

Abstract
A series of salicylanilides was synthesized based on a high-throughput screening hit against Mycobacterium tuberculosis. A free phenolic hydroxyl on the salicylic acid moeity is required for activity, and the structure-activity relationship of the aniline ring is largely driven by the presence of electron withdrawing groups. We synthesized 94 analogs exploring substitutions of both rings and the linker region in this series and we have identified multiple compounds with low micromolar potency. Unfortunately, cytotoxicity in a murine macrophage cell line trends with antimicrobial activity, suggesting a similar mechanism of action. We propose that salicylanilides function as proton shuttles that kill cells by destroying the cellular proton gradient, limiting their utility as potential therapeutics.
Keywords: Tuberculosis, Salicylanilide, Proton gradient, Antibacterial
1. Introduction
Mycobacterium tuberculosis (Mtb) infects an estimated one-third of the world’s population, causing approximately 1.7 million deaths in 2010.1 This, coupled with the rise of multidrug resistant and extensively drug resistant strains, necessitates the search for novel antimycobacterial agents. We conducted a high-throughput screen (HTS) of a subset of compounds from the National Institute of Health’s Chemical Genomics Center (NCGC) library. In this screen we identified N-(3,5-bis(trifluoromethyl)phenyl)-5-chloro-2-hydroxybenzamide (IMD-0354, 1) as a lead compound. IMD-0354 (1) is a potent inhibitor of IκB kinase β, and is currently in Phase I/II clinical trials for both atopic dermatitis and chronic obstructive pulmonary disease.2 The fact that this compound is already in human trials suggested promise for repurposing the drug against Mtb, which lacks IκB kinase β. In this study, we sought to improve on the initial potency of salicylanilide IMD-0354 against Mtb.
Salicylanilides have been shown to be active against many pathogens, including S. aureus,3 E. coli,4 P. aeruginosa,4 A. fumigatus,5 as well as being used as anthelminthics in veterinary practice.6 Salicylanilides have shown activity against M. tuberculosis7 although cytotoxicity in mammalian cells is a common problem. Masking the salicylate hydroxyl moiety with a carbamate linkage decreases cytotoxicity in vitro,8 but as yet it is unknown if this improvement carries into animal models or humans.
Salicylanilides have been suggested to target multiple specific targets in both prokaryotes and eukaryotes, including two-component systems,3 transglycosylase,9 type III secretion systems,10 interleukin 12p40,11 and protein tyrosine kinase.12 Prior to these studies, salicylanilides were known as potent uncouplers of the proton gradient used to generate ATP in mitochondria;13 the precise mode of action in mycobacteria is unclear.
2. Results and Discussion
2.1 SAR of the 5-chlorosalicylate (A) ring
The A ring of the salicylanilide lead (1) was modified in an attempt to improve potency and to decouple potency and cytotoxicity (Table 1). An unsubstituted A ring (4) completely lacked activity while removing only the 5-chloro substituent (2) resulted in a small (2–4 fold) drop in potency. Removal of only the 2-hydroxy substituent (3), however, completely abolished activity. Substitution of the 5-position with other halogens (F, 7; Br, 8; or I, 9) resulted in only small (2-fold) changes in potency, which we initially interpreted to mean that the only requirement for this position was the presence of a weak electron-withdrawing group. Unexpectedly though, the 5-methoxy ether 10 retained activity, although less than the parent compound. The 5-nitrosalicylanilide 13 is likewise inactive suggesting that simply bearing a strong electron-withdrawing substituent is not sufficient for full activity.
Table 1.
