

Abstract
This letter describes the further exploration of two series of M1allosteric agonists, TBPB and VU0357017, previously reported from our lab. Within the TPBP scaffold, either electronic or steric perturbations to the central piperidine ring led to a loss of selective M1 allosteric agonism and afforded pan-mAChR antagonism, which was demonstrated to be mediated via the orthosteric site. Additional SAR around a related M1 allosteric agonist family (VU0357017) identified similar, subtle ‘molecular switches’ that modulated modes of pharmacology from allosteric agonism to pan-mAChR orthosteric antagonism. Therefore, all of these ligands are best classified as bi-topic ligands that possess high affinity binding at an allosteric site to engender selective M1 activation, but all bind, at higher concentrations, to the orthosteric ACh site, leading to non-selective orthosteric site binding and mAChR antagonism.
Keywords: TBPB, M1, Allosteric agonist, Muscarinic receptor
Schizophrenia is a complex psychiatric disorder characterized by a combination of positive and negative symptoms along with significant cognitive dysfunction.1,2 Current antipsychotic therapies can address the positive symptoms, but the negative and cognitive symptoms remain poorly managed, if at all, and are key predictors of functional disability.3–6 A large number of anatomical, molecular, genetic, preclinical behavioral and human clinical studies have provided strong evidence that agents able to enhance cholinergic transmission or activate muscarinic acetylcholine receptors (mAChRs, M1-M5), notably M1, have exciting therapeutic potential for the treatment of the positive, negative and cognitive symptoms of schizophrenia as well as cognitive dysfunction in other CNS disorders.7–16 However, previous compounds developed to selectively activate M1 receptors have failed in clinical development due to a lack of true specificity for this receptor subtype.7–18 Often, many of the compounds bind to the orthosteric ACh binding site, which can result in adverse side effects as a result of M2-M5 activation.7–18 Recently, multiple industrial and academic laboratories, including ours, have targeted less conserved allosteric sites on the M1 receptor in an attempt to develop highly selective M1 activators (both M1 allosteric agonist and M1 positive allosteric modulators, PAMs) and avoid activation of M2-M5.17–39
For example, we have previously reported on TBPB 1, a potent, CNS penetrant and highly selective M1 allosteric agonist that displays robust efficacy in multiple preclinical antipsychotic and cognition models, as well as significant impact on Aβ production.19 Mutagenesis and modeling efforts identified a key H-bond interaction between the central piperidine nitrogen of TBPB and Thr83 of M1, likely contributing to TBPB’s affinity for this M1 allosteric binding site.40 In multiple Letters, we have also described SAR around TBPB 1, and found that halide substitutions were well tolerated on the benzimidazole core 2, as well as amide 3, sulfonamide 4 and urea 5 replacements for the benzyl amine of the distal basic piperidine nitrogen (Fig. 1); however, SAR was ‘shallow’.21 Moreover, these subtle structural modifications could engender D2 antagonism, along with M1 allosteric agonism, affording molecules with an attractive pharmacological profile for the treatment of schizophrenia.21 In this Letter, we describe a more detailed pharmacological profile of 1, along with the as yet unexplored SAR of the central piperidine ring of 1, and the discovery of subtle ‘molecular switches’41 that modulate modes of pharmacology from allosteric agonism to pan-mAChR orthosteric antagonism.
Figure 1.

Structures of the M1 allosteric agonist TBPB (1), highlighting the key H-bond interaction with Thr83 for allosteric binding at M1, and analogs 2–5 of TBPB that retain selective M1 agonism.
Our previous characterization of TBPB revealed that this compound activates M1 receptors.19 Here, we confirm that TBPB is a selective M1 partial agonist and induces responses in CHO cells expressing rM1 receptors, but not rM2-M5Rs (Fig. 2A). Interestingly, TBPB also inhibits ACh-induced responses in cells expressing M2-M5 receptors, suggesting additional activity at an orthosteric site (Fig. 2B). Based on this finding, TBPB can be described as a bi-topic ligand that binds M1 at both an allosteric site and the orthosteric site, the former of which confers functional M1 agonism and the latter of which confers orthosteric antagonism. These findings are similar to previously characterized M1 agonists including AC-42,32,33 77-LH-28-1,36 VU0364572,28,30 and VU035701726,30, which have been characterized as bi-topic ligands.42,43
Figure 2.

