supplementary Material
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (CIHR, Operating Grant MOP 74468 to G.H. and V.L.), the Natural Sciences and Engineering Research Council of Canada (NSERC, Discovery Grants RG298480-04 to V.L., and RG121541 to G.H.), the Canada Foundation for Innovation, and the Ontario Innovation Trust. V.L. is a Canada Research Chair holder, and is a recipient of an Early Researcher Award from the Ontario Ministry of Research and Innovation. M.A. is a recipient of a Doctoral Studentship from the Ministry of Higher Education and Scientific Research of Egypt. We are grateful to Dr. Frances Sharom for the use of her Zetasizer DLS instrument, to Messrs. Jeffery Haines, David Libich, and Abdi Musse for their advice on protein purification and reconstitution, to Mr. Xiaohu Peng for the discussions on spectral analysis and residue assignment, and to Dr. Jim Davis, Ms. Valerie Robertson, and Dr. Jun Gu of the University of Guelph NMR Centre for their assistance.
Abbreviations
- CARA
computer-aided resonance assignment
- CP
cross-polarization
- CPMAS
cross-polarization magic angle spinning
- CSA
chemical shift anisotropy
- CSI
chemical shift index
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DMPG
1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]
- ddH2O
distilled, deionised water
- EDTA
ethylenediamine tetraacetic acid
- EPR
electron paramagnetic resonance
- FID
free induction decay
- GARP
globally optimised alternating phase rectangular pulses
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HSQC
heteronuclear single quantum coherence
- INEPT
insensitive nuclear enhancement of polarization transfer
- LUV
large unilamellar vesicle
- MAS
magic angle spinning
- MBP
myelin basic protein
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser effect spectroscopy
- ppm
parts per million
- rmMBP
recombinant murine MBP
- rpm
revolutions per minute
- SDS
sodium dodecyl sulphate
- SDSL
site-directed spin labelling
- ssNMR
solid-state NMR
- TFE-d2
deuterated 2,2,2-trifluoroethanol (CF3-CD2-OH)
- TOBSY
total through-bond correlation spectroscopy
- TPPI
time-proportional phase incrementation
- TPPM
two-pulse phase modulation
- Tris–HCl
tris(hydroxymethyl)aminomethane, pH adjusted with HCl
- WALTZ
wideband, alternating phase, low-power technique for zero-residual splitting
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbamem.2007.08.013.
References
- 1.Harauz G, Ishiyama N, Hill CMD, Bates IR, Libich DS, Farès C. Myelin basic protein—diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron. 2004;35:503–542. doi: 10.1016/j.micron.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Boggs JM. Myelin basic protein: a multifunctional protein. Cell Mol Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates IR, Boggs JM, Feix JB, Harauz G. Membrane-anchoring and charge effects in the interaction of myelin basic protein with lipid bilayers studied by site-directed spin labeling. J Biol Chem. 2003;278:29041–29047. doi: 10.1074/jbc.M302766200. [DOI] [PubMed] [Google Scholar]
- 4.Bates IR, Feix JB, Boggs JM, Harauz G. An immunodominant epitope of myelin basic protein is an amphipathic alpha-helix. J Biol Chem. 2004;279:5757–5764. doi: 10.1074/jbc.M311504200. [DOI] [PubMed] [Google Scholar]
- 5.Musse AA, Boggs JM, Harauz G. Deimination of membrane-bound myelin basic protein in multiple sclerosis exposes an immunodominant epitope. Proc Natl Acad Sci U S A. 2006;103:4422–4427. doi: 10.1073/pnas.0509158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pointer-Keenan CD, Lee DK, Hallok K, Tan A, Zand R. Ramamoorthy, investigation of the interaction of myelin basic protein with phospholipid bilayers using solid-state NMR spectroscopy. Chem Phys Lipids. 2004;132:47–54. doi: 10.1016/j.chemphyslip.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 8.Dyson HJ, Wright PE. According to current textbooks, a well-defined three-dimensional structure is a prerequisite for the function of a protein. Is this correct? IUBMB Life. 2006;58:107–109. doi: 10.1080/15216540500484376. [DOI] [PubMed] [Google Scholar]
