

Abstract
The mammalian target of rapamycin (mTOR) exerts neuroprotective effects under hypoxic or ischemic conditions. To explore whether mTOR participates in neuroprotective signaling through regulation of hypoxia-inducible factor-1α (HIF-1α), vascular endothelial growth factor (VEGF) and neuronal apoptosis in developing rat brain with hypoxia-ischemia (HI), we operated on postnatal day 10 rats by ligating the common carotid artery followed by exposure to systemic hypoxia. Brains were collected at various intervals to detect the expression of mTOR, phosphorylated mTOR (p-mTOR), HIF-1α, VEGF and cleaved caspase 3 (CC3), using immunohistochemistry and Western blot analysis. We also used terminal deoxynucleotidyl transferase-mediated dUTP-nick end labeling (TUNEL) to detect neuronal apoptosis. The p-mTOR protein expression increased at 2 h after HI, peaked at 8 h, lasted 24 h, and then dropped to the basal level. Also, the expression of HIF-1α and VEGF was significantly enhanced and peaked at 8 h after HI. Up-regulated expression of CC3 was observed at 2 h, peaked at 24 h, and lasted 72 h after HI. Increased neuronal apoptosis is associated with reduced HIF-1α and VEGF expression. Furthermore, pretreatment with rapamycin, a mTOR specific inhibitor, significantly inhibited HIF-1α and VEGF protein after HI. The expression of CC3 and the number of TUNEL-positive cells were up-regulated at 8 h and down-regulated at 24 h after HI in the rapamycin-treated group. Our findings suggest that mTOR may participate in the regulation of HIF-1α, VEGF and neuronal apoptosis, serving neuroprotective functions after HI in developing rat brain.
Keywords: hypoxia-ischemia, mammalian target of rapamycin, hypoxia-inducible factor-1α, vascular endothelial growth factor, apoptosis
1. Introduction
Hypoxia-ischemia (HI) causes serious injury to the body including the hypoxia-ischemia brain damage (HIBD) in neonates, disturbing the development and function of the central nervous system [21]. The pathologic and survival mechanisms of HIBD are of great importance for researchers to probe neuroprotective treatments. We previously found that the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) signaling pathway was involved in the survival process after HIBD through regulating the protein expression of hypoxia-inducible factor-1α (HIF-1α), and its target gene, vascular endothelial growth factor (VEGF) [16]. HIF-1α is an important molecule in maintaining cellular oxygen balance. Hypoxia or ischemia can regulate HIF-1α expression, which then regulates the expression of its target gene VEGF, exerting neuroprotection to the HIBD [17]. Nevertheless, the signaling pathways participating in the regulation of HIF-1α remain unclear.
The mammalian target of rapamycin (mTOR), an atypical Ser/Thr protein kinase, is a main downstream of PI3K/AKT signaling pathway [11, 20, 9]. mTOR functions as a sensor of extracellular signals including stimulations from energy levels and stress, and then regulates angiogenesis, cell growth, apoptosis and autophagy, etc. mTOR can regulate HIF-1α and VEGF in the abnormal proliferative cell types such as tumors or cysts [23,18]. Nevertheless, whether mTOR functions in the developing brain with HI through HIF-1α and VEGF is unknown. Meanwhile, the special inhibitor of mTOR, rapamycin can induce apoptosis in tumor or cyst cells [24, 12], suggesting a potential relationship between mTOR and apoptosis. Apoptosis is reported to play a predominant role in the pathological progress of HIBD [14]. Therefore, we hypothesized that mTOR signaling pathway is involved in the regulation of HIF-1α and VEGF, and thus participates in the survival mechanisms of HIBD through regulating apoptosis. To test this hypothesis, we used postnatal day 10 (P10) rats to simulate neonatal HI model and compared the expression of HIF-1α, VEGF, and apoptosis markers with or without rapamycin.