Pharmacological profile of TBPB. A) rM1-rM5 concentration-response curves (CRCs) of TBPB screened in agonist mode. rM1 EC50 = 79.6 nM (pEC50 = 7.10±0.07), 69.3±6.4% ACh Max, rM2-rM5 >30 μM B) rM2-rM5 CRCs of TBPB screened as antagonists. rM2 IC50 = 1.29 μM (pIC50 = 5.89), rM3 IC50 = 5.48 μM (pIC50 = 5.26), rM4 IC50 = 493 nM (pIC50 = 6.31), rM5 IC50 = 3.97 μM (pIC50 = 5.40), and all reduce an ACh EC80 to baseline. Human CRCs not shown. Screened as agonists, hM1 EC50 = 255 nM (pEC50 = 6.59), 40% ACh Max, M2-M5 >30 μM. Screened as antagonists, hM2 IC50 = 3.5 μM (pIC50 = 5.46), hM3 IC50 = 2.3 μM (pIC50 = 5.63), hM4 IC50 = 734 nM (pIC50 = 6.13), hM5 IC50 = 4.9 μM (pIC50 = 5.31), and all reduce an ACh EC80 to baseline.
As the central piperidine nitrogen of TBPB is thought to be critical (H-bond with Thr83) for allosteric binding,40 we sought to disrupt this key H-bond both electronically and conformationally by installation of a β-fluorine atom (to attenuate basicity and H-bond acceptor ability) and replacement of the piperidine with a [3.3.0] system, respectively.21 Our expectation was that these structural modifications would abolish binding at the allosteric site, and that these analogs would bind solely at the orthosteric site and function as pan-mAChR antagonists. The synthesis was straightforward (Scheme 1), following the synthetic route previously developed.22
Scheme 1.

Synthesis of TBPB analogs 9 (VU0465132) and 10 (VU0546403).
When screened as an agonist, neither 9 nor 10 elicited M1 activation, suggesting that binding at the allosteric site had been abolished by disrupting the key H-bond interaction with Thr83.40 Interestingly, both 9 and 10 proved to be weak, micromolar pan-mAChR antagonists when screened in the presence of an EC80 of ACh against M1-M5. The [3.3.0] analog 10 was a weak M1 antagonist (pIC50 = 5.20±0.04, IC50 = 6.4 μM, −1.07±1.3% ACh Max) and IC50s >10 μM against M2-M5). The β-fluoro analog 9 (Fig. 3A) was slightly more potent at M1 (Rat M1 IC50 = 4.9 μM (pIC50 = 5.31±0.17), 13.6±1.5% ACh Max, M2, M3 M4, and M5 IC50 > 10 μM (pIC50s <5) (Human M1-M5 data not shown, but similar)). Radioligand binding experiments with [3H]-NMS (Fig. 3B) and dissociation kinetic experiments (Fig. 3C) are consistent with 9 acting as an ACh orthosteric site antagonist. Thus, a single fluorine atom serves as ‘molecular switch’ to modulate the pKa of the central piperidine nitrogen atom (from ~11 to 9.4),44 and likely prohibits its ability to accept a key H-bond from Thr83 in the M1 allosteric site, an interaction that may confer M1 functional agonist activity.40
Figure 3.

Pharmacological profile of β-F-TBPB 9 (VU0465132). A) Rat M1-M5 CRCs screened as antagonists. Rat M1 IC50 = 4.9 μM (pIC50 = 5.31±0.17), 13.6±1.5% ACh Max, M2, M3 and M5 IC50 > 10 μM (pIC50s <5), M4 IC50 = 5.2 μM (pIC50 = 5.29±0.09), 26.6±3.4% ACh Max (Human M1-M5 data not shown, but comparable). B) [3H]-NMS competition binding studies. At higher concentrations than the control atropine (Ki = 1.4 nM, pKi = 8.86±0.24), both 9 (Ki = 1.35 μM, pKi = 5.87±0.18) and TBPB (Ki = 0.60 μM, pKi = 6.22±0.14) fully displace [3H]-NMS, consistent with interactions at the orthosteric ACh site. C) Dissociation kinetics with TBPB, 9, atropine, carbachol (CCh) and Gallamine (Gall). In our assays, only the allosteric modulator Gall altered the rate of [3H]-NMS dissociation, providing further evidence that 9 acts as an orthosteric antagonist. Koff rates were as follows ± SEM: Atropine (0.0379±0.0125 min−1), CCh(0.0393±0.0125 min−1), Gall (0.0012±0.0006 min−1), TBPB (0.0335±0.0095 min−1), VU0465132 (0.0308±0.0103 min−1).