SAR of salicylic acid ring (A ring)
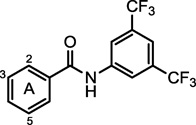 | |||||
|---|---|---|---|---|---|
| Cmpd | 2 | 3 | 5 | MIC(µM) | IC50 (µM) |
| 1 | OH | H | Cl | 3.1 | 5.2 |
| 2 | OH | H | H | 6.3–13 | 12 |
| 3 | H | Cl | H | >50 | >60 |
| 4 | H | H | H | >50 | n.d. |
| 5 | OH | H | 1-pyrrole | 3.1 | 3.1 |
| 6 | OH | H | CH3 | 6.3–13 | 5.4 |
| 7 | OH | H | F | 3.1 | 2.5 |
| 8 | OH | H | Br | 6.3 | 4.9 |
| 9 | OH | H | I | 6.3 | 5.6 |
| 10 | OH | H | OMe | 6.3–13 | 7.7 |
| 11 | OH | Cl | Cl | 13 | >60 |
| 12 | OH | OH | H | >50 | 1.7 |
| 13 | OH | H | NO2 | 50–>50 | >60 |
| 14 | OAc | H | Cl | 1.6–3.1 | >60 |
| 15 | OMe | H | Cl | >50 | >60 |
| 16 | OBn | H | Cl | >50 | >60 |
| 17 | NH2 | H | Cl | >50 | >60 |
| 18 | Cl | H | H | >50 | >1.7 |
n.d.: not determined.
MIC values determined using Mtb H37Rv. IC50 values determined using the murine macrophage cell line J774.1.
In an attempt to improve the solubility of this molecule we also explored substitution with a 1-pyrrole moiety resulting in a more soluble, equipotent, but still cytotoxic molecule (5).
Acetylation at the 2-position was tolerated (14) and cytotoxicity was diminished, most likely because deacetylation occurs in the mycobacterial assay but not the cytotoxicity assay. Methylation or benzylation of the phenol (15 and 16, respectively) resulted in loss of activity, as did substitution with either chlorine (18) or preparation of the corresponding aniline (17). The 3,5-dichlorophenol (11) retained some activity while the 2,3-dihydroxy derivative (12) lost all activity (although notably increased its cytotoxicity presumably due to facile formation of the o-quinone).
2.2 SAR of the linker region
The SAR in the linker region showed very little tolerance for substitution (Table 2). The corresponding amine (19) was inactive as was a derivative where the amide was replaced with a urea (20). Simply reversing the amide linkage (22) resulted in a complete loss of activity, as did replacing the amide with the corresponding ester (21). Replacing the amide with a thioamide (23) resulted in slightly improved activity. A sulfonamide linkage (24) resulted in a complete loss of activity. Substituting the aniline with a benzylamine (25) also resulted in a loss of activity although not as dramatic as the other substitutions.
Table 2.
Effect of linkers on activity
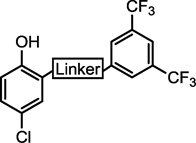 | |||
|---|---|---|---|
| Compd | Linker | MIC (µM) | IC50 (µM) |
| 19 | 50–>50 | 7.8 | |
| 20 | 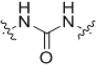 |
50–>50 | 13 |
| 21 | 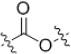 |
>50 | >60 |
| 22 | 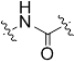 |
>50 | 12 |
| 23 | 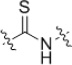 |
1.6 | n.d. |
| 24 | 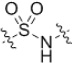 |
50–>50 | 23 |
| 25 | 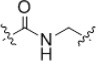 |
6.3–13 | 27 |
n.d.: not determined
2.3 SAR of the 3,5-bis(trifluoromethyl)aniline (B) ring
The potency on the B ring seemed to be largely driven by the presence of electron withdrawing groups (Table 3). Removal of one of the trifluoromethyl groups from the lead compound 1 had only a slight effect on activity (27), whereas removing both decreased potency 4–8 fold (26). Changing the position of a single trifluoromethyl from meta- to ortho resulted in a 4–8 fold loss in activity (28), possibly attributable to conformational change at the amide. Installing a single trifluoromethyl group in the para position resulted in a slight enhancement in potency (29). Amongst the bis(trifluoromethyl) substitution patterns that were synthetically accessible, potency was found to follow: 3,4- (30) > 3,5- (1) = 2,5- (32) > 2,3- (31) positions with compound 30 being the most potent compound identified in this series showing a 0.8–1.6 µM MIC.
Table 3.