2. Experimental procedures
2.1. Animal protocols
All animal research was approved by Sichuan University Committee on Animal Research. Male Sprague-Dawley rats (18–22g) with litters of mixed gender were acquired from the Animal Center of Sichuan University. Vannucci method [7] was used to simulate the HIBD model using P10 rats. After anesthetized with halothane, the right common carotid artery (CCA) of the pups was isolated and permanently double-ligated with a 7-0 silk suture. After recovering from anesthesia for 1 h, pups were returned for 2.5 h of hypoxia (8% O2/92% N2) in a box to produce HI injury. Sham controls only received exposure of the CCA. Rats’ brains from sham controls and from HI groups at 2, 4, 8, 12, 24, and 72 h were collected. For the rapamycin-treated group, pups received intraperitoneal injections of rapamycin (Calbiochem, EMD Chemicals Inc., Germany) four times (total 2 mg/kg, each time 0.5 mg/kg, an interval of 4 h per injection). The last injection was finished 1 h before HI, while the control group was injected with DMSO.
2.2. Immunohistochemistry
Six micrometers paraffin embedded sections were deparaffinized and rehydrated, and then washed with 0.05 M phosphatebuffered saline (PBS). After immersed in antigen unmasking solution, endogenous peroxidase was inhibited with 0.3% hydrogen peroxide in methanol at room temperature for 20 min. Then sections were treated with the following primary antibodies in blocking solution at 4 °C overnight: rabbit anti-mTOR (1:100, Cell Signaling Technology, USA); rabbit anti-phosphorylated mTOR (p-mTOR) (1:50, Ser2448, Cell Signaling Technology, USA); rabbit anti-HIF-1α (1:50, Santa Cruz Biotechnology, USA); rabbit anti-VEGF (1:100, Santa Cruz Biotechnology, USA); and rabbit anti-cleaved caspase 3 (1:200, Cell Signaling Technology, USA). After washing in PBS, the sections were incubated with biotin-conjugated goat anti-rabbit IgG or goat anti-mouse IgG as secondary antibodies for 30 min at 37 °C, and then placed in an avidin conjugated horseradish peroxidase complex solution for 30 min. Staining was visualized by dipping 3-3′ diaminobenzidine (DAB) (KPL, USA) for 5 min and 0.03% H2O2 for 5 min. After counterstaining with hematoxylin, sections were cover slipped and observed under a microscope (Leica, CM 2000). Application of control serum instead of the primary antibody on alternative sections of the same brain provided negative controls.
2.3. Western blot analysis
The isolated ischemic hemispheres at different time points as described above were homogenized in ice-cold lysis buffer containing cytosol extraction and Ser/Thr phosphokinase inhibitor cocktail 20 μl/ml (Calbiochem, EMD Chemicals Inc., Germany). Lysates were centrifuged at 14,000 rpm for 30 min at 4 °C. Cytosol and nuclear proteins were purified as described before [19]. Nuclear proteins and cytosol proteins were used to detect the expression of HIF-1α and VEGF, respectively. Total proteins were used to detect the expression of CC3, mTOR, and p-mTOR. Equivalent quantity protein (100 μg per lane) lysates was electrophoresed in 6–12% SDS-polyacrylamide gels and transferred to PVDF membranes (Roche, Switzerland). After blocked in 5% bovine serum albumin, the membranes were incubated for 1 h at room temperature and then overnight at 4 °C with the following primary antibodies: rabbit anti-mTOR polyclonal antibody (1:500, Cell Signaling Technology, USA); rabbit anti-p-mTOR (Ser2448) polyclonal antibody (1:200, Cell Signaling Technology, USA); rabbit anti-HIF-1α monoclonal antibody (1:200, Santa Cruz Biotechnology, USA); rabbit anti-VEGF polyclonal antibody (1:400, Santa Cruz Biotechnology, USA); rabbit anti-CC3 polyclonal antibody (1:100, Cell Signaling Technology, USA) or rabbit anti-β-actin polyclonal antibody (1:3000, Sigma, USA). Membranes were then incubated with peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (1:3000, Santa Cruz Biotechnology, USA) for 1 h. The blots were visualized by enhanced chemiluminescence (ECL; Millipore, USA) using a gel imaging system (Bio-RAD, USA). Protein levels were normalized to β-actin as a loading control. Gel-pro analyzer was used to measure the relative optical density of protein bands. All experiments were repeated at least three times to ensure reproducibility of the results.