Interestingly, our dissociation kinetics experiments with TBPB indicate that this compound also does not alter the off-rate of [3H]-NMS. These data are inconsistent with those of Jacobson et al., 2010, where TBPB was shown to slow the off-rate of [3H]-NMS.40 The most likely explanation for the discrepancy between our results and those of Jacobson et al., 2010 is a difference in our assay protocols. Jacobson et al. allow 1 hour for equilibrium to occur between atropine and [3H]-NMS at room temperature for their dissociation kinetic studies whereas we allow 3 hours at room temperature for this equilibrium to occur. Traditionally, when a 1 hour equilibrium is utilized for M1 dissociation studies, the studies are performed at 37°C.43 Incubation for only 1 hour at room temperature may not allow enough time for a proper equilibrium to be established between atropine and [3H]-NMS and thus Jacobson et al. may have performed their TBPB dissociation studies under non-equilibrium conditions.
While ‘molecular switches’ that modulate modes of pharmacology and subtype selectivity are common in Family C GPCRs (eg., mGlu receptors),41 we have rarely encountered them in Family A GPCRs. Within the mAChRs, we had previously only encountered ‘molecular switches’ that modulate mAChR subtype selectivity.27,41 We recently reported on a chemically distinct series of M1 allosteric agonists, represented by 11 and 12 (Fig. 4), where mutagenesis work indicated a novel allosteric binding site in the third extracellular loop of M1.26 Serendipitously, when preparing additional analogs around 12, a chemical vendor sent the incorrect starting material, leading to the synthesis of the alternative regioisomer of 12, compound 13.13 proved to be a potent antagonist of M1 (IC50 = 273 nM, pIC50 = 6.32±0.11), as well as M2-M5, and detailed molecular pharmacology showed that 13 was an orthosteric mAChR antagonist. Note, the modulation from M1 allosteric agonist to pan-mAChR orthosteric antagonist was due to major structural modifications (ie., 3° to 2° amine and 2° to 3° amide).
Figure 4.

Structures and activities of M1 allosteric agonists 11 and 12, and a mAChR orthosteric antagonist 13 (regioisomer of 12).
These data, coupled with the results from TBPB and 9, led us to scrutinize additional analogs of 12, in an effort to discover more subtle ‘molecular switches’ that can modulate modes of pharmacology within the VU0364572 bi-topic ligand scaffold. One of our initial efforts led to the development of VU0419757 (14) which contains a 3-bromophenyl amide (Fig. 5) and proved to be a weak partial M1 allosteric agonist due to the addition of a 3-bromophenyl amine (EC50 = 2.0 μM, pEC50 = 5.69±0.08, 37.4±8.7% ACh Max). Modification with an additional bromine atom to the 5-position afforded VU0430613 (15), an equipotent M1 antagonist (IC50 = 3.4 μM, pIC50 = 5.47±0.03). Furthermore, when the aryl amides in 12 were replaced with heterocyclic amides, pan-mAChR antagonists resulted. Even when fluorine atoms were employed to attenuate the basicity of the heterocycles, M1 allosteric agonism was lost. For example, a 6-trifluomethoxy nicotinamide derivative, VU0362058 (16), proved to be another pan-mAChR antagonist (Fig. 6), favoring inhibition of M2 (IC50 = 1.1 μM, pIC50 = 5.97) over M1 (IC50 = 3.2 μM, pIC50 = 5.50±0.06) and M3-M5 (IC 50s >10 μM).
Figure 5.

Structures and activities of M1 allosteric agonist 14 and a 3,5-dibromo congener 15, an M1 antagonist.
Figure 6.

A) Structure of VU0362058 (16) and full M1-M5 CRCs, highlighting the pan-mAChR antagonism via a subtle modification to the M1 allosteric agonist 12. B) Representative additional amide analogs of 12 (17) where the nature of substitutions on the aromatic ring conferred M1 antagonism as opposed to M1 agonism. While some SAR trends are evident, other switches in the mode of M1 pharmacology are random and confound chemical optimization efforts.
In summary, we have explored additional SAR around two M1 allosteric agonists TBPB and VU0364572 (12). Within the TBPB scaffold, either electronic or steric perturbations to the benzimidazolone piperidine ring led to a loss of selective M1 allosteric agonism and afforded pan-mAChR orthosteric antagonists. Based on mutagenesis and modeling studies with TBPB, the electronic and steric perturbations disrupted a key H-bond with Thr83 in the allosteric site on the M1 receptor. Similar subtle ‘molecular switches’ were found within the VU0364572 series, converting potent M1 agonists into pan-mAChR antagonists. These findings, coupled with our recent studies showing brain-region-dependent and receptor reserve-dependent pharmacology of functionally selective M1 bi-topic agonists42 show the challenges associated with development of selective M1 agonists. Therefore, we propose that positive allosteric modulation of M1 receptors, as opposed to allosteric agonism, may be the preferred mechanism by which to develop selective M1 activators as therapeutic agents.