- 9.Receveur-Bréchot V, Bourhis JM, Uversky VN, Canard B, Longhi S. Assessing protein disorder and induced folding. Proteins. 2006;62:24–45. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 10.Libich DS, Robertson VJ, Monette MM, Harauz G. Backbone resonance assignments of the 18.5 kDa isoform of murine myelin basic protein (MBP) J Biomol NMR. 2004;29:545–546. doi: 10.1023/B:JNMR.0000034348.99658.d7. [DOI] [PubMed] [Google Scholar]
- 11.Libich DS, Monette MM, Robertson VJ, Harauz G. NMR assignment of an intrinsically disordered protein under physiological conditions: the 18.5 kDa isoform of murine myelin basic protein (BMRB 15131) J Biomol NMR Assignments. 2007;1:61–63. doi: 10.1007/s12104-007-9016-1. [DOI] [PubMed] [Google Scholar]
- 12.Farès C, Libich DS, Harauz G. Solution NMR structure of an immunodominant epitope of myelin basic protein. conformational dependence on environment of an intrinsically unstructured protein. FEBS J. 2006;273:601–614. doi: 10.1111/j.1742-4658.2005.05093.x. [DOI] [PubMed] [Google Scholar]
- 13.Straus SK, Bremi T, Ernst RR. Experiments and strategies for the assignment of fully C-13/N-15-labelled polypeptides by solid state NMR. J Biomol NMR. 1998;12:39–50. doi: 10.1023/a:1008280716360. [DOI] [PubMed] [Google Scholar]
- 14.Hong M. Resonance assignment of C-13/N-15 labeled solid proteins by two- and three-dimensional magic-angle-spinning NMR. J Biomol NMR. 1999;15:1–14. doi: 10.1023/a:1008334204412. [DOI] [PubMed] [Google Scholar]
- 15.Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and side-chain C-13 and N-15 signal assignments of the alpha-spectrin Sh3 domain by magic angle spinning solid-state NMR at 17.6 Tesla. ChemBiochem. 2001;2:272–281. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 16.Bockmann A, Lange A, Galinier A, Luca S, Giraud N, Juy M, Heise H, Montserret R, Penin F, Baldus M. Solid state NMR sequential resonance assignments and conformational analysis of the 2×10.4 kDa dimeric form of the Bacillus subtilis protein Crh. J Biomol NMR. 2003;27:323–339. doi: 10.1023/a:1025820611009. [DOI] [PubMed] [Google Scholar]
- 17.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 18.Igumenova TI, McDermott AE, Zilm KW, Martin RW, Paulson EK, Wand AJ. Assignments of carbon NMR resonances for microcrystalline ubiquitin. J Am Chem Soc. 2004;126:6720–6727. doi: 10.1021/ja030547o. [DOI] [PubMed] [Google Scholar]
- 19.Franks WT, Zhou DH, Wylie BJ, Money BG, Graesser DT, Frericks HL, Sahota G, Rienstra CM. Magic-angle spinning solid-state NMR spectroscopy of the Beta 1 immunoglobulin binding domain of protein G (Gb1): N-15 and C-13 chemical shift assignments and conformational analysis. J Am Chem Soc. 2005;127:12291–12305. doi: 10.1021/ja044497e. [DOI] [PubMed] [Google Scholar]
- 20.Marulanda D, Tasayco ML, McDermott A, Cataldi M, Arriaran V, Polenova T. Magic angle spinning solid-state NMR spectroscopy for structural studies of protein interfaces. Resonance assignments of differentially enriched Escherichia coli thioredoxin reassembled by fragment complementation. J Am Chem Soc. 2004;126:16608–16620. doi: 10.1021/ja0464589. [DOI] [PubMed] [Google Scholar]
- 21.Siemer AB, Arnold AA, Ritter C, Westfeld T, Ernst M, Riek R, Meier BH. Observation of highly flexible residues in amyloid fibrils of the Het-S prion. J Am Chem Soc. 2006;128:13224–13228. doi: 10.1021/ja063639x. [DOI] [PubMed] [Google Scholar]
- 22.Siemer AB, Ritter C, Steinmetz MO, Ernst M, Riek R, Meier BH. C-13, N-15 resonance assignment of parts of the Het-S prion protein in its amyloid form. J Biomol NMR. 2006;34:75–87. doi: 10.1007/s10858-005-5582-7. [DOI] [PubMed] [Google Scholar]
- 23.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rienstra CM, Tucker-Kellogg L, Jaroniec CP, Hohwy M, Reif B, McMahon MT, Tidor B, Lozano-Perez T, Griffin RG. De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2002;99:10260–10265. doi: 10.1073/pnas.152346599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2004;101:711–716. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baldus M. Spectral assignment of (membrane) proteins under magic-angle-spinning. In: Ramamoorthy A, editor. NMR Spectroscopy of Biological Solids. Taylor & Francis Group, LLC; Boca Raton, FL: 2006. [Google Scholar]