2.4. TUNEL
Apoptotic cell death was detected using In Situ Cell Death Detection Kit (Roche, Switzerland) according to the manufacturer’s instructions. The sections were deparaffinized and treated with 0.1 M citrate solution and 3% hydrogen peroxide at room temperature for 10 min. They were then treated with proteinase k (20μg/mL) for 7 min at room temperature, and re-washed with PBS. After incubated at 37 °C for 1 h with biotinylated nucleotide and the Terminal Deoxynucleotidyl Transferase, Recombinant (rTdT) enzyme, sections were washed and incubated with streptavidin HRP solution and detected with 0.05% DAB. As negative controls, alternate sections were processed in parallel without rTdT enzyme.
2.5. Statistical analysis
Data were expressed as means ± standard deviation (SD). Statistical differences between sham control and each group were compared using one-way ANOVA with Bonferroni/Dunnett post hoc tests. Statistical differences between two groups were compared using Least Significant Difference test. Statistical significance was defined as *p < 0.05 and **p < 0.01.
3. Results
3.1. Expression and distribution of mTOR, p-mTOR, HIF-1α, VEGF and CC3 after HI
To detect the expression of mTOR, p-mTOR, HIF-1α, VEGF and CC3 after HI in neonatal rat brain, we used immunohistochemistry on paraffin embedded sections from the sham controls, 2, 4, 8, 24 and 72 h after HI (each group n=4). The expression of total mTOR was stable in all the groups, while the p-mTOR expression increased at 2 and 4 h (data not shown), and peaked at 8 h after HI (Fig. 1). HIF-1α and VEGF expressions were significantly up-regulated and peaked at 8 h after HI (Fig. 1). The expression of CC3 facilitated at 2 h (data not shown), peaked at 24 h, and lasted 72 h after HI (Fig. 1). The mTOR, p-mTOR and VEGF-positive staining signals were mainly localized in the cytoplasm, HIF-1α-positive staining signals in the cellular nucleus, and CC3-positive staining signals in the cellular nucleus and cytoplasm.
Fig. 1.

Immunohistochemistry of mTOR, p-mTOR, HIF-1α, VEGF and CC3 expression in P10 rats brain after HI. The total mTOR-positive cells were not obviously changed compared with the sham control. The p-mTOR, HIF-1α, VEGF and CC3-positive cells were weakly detected in sham controls. By contrast, p-mTOR, HIF-1α and VEGF-positive cells were significantly increased and peaked at 8 h in cortex after HI. CC3-positive cells were significantly increased and peaked at 24 h, lasted 72 h after HI. Arrows show the positive staining cells (n=4). 400× (HI, hypoxia-ischemia).
3.2. Protein expression of mTOR, p-mTOR, HIF-1α, VEGF and CC3 after HI
Western blot analysis was used to quantify mTOR, p-mTOR, HIF-1α, VEGF and CC3 expression after HI. Total protein was segregated using the ischemic hemisphere from both the sham controls and HIgroups at 2, 4, 8, 12, 24, and 72 h (each group n=4). The total mTOR protein was not obviously changed at any time point compared to the sham controls. The p-mTOR protein was significantly induced at 8 h, and maintained a high level at 24 h, and then declined to basal level at 72 h (Fig. 2A). We also found HIF-1α and VEGF was significantly up-regulated and peaked at 8 h, and then reduced at 24 h after HI compared with sham controls (Fig. 2C). The expression of CC3 increased at 2 h, peaked at 24 h, and lasted 72 h after HI (Fig. 2C).
Fig. 2.

mTOR, p-mTOR, HIF-1α, VEGF and CC3 proteins expression in P10 rat brain after HI detected by Western blot analysis. Equal amount of protein samples (100 μg) was loaded, and β-actin served as loading control. Values are expressed in relative optical density and represented as mean ± SD (n=4). Total mTOR protein was not obviously changed at different time points compared with sham controls (A). However, p-mTOR protein was induced in ischemia hemisphere, peaked at 8 h, and maintained a high level at 24 h, and then declined to baseline at 72 h (A). HIF-1α and VEGF proteins were obviously induced and peaked at 8 h, then reduced at 24 h after HI (C). CC3 protein was induced at 2 h, peaked at 24 h and maintained a high level at 72 h after HI (C). mTOR, p-mTOR, HIF-1α, VEGF and CC3 expression in HI groups and sham controls were quantified. Data were obtained by densitometry and normalized by β-actin. *p < 0.05, **p < 0.01 compared with the sham control (B and D) (HI, hypoxia-ischemia).