SAR of aniline ring (B ring)
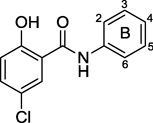 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | 2 | 3 | 4 | 5 | 6 | MIC (µM) | IC50 (µM) |
| 1 | H | CF3 | H | CF3 | H | 3.1 | 5.2 |
| 26 | H | H | H | H | H | 13–25 | 22 |
| 27 | H | CF3 | H | H | H | 3.1–6.3 | 5.9 |
| 28 | CF3 | H | H | H | H | 13–25 | 19 |
| 29 | H | H | CF3 | H | H | 1.6–3.1 | 2.8 |
| 30 | H | CF3 | CF3 | H | H | 0.8–1.6 | 4.9 |
| 31 | CF3 | CF3 | H | H | H | 6.3–13 | 8.1 |
| 32 | CF3 | H | H | CF3 | H | 3.1 | 6.6 |
| 33 | SO2Ph | H | H | H | H | >50 | >60 |
| 34 | H | F | H | H | H | 6.3–13 | 15 |
| 35 | H | Br | H | H | H | 6.3–13 | 5.7 |
| 36 | H | CN | H | H | H | 25–50 | 10 |
| 37 | H | CO2H | H | H | H | >50 | >60 |
| 38 | H | CO2Et | H | H | H | 13 | 17 |
| 39 | H | CONH2 | H | H | H | >50 | >60 |
| 40 | H | CONHPh | H | H | H | >50 | >60 |
| 41 | H | H | F | H | H | 13–25 | 17 |
| 42 | H | H | OCF3 | H | H | 3.1–6.3 | 7.3 |
| 43 | H | H | CN | H | H | 13–25 | 11 |
| 44 | H | H | OPh | H | H | 13 | 43 |
| 45 | H | H | t-Bu | H | H | 13–25 | 19 |
| 46 | H | H | OMe | H | H | >50 | >60 |
| 47 | H | H | 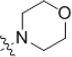 |
H | H | >50 | n.d. |
| 48 | H | H | CONH2 | H | H | >50 | >60 |
| 49 | H | H | CONHPh | H | H | >50 | >60 |
| 50 | H | H | SO2NHPh | H | H | >50 | >60 |
| 51 | H | H | 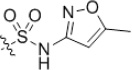 |
H | H | >50 | >60 |
| 52 | Me | CF3 | H | H | H | 3.1–6.3 | 8.0 |
| 53 | F | CF3 | H | H | H | 3.1–6.3 | 5.3 |
| 54 | F | F | H | H | H | 13 | 13 |
| 55 | CF3 | H | F | H | H | 6.3–13 | 13 |
| 56 | CF3 | H | Br | H | H | 6.3–13 | 11 |
| 57 | Cl | H | CF3 | H | H | 0.8–1.6 | 2.4 |
| 58 | F | H | F | H | H | 13 | 8.5 |
| 59 | Cl | H | H | CF3 | H | 1.6 | 3.5 |
| 60 | F | H | H | CF3 | H | 1.6–3.1 | 2.6 |
| 61 | Br | H | H | CF3 | H | 0.8–1.6 | 2.9 |
| 62 | CF3 | H | H | OMe | H | 6.3–13 | 4.8 |
| 63 | Cl | H | H | F | H | 3.1–6.3 | 3.2 |
| 64 | F | H | H | NO2 | H | 6.3 | 8.5 |
| 65 | CF3 | H | H | H | Cl | >50 | 23 |
| 66 | F | H | H | H | F | >50 | >60 |
| 67 | H | CF3 | F | H | H | 1.6–3.1 | 5.3 |
| 68 | H | CF3 | Cl | H | H | 1.6–3.1 | 4.5 |
| 69 | H | CF3 | Br | H | H | 3.1 | 4.5 |
| 70 | H | CF3 | CN | H | H | 6.3 | 5.8 |
| 71 | H | Cl | F | H | H | 3.1 | 4.4 |
| 72 | H | F | F | H | H | 3.1–6.3 | 7.7 |
| 73 | H | I | Me | H | H | 6.3 | 6.3 |
| 74 | H | Br | H | CF3 | H | 1.6 | 5.8 |
| 75 | H | CF3 | H | OMe | H | 6.3–13 | 18 |
| 76 | H | Cl | H | Cl | H | 1.6 | 3.1 |
| 77 | H | F | H | F | H | 3.1–6.3 | 6.1 |
| 78 | H | t-Bu | H | t-Bu | H | 6.3–13 | 4.1 |
| 79 | H | Me | H | Me | H | 50–>50 | 21 |
| 80 | H | CF3 | Me | CF3 | H | 0.8 | <1.9 |
| 81 | Me | CF3 | H | CF3 | H | 3.1 | 3.2 |
| 82 | H | F | Br | F | H | 1.6–3.1 | 4.3 |
| 83 | H | F | F | F | H | 1.6 | 4.7 |
| 84 | H | Cl | Cl | Cl | H | 1.6 | 5.4 |
| 85 | F | H | F | F | H | 3.1–6.3 | 6.2 |
| 86 | F | F | F | H | H | 6.3 | 6.7 |
| 87 | Cl | H | Cl | OMe | H | 13–25 | 8.0 |
| 88 | Cl | H | Cl | OH | H | >50 | 32 |
n.d.: not determined