- 27.Lange A, Giller K, Hornig S, Martin-Eauclaire MF, Pongs O, Becker S, Baldus M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–962. doi: 10.1038/nature04649. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Berthold DA, Frericks HL, Gennis RB, Rienstra CM. Partial 13C and 15N chemical-shift assignments of the disulfide-bond-forming enzyme Dsbb by 3D magic-angle spinning NMR spectroscopy. Chem-Biochem. 2007;8:434–442. doi: 10.1002/cbic.200600484. [DOI] [PubMed] [Google Scholar]
- 29.Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by Solid-State NMR Spectroscopy. Angewandte Chemie-International Edition. 2007;46:459–462. doi: 10.1002/anie.200602139. [DOI] [PubMed] [Google Scholar]
- 30.Frericks HL, Zhou DH, Yap LL, Gennis RB, Rienstra CM. Magic-angle spinning solid-state NMR of a 144 kDa membrane protein complex: E. coli cytochrome bo3 oxidase. J Biomol NMR. 2006;36:55–71. doi: 10.1007/s10858-006-9070-5. [DOI] [PubMed] [Google Scholar]
- 31.Durr UHN, Yamamoto K, Im SC, Waskell L, Ramamoorthy A. Solid-state NMR reveals structural and dynamical properties of a membrane-anchored electron-carrier protein, cytochrome b(5) J Am Chem Soc. 2007;129:6670–6671. doi: 10.1021/ja069028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fraser PE, Deber CM. Surface accessibility of C-13-labeled lysine residues in membrane-bound myelin basic-protein. J Biol Chem. 1984;259:8689–8692. [PubMed] [Google Scholar]
- 33.Mendz GL, Miller DJ, Ralston GB. Interactions of myelin basic-protein with palmitoyllysophosphatidylcholine—characterization of the complexes and conformations of the protein. Eur Biophys J. 1995;24:39–53. doi: 10.1007/BF00216829. [DOI] [PubMed] [Google Scholar]
- 34.Morris GA, Freeman R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J Am Chem Soc. 1979;101:760–762. [Google Scholar]
- 35.Chen PS, Jr, Toribara TY, Warner H. Microdetermination of phosphorus. Anal Chem. 1956;28:1756–1758. [Google Scholar]
- 36.Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. J Biol Chem. 1925;66:374–389. [Google Scholar]
- 37.Wiechelman KJ, Braun RD, Fitzpatrick JD. Investigation of the bicinchoninic acid protein assay—identification of the groups responsible for color formation. Anal Chem. 1988;175:231–237. doi: 10.1016/0003-2697(88)90383-1. [DOI] [PubMed] [Google Scholar]
- 38.Bates IR, Matharu P, Ishiyama N, Rochon D, Wood DD, Polverini E, Moscarello MA, Viner NJ, Harauz G. Characterization of a recombinant murine 18.5 kDa myelin basic protein. Protein Expr Purif. 2000;20:285–299. doi: 10.1006/prep.2000.1307. [DOI] [PubMed] [Google Scholar]
- 39.Bates IR, Libich DS, Wood DD, Moscarello MA, Harauz G. An Arg/Lys→Gln mutant of recombinant murine myelin basic protein as a mimic of the deiminated form implicated in multiple sclerosis. Protein Expr Purif. 2002;25:330–341. doi: 10.1016/s1046-5928(02)00017-7. [DOI] [PubMed] [Google Scholar]
- 40.Kaur J, Libich DS, Campagnoni CW, Wood DD, Moscarello MA, Campagnoni AT, Harauz G. Expression and properties of the recombinant murine golli-myelin basic protein isoform J37. J Neurosci Res. 2003;71:777–784. doi: 10.1002/jnr.10547. [DOI] [PubMed] [Google Scholar]
- 41.Jo EJ, Boggs JM. Aggregation of acidic lipid vesicles by myelin basic-protein—dependence on potassium concentration. Biochemistry. 1995;34:13705–13716. doi: 10.1021/bi00041a053. [DOI] [PubMed] [Google Scholar]
- 42.Boggs JM, Yip PM, Rangaraj G, Jo E. Effect of posttranslational modifications to myelin basic protein on its ability to aggregate acidic lipid vesicles. Biochemistry. 1997;36:5065–5071. doi: 10.1021/bi962649f. [DOI] [PubMed] [Google Scholar]
- 43.Smith R, Cornell BA. Myelin basic protein induces hexagonal phase formation in dispersions of diacylphosphatidic acid. Biochim Biophys Acta. 1985;818:275–279. doi: 10.1016/0005-2736(85)90569-3. [DOI] [PubMed] [Google Scholar]
- 44.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRpipe: a multidimensional spectral processing system based on unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 45.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. J Magn Reson. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 46.Keller R. The Computer Aided Resonance Assignment Tutorial. 1. CANTINA Verlag; 2004. [Google Scholar]