3.3. The involvement of mTOR in the regulation of HIF-1α, VEGF, CC3
To disclose the potential role of mTOR in regulating HIF-1α, VEGF, and CC3, we used rapamycin, a special mTOR inhibitor, to suppress mTOR function. Total protein was segregated using the ischemic hemisphere from both the sham controls and HI groups at 8 and 24 h of rapamycin- or DMSO-treated rats (each group n=4). Western blot analysis revealed that total mTOR expression was not obviously changed while p-mTOR, HIF-1α and VEGF expressions were significant inhibited by rapamycin treatment in contrast to the DMSO group at 8 and 24 h (Fig. 3A and C). CC3 expression in rapamycin-treated group was increased at 8 h but decreased at 24 h after HI (Fig. 3C).
Fig. 3.

mTOR involvement in the regulation of HIF-1α, VEGF, and CC3 after HI in P10 rat brains. Western blot was used to analyse mTOR, p-mTOR, HIF-1α, VEGF, and CC3 expression in rapamycin- and DMSO-treated rats (A and C). In rapamycin-treated rats, p-mTOR was significantly reduced but the total mTOR remained unchanged compared with DMSO-treated rats (A). HIF-1α and VEGF protein expression were significantly inhibited by rapamycin. CC3 protein expression was increased at 8 h but reduced at 24 h after HI in rapamycin-treated rats (C). β-actin served as a loading control. The values are expressed in relative optical density and represented as mean ± SD (n=4). *p < 0.05, **p < 0.01 DMSO-treated group compared with the rapamycin-treated group at the same time points (B and D) (HI, hypoxia-ischemia; DM, DMSO; RA, rapamycin).
3.4. The effect of mTOR signaling on neuronal apoptosis
To investigate the effect of mTOR signaling on HI-induced neuronal apoptosis, we used rapamycin to block mTOR signaling and TUNEL labeling to detect neuronal apoptosis. Very few TUNEL positive cells were detected in the sham controls (data not shown), while the number of positive cells was increased at 8 and 24 h after HI in the DMSO-treated group. Nevertheless, TUNEL positive cells after rapamycin administration were up-regulated at 8 h and down-regulated at 24 h after HI (Fig. 4), which was in accordance with CC3 expression as shown by Western blot in Fig. 3.
Fig. 4.
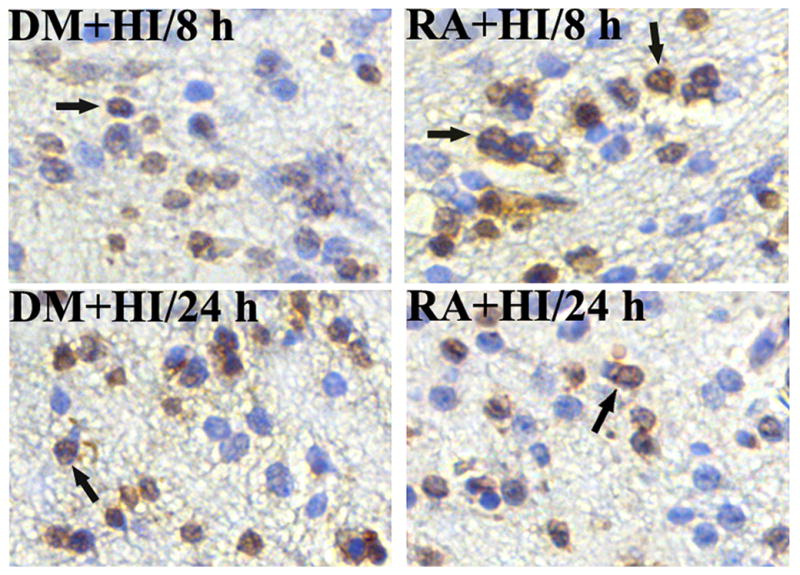
The effect of mTOR on apoptosis after HI in P10 rat brains. TUNEL-positive cells were hardly detected in the sham control (data not shown), but significantly induced at 8 and 24 h after HI in the DMSO-treated group. The number of TUNEL positive cells increased at 8 h but decreased at 24 h after HI in rapamycin-treated rats (n=4). Arrows show the positive staining cells. 400× (HI, hypoxia-ischemia; DM, DMSO; RA, rapamycin).