Amongst mono-substitutions on the B ring, we explored only one further substitution in the ortho position, the phenyl sulfoxide 33, which completely lacked activity. The meta position was somewhat more tolerant with simple halogens (F, 34; Br, 35) retaining some activity (albeit 2–4 fold less than the parent 3,5-bis(trifluoromethyl)) 1 while the corresponding nitrile (36), acid (37), ethyl ester (38), unsubstituted (39) and monosubstituted (40) amides all lost activity. The para monosubstituted anilines also showed some interesting trends with p-CF3 > p-OCF3 > p-F (29, 42, and 41, respectively). Para-cyano (43), phenoxy (44) and t-butyl (45) all showed weak activity while methoxy (46), morpholino (42), carboxamido (48), phenylcarboxamido (49), and phenylsulfonamido (50) all lacked activity.
Amongst the 2,3-disubstituted anilines that were synthesized there was a slight preference against CF3 in the 2-position (compare 31 with either methyl (52) or fluoro (53) substituents). Replacing both the 2 and 3 positions with fluorine (54) resulted in a further loss of activity. The 2,4-disubstitution pattern yielded some highly potent compounds, including 2-chloro-4-trifluoromethyl (57) which showed an MIC of 0.8–1.6 µM. The 2,4-disubstitution pattern yielded some highly potent compounds, including 2-chloro-4-trifluoromethyl (57) which showed an MIC of 0.8–1.6 µM. Again there appeared to be a preference against CF3 in the ortho position with two 4-halo analogs carrying 2-CF3 groups showing poor activity (4-F, 55; and 4-Br, 56). Notably, the 2,4-difluoro analog (58) was the weakest in the 2,4-disubstituted series. The 2,5-disubstitution pattern also yielded some highly potent compounds, with 2-Br, 5-CF3 (61) showing submicromolar MIC. Interestingly, among the series of three 2-halo-5-CF3 substituted salicylanilides potency was inversely related to their electronegativity with Br (61) > Cl (59) > F (60). The preference at the 5-position for CF3 was not solely due to the electron withdrawing character of this moiety since the 5-nitro (64) derivative showed about the same weak activity as the methoxy derivative (62). In the 2,6-disubstitution pattern only two molecules were synthesized, 2-CF3-6-Cl (65) and 2,6-difluoro (66) both of which were completely inactive.
We synthesized four 3-CF3, 4-X disubstituted compounds which showed an activity trend of 3-CF3, 4-F (67) = 3-CF3, 4-Cl (68) > 3-CF3, 4-Br (69) > 3-CF3, 4-CN (70) although the differences in activity were only two-fold. Substitution of both groups with either di-fluoro (72) or 3-chloro-4-fluoro (71) resulted in a slight loss of activity as did replacement with iodine at the 3-position with substitution of a methyl group at the 4-position (73). Amongst the 3,5-disubstituted series various combinations of electron withdrawing groups (3-Br, 5-CF3 (74); 3,5-dichloro (76); 3,5-difluoro (77) were active in the range of 1.6–6.3 µM. Combining a 3-trifluoromethyl with a 5-methoxy group (75) resulted in a loss of activity (compare 75 to 1) while replacing both trifluoromethyls with either 3,5-dimethyl (79) or 3,5-di-t-butyl (78) either decreased (78) or eliminated (79) activity altogether.