- 47.Hahn EL. Spin Echoes. Phys Rev. 1950;80:580–594. [Google Scholar]
- 48.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 49.Burum DP, Ernst RR. Net polarization transfer via a J-ordered state for signal enhancement of low-sensitivity nuclei. J Magn Reson. 1980;39:163–168. [Google Scholar]
- 50.Hardy EH, Verel R, Meier BH. Fast MAS total through-bond correlation spectroscopy. J Magn Reson. 2001;148:459–464. doi: 10.1006/jmre.2000.2258. [DOI] [PubMed] [Google Scholar]
- 51.Baldus M, Iuliucci RJ, Meier BH. Probing through-bond connectivities and through-space distances in solids by magic-angle-spinning nuclear magnetic resonance. J Am Chem Soc. 1997;119:1121–1124. [Google Scholar]
- 52.Marion D, Ikura M, Tschudin R, Bax A. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J Magn Reson. 1989;85:393–399. [Google Scholar]
- 53.Shaka AJ, Keeler J, Freeman R. Evaluation of a new broadband decoupling sequence: Waltz-16. J Magn Reson. 1983;53:313–340. [Google Scholar]
- 54.Shaka AJ, Keeler J, Frenkiel T, Freeman R. An improved sequence for broadband decoupling: Waltz-16. J Magn Reson. 1983;52:335–338. [Google Scholar]
- 55.Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys. 1979;71:4546–4553. [Google Scholar]
- 56.Wagner G, Wüthrich K. Sequential resonance assignments in protein 1H nuclear magnetic resonance spectra: basic pancreatic trypsin inhibitor. J Mol Biol. 1982;155:347–366. doi: 10.1016/0022-2836(82)90009-2. [DOI] [PubMed] [Google Scholar]
- 57.Grzesiek S, Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J Magn Reson. 1992;96:432–440. [Google Scholar]
- 58.Cavanagh J, Fairbrother WJ, Palmer AG, III, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Academic Press; San Diego: 1996. [Google Scholar]
- 59.Mac Millan SV, Ishiyama N, White GF, Palaniyar N, Hallett FR, Harauz G. Myelin basic protein component C1 in increasing concentrations can elicit fusion, aggregation, and fragmentation of myelin-like membranes. Eur J Cell Biol. 2000;79:327–335. doi: 10.1078/S0171-9335(04)70036-9. [DOI] [PubMed] [Google Scholar]
- 60.Pines A, Gibby MG, Waugh JS. Proton-enhanced NMR of dilute spins in solids. J Chem Phys. 1973;59:569–590. [Google Scholar]
- 61.Andrew ER, Bradbury A, Eads RG. Nuclear magnetic resonance spectra from a crystal rotated at high speed. Nature (London) 1959;183:1802. [Google Scholar]
- 62.Dyson HJ, Wright PE. Nuclear magnetic resonance methods for elucidation of structure and dynamics in disordered states. Methods Enzymol. 2001;339:258–270. doi: 10.1016/s0076-6879(01)39317-5. [DOI] [PubMed] [Google Scholar]
- 63.Dyson HJ, Wright PE. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv Protein Chem. 2002;62:311–340. doi: 10.1016/s0065-3233(02)62012-1. [DOI] [PubMed] [Google Scholar]
- 64.Otting G, Liepinsh E, Wüthrich K. Protein hydration in aqueous solution. Science. 1991;254:974–980. doi: 10.1126/science.1948083. [DOI] [PubMed] [Google Scholar]
- 65.Andronesi OC, Becker S, Seidel K, Heise H, Young HS, Baldus M. Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J Am Chem Soc. 2005;127:12965–12974. doi: 10.1021/ja0530164. [DOI] [PubMed] [Google Scholar]
- 66.Wishart DS, Sykes BD, Richards FM. The chemical-shift index—a fast and simple method for the assignment of protein secondary structure through NMR-spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 67.Schwarzinger S, Kroon GJA, Foss TR, Chung J, Wright PE, Dyson HJ. Sequence-dependent correction of random coil NMR chemical shifts. J Am Chem Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 68.Wishart DS, Sykes BD. The C-13 chemical-shift index—a simple method for the identification of protein secondary structure using C-13 chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 69.Emsley L, Bodenhausen G. Optimization of shaped selective pulses for NMR using a quaternion description of their overall propagators. J Magn Reson. 1992;97:135–148. [Google Scholar]
- 70.Shaka AJ, Barker PB, Freeman R. Computer-optimized decoupling scheme for wideband applications and low-level operation. J Magn Reson. 1985;64:547–552. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.