4. Discussion
We have previously shown that HIF-1α may play a neuroprotective role in the repair process of HIBD [14, 16, 17]. HIF-1α can promote the expression of its target gene VEGF, and then stimulate angiogenesis to meet the demand of repair. In addition, HIF-1α may support the survival mechanism by influencing apoptosis in neonatal HIBD [14]. In this study, we first showed that mTOR signaling may be involved in the regulation of HIF-1α, VEGF, and apoptosis in the developing rat brain with HI.
Our results showed that the p-mTOR expression increased at 2 h after HI, peaked at 8 h, the same trend as that of HIF-1α. On the other hand, the increased expression of HIF-1α and VEGF is reduced by a mTOR inhibitor, rapamycin. Nevertheless, the increased p-mTOR lasted 24 h after HI while the up-regulated HIF-1α only lasted 8 h. It is possible that the positive regulation of mTOR on HIF-1α is not sufficient to counteract its degradation in the later phase after HI.
In developing brains under HI, shortage of oxygen is the stimulus of angiogenesis to form a proper blood vessel network [5], which is regulated through HIF-mediated transcription of angiogenic factors [13]. The sensitive hypoxia sensor mTOR can integrate the signal and transmit to the nucleus, and then activate its two main down-stream targets, eukaryotic initiation factor 4E binding protein 1 (4EBP1) and p70 ribosomal protein S6 kinases 1 (p70S6K). 4EBP1, phosphorylated by mTOR, lead to eukaryotic initiation factor 4E release from 4EBP1, inducing protein synthesis. p70S6K, activated by mTOR, phosphorylates S6 ribosomal protein to regulate translation of 5′-TOP mRNA and ribosome biogenesis [22]. In addition, mTOR can promote the adaptive survival mechanism under hypoxia resulting from the rapid proliferation in tumors by regulating HIF-1α and VEGF expression levels [13]. p70S6K and 4EBP1 account for the regulation of mTOR on HIF-1α and VEGF [4,6]. These studies strongly support that mTOR can participate in the signaling pathway to regulate HIF-1α and VEGF under HI.
mTOR functions as an important regulator of HIF-1α or VEGF in hypoxia. mTOR could be a positive regulator of HIF-1α-dependent gene transcription, participating in the mechanism where by hypoxia promotes angiogenesis [10]. Moreover, HIF-1α is a partial effecter of mTOR-dependent, hypoxia-induced proliferation [15]. mTOR inhibition by rapamycin is reported to modulate HIF-1α activity via a Von Hippel-Lindau (VHL)-independent mechanism without affecting its stability [13]. In our study, rapamycin reduced HIF-1α protein expression as well as VEGF, which supports that mTOR can regulate HIF-1α and VEGF expression under HI in developing rat brain.
Dying neurons in HI may experience various morphologic changes, through apoptosis, autophagy and necrosis cell death pathways [2]. mTOR can participate in the regulation of neuronal death. mTOR inhibition by rapamycin can suppress the activation of cyclin-dependent kinases and then inhibit the cell cycle progress; suppress cell proliferation; and finally result in cell apoptosis[8]. The increased expression of CC3 protein is accepted to be a marker of neuronal apoptosis after HI [14]. In this study, rapamycin increased CC3 expression at 8 h after HI, consistent with rapamycin’s apoptosis-promoting function. In addition, this pro-apoptosis mechanism was possibly related to the reduced expression of HIF-1α by rapamycin, because HIF-1α could protect neurons from apoptosis under HI [14]. However, we also found that the expression of CC3 and neuronal apoptosis were decreased with rapamycin treatment at 24 h after HI. This decrease could be explained by increased autophagy after HI. Autophagy can precede apoptosis and play a protective role in neuronal death [3]. In the same HIBD model, Balduini et al. [1] has found that blocking mTOR with rapamycin led to enhanced autophagy (the up-regulating of autophagy marker Beclin 1) and reduced apoptosis (the downregulating of apoptosis marker CC3) at 24 h after HI. All these studies suggest a dual role of rapamycin in neonatal HIBD by interfering with the function of mTOR.