We also prepared a number of trisubstituted B ring analogs related to both the 3,5-disubstituted series and the 2,4-disubstituted series. 3,4,5-trifluoro (83), 3,4,5-trichloro (84) and 3,5-difluoro-4-bromo (82) were all approximately the same potency as the corresponding 3,5-dihalo analogs. Introducing a methyl group in the ortho position of the 3,5-bis-CF3 parent (81) had no effect on potency, while introducing a methyl group in the para position (80) improved potency slightly, to less than 1 µM. The 2,4,5-trifluoro aniline derivative (85) had potency similar to the 3,4-difluoro compound 72, improved compared to the 2,4-difluoro analog 54. The 2,3,4-trifluoro analog (86), in contrast, was slightly improved compared to either the 2,3-difluoro 54 or the 2,4-difluoro 58. In the 2,4-dichloro aniline bearing series addition of a 5-hydroxy (88) resulted in complete loss of activity which was only slightly rescued by methylation (87).
We synthesized six additional compounds combining a 5-F salicylate ring with electron withdrawing character on the aniline ring (89–94 as shown in Table 4). While these compounds maintain fairly strong potency, none offers an improvement over the parent compound in cytotoxicity.
Table 4.
5-F-salicylic acid: aniline modifications
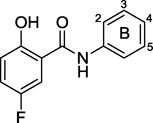 | ||||||
|---|---|---|---|---|---|---|
| Compd | 2 | 3 | 4 | 5 | MIC (µM) | IC50 (µM) |
| 89 | H | CF3 | H | Br | 3.1–6.3 | 4.7 |
| 90 | Cl | H | H | CF3 | 3.1–6.3 | 6.1 |
| 91 | H | F | H | F | 6.3 | 9.6 |
| 92 | H | H | CF3 | H | 3.1–6.3 | 5.4 |
| 93 | F | H | H | CF3 | 6.3 | 5.1 |
| 94 | H | CF3 | H | H | 6.3–13 | 6.1 |
2.4 Summary of cytotoxicity data
As mentioned above, compounds were tested for cytotoxicity in a murine macrophage cell line (J774.1) as a prelude to treatment studies in this model (Tables 1–4). These compounds displayed high cytotoxicity, with IC50 values often in the low micromolar range. Overall, the data showed that the potency in Mtb (MIC) was modestly positively correlated to the cytotoxicity (IC50) in J774.1 cells (Figure 1). This suggests that there may be a common mechanism of toxicity in both a eukaryotic cell line and Mtb. A notable exception is compound 14, in which an acetyl group protects the salicylate 2-OH. This suggests that the bacteria cleave this acetyl group while J774.1 cells cannot.
Figure 1.

Correlation between Mtb MIC and IC50 in J774.1 macrophage cell line. Compounds with off-scale MIC (>50 µM) or IC50 (>60 µM) values were not included.
2.5 Possible mechanism
Despite repeated attempts to generate spontaneous mutants of Mtb by selecting on media containing these compounds we were never able to identify colonies with significant levels of resistance. This result suggests a complex mode of action without a specific receptor target. The inability to generate salicylanilides specific for Mtb over J774.1 cells also suggested that there may be a complex mechanism of action. A possible mechanism consistent with the absolute requirement for a free phenolic hydroxyl is that the salicylanilides are decoupling the proton gradient in both Mtb and the mitochondria of J774.1 cells by acting as a proton shuttle. Indeed, salicylanilides have previously been suggested to destroy the proton gradient in mitochondria.13 The determinants for strongly depolarizing salicylanilides parallel the SAR observed with salicylanilides against Mtb: electron withdrawing groups on the B ring and somewhat lipophilic groups on the A ring,14 although the trend of lipophilicity has been previously shown to be less relevant to the SAR of Mtb inhibitors.15 The salicyl phenol proton has a pKa near physiological range. In the slightly basic interior of the Mtb cell, the phenol could become deprotonated (Figure 2). Stabilization of the phenolate by a hydrogen bond to the amide NH would allow the compound to cross the membrane to the extracellular space, assisted by the positive electrochemical gradient. There, in the lower pH of the media, the phenolate would become protonated and the protonated form could transit the membrane and deposit the acquired proton within the cell, thereby disrupting the proton gradient.
Figure 2.