Acknowledgments
This work was supported by grants from the NIH and NIMH (1RO1MH082867). Vanderbilt is a Specialized Chemistry Center within the Molecular Libraries Probe Centers Network (U54 MH84659).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Mol Psychiatry. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg TE, Gold JM, Greenberg R, Griffin S, Schulz SC, Pickar D. Am J Psychiatry. 1993;150:1355–1362. doi: 10.1176/ajp.150.9.1355. [DOI] [PubMed] [Google Scholar]
- 3.Weickert TW, Goldberg TE, Gold JM, Bigelow LB, Egan MF, Weinberg DR. Arch Gen Psychiatry. 2000;57:907–913. doi: 10.1001/archpsyc.57.9.907. [DOI] [PubMed] [Google Scholar]
- 4.Siris SG. Schizophrenia. Blackwell Science Ltd; Oxford: 1995. pp. 128–146. [Google Scholar]
- 5.Green MF. Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- 6.Carlsson A. Neuropsychopharmacology. 1988;1:179–186. doi: 10.1016/0893-133x(88)90012-7. [DOI] [PubMed] [Google Scholar]
- 7.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Am J Psych. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 8.Mirza NR, Peters D, Sparks RG. CNS Drug Rev. 2003;9(2):159–186. doi: 10.1111/j.1527-3458.2003.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sur C, Mallorga PJ, Wittmann M, Jacobson MA, Pascarella D, Williams JB, Brandish PE, Pettibone DJ, Scolnick EM, Conn PJ. PNAS. 2003;100:13674–13679. doi: 10.1073/pnas.1835612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pakrasi S, Colloby SJ, Firbank MJ, Perry EK, Wyper DJ, Owens J, McKeith IG, Williams ED, O’Brien JT. J Neurol. 2003;254:907–913. doi: 10.1007/s00415-006-0473-8. [DOI] [PubMed] [Google Scholar]
- 11.Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM. Arch Neurol. 1997;54:465–473. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- 12.Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Mol Psych. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 14.Fisher A. Neurodegen Diseases. 2008;5:237–240. [Google Scholar]
- 15.Conn PJ, Jones C, Lindsley CW. Trends in Pharm Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conn PJ, Tamminga C, Schoepp DD, Lindsley C. Mol Intervent. 2008;8:99–105. doi: 10.1124/mi.8.2.7. [DOI] [PubMed] [Google Scholar]
- 17.Conn PJ, Christopolous A, Lindsley CW. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane A, Bridges TM, Kennedy JP, Bradley SR, Peterson T, Baldwin RM, Kessler R, Deutch A, Lah JL, Levey AI, Lindsley CW, Conn PJ. J Neurosci. 2008;28(41):10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marlo JE, Niswender CM, Luo Q, Brady AE, Shirey JK, Rodriguez AL, Bridges TM, Williams R, Days E, Nalywajko NT, Austin C, Williams M, Xiang Y, Orton D, Brown HA, Kim K, Lindsley CW, Weaver CD, Conn PJ. Mol Pharm. 2009;75:577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
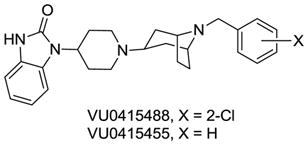
21.In addition to the [3.3.0] system, we also synthesized and evaluated tropane replacements for the piperidine ring, and these also lost M1 agonism and afforded M1 antagonism (IC50s 950 nM to 1.6 μM).