5. Conclusion
mTOR functions as an important oxygen sensor in the pathologic and survival mechanisms responding to HI stress. Our study suggests that mTOR may participate in the repair through mTOR/HIF-1α/VEGF axis, as well as apoptosis through mTOR/CC3, via its function on promoting protein synthesis. How to regulate the mTOR signaling pathway is an important issue that needs further study.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 30825039, No. 31171020 and No. 30973236 to Dezhi Mu; and No. 81172174 to Yi Qu), and Program for Changjiang Scholars and Innovative Research Team in University (IRT0935).
Abbreviations
- HI
Hypoxia-ischemia
- mTOR
mammalian target of rapamycin
- HIF-1α
hypoxia-inducible factor-1α
- VEGF
vascular endothelial growth factor
- P10
postnatal day 10
- CC3
cleaved caspase 3
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP-nick end labeling
- HIBD
hypoxia-ischemia brain damage
- PI3K
phosphatidylinositol 3-kinase
- AKT
protein kinase B
- CCA
common carotid artery
- PBS
phosphate-buffered saline
Footnotes
Conflict of interest statement
All of the authors involved in preparation of the above manuscript declare no conflict of interest in any form.
References
- 1.Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia-ischemia induced brain injury: evidence and speculations. Autophagy. 2009;5(2):221–223. doi: 10.4161/auto.5.2.7363. [DOI] [PubMed] [Google Scholar]
- 2.Blomgren K, Leist M, Groc L. Pathological apoptosis in the developing brain. Apoptosis. 2007;12(5):993–1010. doi: 10.1007/s10495-007-0754-4. [DOI] [PubMed] [Google Scholar]
- 3.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis. 2008;32(3):329–339. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 4.Del Bufalo D, Ciuffreda L, Trisciuoglio D, Desideri M, Cognetti F, Zupi G, Milella M. Antiangiogenic potential of the mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006;66(11):5549–5554. doi: 10.1158/0008-5472.CAN-05-2825. [DOI] [PubMed] [Google Scholar]
- 5.Fahling M. Cellular oxygen sensing, signalling and how to survive translational arrest in hypoxia. Acta Physiol (Oxf) 2009;195(2):205–230. doi: 10.1111/j.1748-1716.2008.01894.x. [DOI] [PubMed] [Google Scholar]
- 6.Garcia Maceira P, Mateo J. Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene. 2009;28(3):313–324. doi: 10.1038/onc.2008.398. [DOI] [PubMed] [Google Scholar]
- 7.Ginet V, Puyal J, Clarke PG, Truttmann AC. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol. 2009;175(5):1962–1974. doi: 10.2353/ajpath.2009.090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu L, Gao J, Li Q, Zhu YP, Jia CS, Fu RY, Chen Y, Liao QK, Ma Z. Rapamycin reverses NPM-ALK-induced glucocorticoid resistance in lymphoid tumor cells by inhibiting mTOR signaling pathway, enhancing G1 cell cycle arrest and apoptosis. Leukemia. 2008;22:2091–2096. doi: 10.1038/leu.2008.204. [DOI] [PubMed] [Google Scholar]
- 9.Gunn RM, Hailes HC. Insights into the PI3-K-PKB-mTOR signalling pathway from small molecules. J Chem Biol. 2008;1(1–4):49–62. doi: 10.1007/s12154-008-0008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16(8):771–780. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 11.Kang J, Rychahou PG, Ishola TA, Mourot JM, Evers BM, Chung DH. Nmyc is a novel regulator of PI3K-mediated VEGF expression in neuroblastoma. Oncogene. 2008;27(28):3999–4007. doi: 10.1038/onc.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotulska K, Larysz-Brysz M, Grajkowska W, Jozwiak J, Wlodarski P, Sahin M, Lewin-Kowalik J, Domanska-Pakiela D, Jozwiak S. Cardiac rhabdomyomas in tuberous sclerosis complex show apoptosis regulation and mTOR pathway abnormalities. Pediatr Dev Pathol. 2009;12:89–95. doi: 10.2350/06-11-0191.1. [DOI] [PubMed] [Google Scholar]