Proton shuttling mechanism for salicylanilides in killing Mtb. Through repeated cycles, the salicylanilide disrupts the proton gradient. pH values from Vandal et al.16
The electron poor B ring of the salicylanilides appears to stabilize this process by modulating the pKa of the amide proton. This explanation is further supported by the disruption of membrane potential and pH homeostasis recently observed in Mtb using the salicylanilide niclosamide.17 Support for the existence of these two conformers of salicylanilides has been previously published from NOESY NMR, IR and fluorescence spectral changes of these compounds as a function of acid concentration18.
Disruption of the proton gradient may be a common mechanism of salicylanilide action against diverse cells even if the salicylanilides also targeted specific enzymes. Extensive SAR experiments on both the enzymatic target, in vitro, and whole-cell organism would be required to establish unique modes of action (as was elegantly shown in Garner et al.).19 In the case of Mtb, the shared SAR between mammalian cytotoxicity values and Mtb MIC values that parallels known SAR for strong proton gradient decouplers argues for disruption of the proton gradient. Additional experiments to confirm this mechanism by measuring the proton motive force and examining transcriptional responses are underway.
3. Conclusion
We have synthesized a series of salicylanilides based on a bis(trifluoromethyl) salicylanilide hit from a high-throughput screen. The electron withdrawing character of substituents on the B ring appears to be the major driver of activity, with many compact electron-withdrawing groups tolerated. Despite synthesizing a rather large number of molecules we have been unable to separate MIC activity from cytotoxicity in a macrophage cell line, suggesting a common mode of action. We propose that this mechanism involves the decoupling of the proton motive force by proton shuttling mediated by the salicylanilides, consistent with the observed SAR. This general mechanism likely explains the cytotoxicity of various salicylanilides against other microbes and suggests that further development may be challenging.
4. Materials and methods
Parent salicylanilide 1 was identified from high-throughput screening of the NIH Chemical Genomics Center (NCGC) library using Mtb (H37Rv) expressing green fluorescent protein (GFP, pMSP12 plasmid), using GFP fluorescence as readout.
All chemicals used were of analytical grade and commercially available. All solvents were dried and freshly distilled prior to use according to standard procedures. Solvents are reported as a volume ratio. 1H NMR spectra were recorded on a Bruker spectrometer (300 MHz) in d6-DMSO or CDCl3 as specified below. Melting points were performed on a Mettler Toledo MP50 melting point apparatus. IR spectra were recorded on a Smiths Detection IdentifyIR FT-IR spectrometer(ATR). HRMS was obtained by APCI accurate mass on a Waters LCT Premier time-of-flight mass spectrometer (negative mode).
General procedures
Procedure A
The salicylic acid (1.2 equiv) was added to a mixture of toluene (0.3 M), aniline (1.0 equiv), phosphorus trichloride (1.1 equiv), and pyridine (0.05 equiv) in a Radley’s Carousel reaction tube (modified from Itai et al.20). The mixture was refluxed under nitrogen for 12 h then cooled to room temperature. Aqueous sodium bicarbonate was added dropwise to attain pH 6–7. The resultant mixture was extracted with EtOAc. The organic extracts were combined, dried (MgSO4), and concentrated under vacuum. After chromatography (1:10 EtOAc:Hex) compounds were recrystallized (EtOAc/Hex).
Procedure B
5-chlorosalicylic acid (20 g, 116 mmol, 1.0 equiv), pyridine (25 mL, 310 mmol, 3.0 equiv), and benzoyl chloride (25 mL, 311 mmol, 3.0 equiv) in Et2O (0.5 M) were combined and stirred at room temperature (5 h). 1 N HCl was added dropwise until the reaction reached pH 2–3, followed by extraction with EtOAc. The organic extracts were combined, dried (MgSO4), and concentrated under vacuum. Chromatography (1:5 EtOAc:Hex) yielded 2-(benzoyloxy)-5-chlorobenzoic acid (18 g, 53%). 1H NMR (300 MHz, d6-DMSO) δ 7.44 (d, J = 8.6 Hz, 1H), 7.58–7.63 (m, 2H), 7.72–7.79 (m, 2H), 7.94 (d, J = 8.6 Hz, 1H), 8.11 (m, 2H).