- 22.a) Miller NR, Daniels NR, Bridges TM, Brady AE, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:5443–5446. doi: 10.1016/j.bmcl.2008.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bridges TM, Brady AE, Kennedy JP, Daniels NR, Miller NR, Kim K, Breininger ML, Gentry PR, Brogan JT, Jones JK, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:5439–5442. doi: 10.1016/j.bmcl.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma L, Seager M, Wittman M, Bickel N, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danzinger A, Regan C, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Jacobson M, Sur C, Kinney G, Seabrook GR, Ray WJ. Proc Natl Acad Sci USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Jadhav SB, Menon U, Christain EP, Doherty JJ, Quirk MC, Snyder DH, Levey AI, Watson ML, Nicolle MM, Lindsley CW, Conn PJ. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittman M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Bioorg Med Chem Lett. 2010;20:531–536. doi: 10.1016/j.bmcl.2009.11.100. [DOI] [PubMed] [Google Scholar]
- 26.Lebois EP, Bridges TM, Dawson ES, Kennedy Jp, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. ACS Chemical Neurosci. 2010;1:104–121. doi: 10.1021/cn900003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeBois EP, Sheffler DJ, Digby GJ, Melancon BJ, Tarr JC, Cho HP, Morrison R, Bridges TM, Xiang Z, Daniels JS, Wood MR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2011;21:6451–6455. doi: 10.1016/j.bmcl.2011.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid PR, Bridges TM, Sheffler DA, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Bioorg Med Chem Lett. 2011;21:2697–2701. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Digby GJ, Noetzel MJ, Bubser M, Utley TJ, Walker AG, Byun NB, LeBois EP, Xiang Z, Sheffler DJ, Niswender CM, Plumley HC, Davis AA, Nemirovsky NE, Mennenga S, Camp BW, Bimonte-Nelson HA, Morrison R, Daniels S, Lindsley CW, Olive MF, Conn PJ. J Neurosci. 2012;32:8532–8544. doi: 10.1523/JNEUROSCI.0337-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuduk SD, Chang RK, DiMarco CN, Ray WJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. ACS Med Chem Lett. 2010;1:263–267. doi: 10.1021/ml100095k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spalding TA, Trotter C, Skaaerbaek N, Meissner TL, Curreier EA, Burstein ES, LI D, Hacksell U, Brann MR. Mol Pharmacol. 2002;61:1297–1302. doi: 10.1124/mol.61.6.1297. [DOI] [PubMed] [Google Scholar]
- 33.Spalding TA, Ma JN, Ott TR, Friberg M, Bajpai A, Bradley SR, Davis RE, Brann MR, Burstein ES. Mol Pharmacol. 2006;70:1974–1983. doi: 10.1124/mol.106.024901. [DOI] [PubMed] [Google Scholar]
- 34.Sur C, Mallorga PJ, Wittman M, Jacobson MA, Pascarella D, Williams JB, Brandish PE, Pettibone DJ, Scolnick EM, Conn PJ. Proc Natl Acad Sci USA. 2003;100:13674–13679. doi: 10.1073/pnas.1835612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuduk SD, Chang RK, DiMarco CN, Pitts DR, Greshok TJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT, Ray W. J Med Chem. 2011;54:4773–4780. doi: 10.1021/jm200400m. [DOI] [PubMed] [Google Scholar]
- 36.Langmead CJ, Austin NE, Branch CL, Buchanan KA, Davies CH, Fores IT, Fry VAH, Hagan JJ, et al. Br J Pharmacol. 2008;154:1104–1115. doi: 10.1038/bjp.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuduk SD, Chang RK, DiMArco CN, Ray WJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MD. Bioorg Med Chem Lett. 2011;21:1710–1715. doi: 10.1016/j.bmcl.2011.01.094. [DOI] [PubMed] [Google Scholar]
- 38.Kuduk SD, DiMarco CN, Cofre V, Pitts DR, Ray WJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MD. Bioorg Med Chem Lett. 2010;20:657–661. doi: 10.1016/j.bmcl.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 39.Budzik B, Garzya V, Shi D, Walker G, Lauchart Y, Lucas AJ, Rivero RA, Langmead CJ, Watson J, Wu Z, Forbes IT, Jin J. Bioorg Med Chem Lett. 2010;20:3545–3549. doi: 10.1016/j.bmcl.2010.04.127. [DOI] [PubMed] [Google Scholar]
- 40.Jacobson MA, Kreatsoulas C, Pascarella DM, O’Brien JA, Sur C. Mol Pharmacol. 2010;78:648–657. doi: 10.1124/mol.110.065771. [DOI] [PubMed] [Google Scholar]
- 41.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Biochemistry. 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Digby GJ, Utley TJ, Lamsal A, Sevel C, Sheffler DJ, Lebois EP, Bridges TM, Wood MR, Niswender CM, Lindsley CW, Conn PJ. ACS Chem Neurosci. doi: 10.1021/cn300103e. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alvani VA, Langmead CJ, Guida E, Wood MD, Tehan BG, Hedron HJ, Watson JM, Sexton PM, Christopoulos A. Mol Pharmacol. 2010;78:94–104. doi: 10.1124/mol.110.064345. [DOI] [PubMed] [Google Scholar]
- 44.Fadeyi O, Lindsley CW. Org Lett. 2009;11:943–946. doi: 10.1021/ol802930q. [DOI] [PMC free article] [PubMed] [Google Scholar]