- 13.Land SC, Tee AR. Hypoxia-inducible factor-1 alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282(28):20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Qu Y, Li J, Xiong Y, Mao M, Mu D. Relationship between HIF-1α expression and neuronal apoptosis in neonatal rats with hypoxia-ischemia brain injury. Brain Res. 2007;1180:133–139. doi: 10.1016/j.brainres.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 15.Li W, Petrimpol M, Molle KD, Hall MN, Battegay EJ, Humar R. Hypoxiainduced endothelial proliferation requires both mTORC1 and mTORC2. Circ Res. 2007;100(1):79–87. doi: 10.1161/01.RES.0000253094.03023.3f. [DOI] [PubMed] [Google Scholar]
- 16.Li L, Qu Y, Mao M, Xiong Y, Mu D. The involvement of phosphoinositid 3-kinase/Akt pathway in the activation of hypoxia-inducible factor-1 in the developing rat brain after hypoxia-ischemia. Brain Res. 2008;1197:152–158. doi: 10.1016/j.brainres.2007.12.059. [DOI] [PubMed] [Google Scholar]
- 17.Li L, Xiong Y, Qu Y, Mao M, Mu W, Wang H, Mu D. The requirement of extracellular signal-related protein kinase pathway in the activation of hypoxia inducible factor 1 in the developing rat brain after hypoxia-ischemia. Acta Neuropathol. 2008;115(3):297–303. doi: 10.1007/s00401-008-0339-5. [DOI] [PubMed] [Google Scholar]
- 18.Miyazawa M, Yasuda M, Fujita M, Hirabayashi K, Hirasawa T, Kajiwara H, Muramatsu T, Miyazaki S, Harasawa M, Matsui N, Ogane N, Murakami M, Mikami M, Yanase T, Osamura RY. Granulosa cell tumor with activated mTORHIF-1alpha-VEGF pathway. J Obstet Gynaecol Res. 2010;36(2):448–453. doi: 10.1111/j.1447-0756.2009.01127.x. [DOI] [PubMed] [Google Scholar]
- 19.Mu D, Chang YS, Vexler ZS, Ferriero DM. Hypoxia-inducible factor 1alpha and erythropoietin upregulation with deferoxamine salvage after neonatal stroke. Exp Neurol. 2005;195(2):407–415. doi: 10.1016/j.expneurol.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura K, Martin KC, Jackson JK, Beppu K, Woo CW, Thiele CJ. Brainderived neurotrophic factor activation of TrkB induces vascular endothelial growth factor expression via hypoxia-inducible factor-1 alpha in neuroblastoma cells. Cancer Res. 2006;66(8):4249–4255. doi: 10.1158/0008-5472.CAN-05-2789. [DOI] [PubMed] [Google Scholar]
- 21.Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol. 2011;69(5):743–758. doi: 10.1002/ana.22419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park KK, Liu K, Hu Y, Kanter JL, He Z. PTEN/mTOR and axon regeneration. Exp Neurol. 2010;223(1):45–50. doi: 10.1016/j.expneurol.2009.12.032. [DOI] [PubMed] [Google Scholar]
- 23.Shackelford DB, Vasquez DS, Corbeil J, Wu S, Leblanc M, Wu CL, Vera DR, Shaw RJ. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci U S A. 2009;106(27):11137–11142. doi: 10.1073/pnas.0900465106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology. 2010;51(5):1778–1788. doi: 10.1002/hep.23511. [DOI] [PMC free article] [PubMed] [Google Scholar]