Oxalyl chloride (3.2 mL, 36 mmol, 2.0 equiv) at 0 °C, 2-(benzoyloxy)-5-chlorobenzoic acid (5 g, 18 mmol, 1 equiv), and DMF (2 drops) were combined in dichloromethane (0.3 M). The mixture warmed to room temperature with stirring (4 h). The solvent was removed under reduced pressure, yielding 4-chloro-2-(chlorocarbonyl)phenyl benzoate as a thick yellow oil (17 g), which was used without further purification. 1H NMR (300 MHz, CDCl3): δ 7.21 (d, J = 8.6 Hz, 1H), 7.46–7.51 (m, 2H), 7.59–7.64 (m, 2H), 8.07–8.09 (m, 1H), 8.16 (d, J = 8.1 Hz, 2H).
To a solution of 4-chloro-2-(chlorocarbonyl)phenyl benzoate (1 mmol, 1.4 equiv) in DCM (0.2 M), amine (0.7 mmol, 1 equiv) and triethylamine (0.6 mL, 1.2 mmol, 1.7 equiv) were added at 0 °C. The mixture was stirred at room temperature (16 h) and extracted with DCM. The organic layer was washed with water and brine, dried (MgSO4), and the solvent was removed under vacuum. The residue was purified by flash column chromatography.
Potassium carbonate (18 mg, 0.13 mmol, 1.2 equiv) was added to a solution of crude product from the previous step (0.11 mmol, 1.0 equiv) in 1:1 MeOH:1,4-dioxane (0.1 M). The reaction mixture was stirred at room temperature (2 h) and acidified with 1 N HCl, followed by extraction with EtOAc and DCM. The organic layer was washed with water and brine, dried (MgSO4), and the solvent was removed under vacuum. The residue was purified by column chromatography (1:19 MeOH:CHCl3), and recrystallized from Et2O/Hex.
Compound purity
Compound purity was assessed by LC/MS comparison of the total ion current with the peak representing the desired product on reversed-phase chromatography as well as by the presence of extraneous peaks in the 1H-NMR. All compounds submitted for biological testing were >90% pure.
N-(3,5-Bis(trifluoromethyl)phenyl)-5-chloro-2-hydroxy-benzamine (1)20
Prepared by general procedure A. White solid (320mg, 28%). mp 172–173 °C. IR (neat): 3082, 1639, 1580, 1379, 1276, 1137, 888 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)-2-hydroxybenzamide (2)20
Prepared by general procedure A. White solid, mp 181–182 °C [lit20: mp 179–180 °C]. IR (neat): 3322, 3103, 1623, 1568, 1381, 1272, 1155, 1125, 879 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)-3-chlorobenzamide (3)20
Prepared by general procedure A. White solid. mp 189–190 °C. Yield 87%. 1H NMR (300 MHz, d6-DMSO): δ 7.61 (dd, J1 = 8.1 Hz, J2 = 8.0 Hz, 1H), 7.72 (d, J = 7.7 Hz, 1H), 7.84 (s, 1H), 7.95 (d, J = 7.8 Hz, 1H), 8.06 (s, 1H), 8.50 (s, 2H), 10.90 (s, 1H). HRMS (APCI), m/z calcd for C15H8ClF6NO-H: 366.0120, found 366.0112.
N-(3,5-Bis(trifluoromethyl)phenyl)-benzamide (4)
Prepared by general procedure A. White solid. mp 186–187 °C. Yield 39%. 1H NMR (300 MHz, d6-DMSO): δ 7.59–7.66 (m, 3H), 7.83 (s, 1H), 8.01 (d, J = 6.9 Hz, 2H), 8.54 (s, 2H), 10.85 (s, 1H). IR (neat): 3205, 1653, 1564, 1381, 1274, 1118, 879 cm−1. HRMS (APCI), m/z calcd for C15H9F6NO-H: 332.0510, found 332.0504.
N-(3,5-Bis(trifluoromethyl)phenyl)-2-hydroxy-5-(1H-pyrrol-1-yl)benzamide (5) 20
Prepared by general procedure A. Brown solid. mp >230 °C dec. IR (neat): 3231,1637, 1552, 1520, 1379, 1274, 1128, 742 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)-2-hydroxy-5-methylbenzamide (6)20
Prepared by general procedure A. White solid, mp 145–147 °C. Yield 22%. 1H NMR (300 MHz, d6-DMSO): δ 2.28 (s, 3H), 6.90 (d, J = 5.0 Hz, 1H), 7.27 (dd, J1 = 8.3 Hz, J2 = 2.1 Hz, 1H), 7.69 (d, J = 1.7 Hz, 1H), 7.83 (s, 1H), 8.46 (s, 2H), 10.81 (s, 1H), 10.86 (s, br, 1H). IR (neat): 3290, 1655, 1571, 1383, 1274, 1114, 881 cm−1. HRMS (APCI), m/z calcd for C16H11F6NO2-H: 362.0616, found 362.0621.
N-(3,5-Bis(trifluoromethyl)phenyl)-5-fluoro-2-hydroxy-benzamide (7)20
Prepared by general procedure A. White solid, mp 187–178 °C. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)-5-bromo-2-hydroxybenzamide (8)20
Prepared by general procedure A. Slightly brown solid, mp 194–195 °C. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)-2-hydroxy-5-iodobenzamide (9)20
Prepared by general procedure A. White solid. mp 216–219 °C. IR (neat): 3318, 3194, 1639, 1552, 1381, 1172, 1128, 881, 810 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)-2-hydroxy-5-methoxybenz-amide (10) 20
Prepared by general procedure A. White solid, mp 208–210 °C. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)3,5-dichloro-2-hydroxybenz-amide (11)24
Prepared by general procedure A. White solid. mp 141–143 °C. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis-trifluoromethyl-phenyl)-2,3-dihydroxy-benzamide (12)24
Prepared by general procedure A. White solid. mp 154–156 °C. IR (neat): 3507, 3343, 3137, 1657, 1272, 1141, 1073, 885 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis-trifluoromethyl-phenyl)-2-hydroxy-5-nitro-benzamide (13)20
Prepared by general procedure A. Slightly yellow solid, mp 225–226 °C. IR (neat): 3388, 3338, 3096, 1568, 1470, 1340, 1276, 1164, 1118, 1075 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
Acetic acid 2-(3,5-bis-trifluoromethyl-phenylcarbamoyl)-4-chloro-phenyl ester (14)20
Prepared by general procedure A. White solid. mp 117–118 °C. Characterization (NMR, MS) was in accordance with previously reported values.
N-(3,5-Bis(trifluoromethyl)phenyl)-5-chloro-2-methoxy-benzamide (15)27
Prepared by general procedure A. White solid. mp 148–150 °C. Characterization (NMR, MS) was in accordance with previously reported values.
2-Benzyloxy-N-(3,5-bis-trifluoromethyl-phenyl)-5-chlorobenzamide (16)
To a solution of compound 1 (192 mg, 0.5 mmol, 1.0 equiv) in DMF (1.5 mL, 0.3 M), benzyl bromide (0.07 mL, 0.55 mmol, 1.1 equiv) and potassium carbonate (83 mg, 0.6 mmol, 1.2 equiv) were added. The solution was stirred at room temperature (4 h). The mixture was concentrated under reduced pressure and the residue was dissolved in EtOAc (20 mL) and then washed with brine. The organic layer was dried (MgSO4) and concentrated under vacuum. The crude product was purified by column chromatography (1:15 EtOAc:Hex) to give product 16 (220 mg, 93%). White solid. mp 178–180 °C. 1H NMR (300 MHz, d6-DMSO): δ 5.23 (s, 2H), 7.30–7.34 (m, 4H), 7.47–7.48 (m, 2H), 7.60 (dd, J1 = 11.6 Hz, J2 = 2.7 Hz, 1H), 7.68(d, J = 2.7 Hz, 1H), 7.80 (s, 1H), 8.26 (s, 1H), 10.84 (s, 1H). IR(neat): 3318, 2925, 1669, 1561, 1472, 1381, 1269, 1166, 1121 cm−1. HRMS (APCI), m/z calcd for C22H14ClF6NO2-H: 472.0539, found 472.0542.
2-Amino-N-(3,5-bis(trifluoromethyl)phenyl)5-chlorobenzamide (17)
2-amino-5-chlorobenzoic acid (1 g, 6 mmol, 1.0 equiv) and DMF (2 drops) were added to thionyl chloride (4.4 mL, 60 mmol, 10 equiv) in 1,2-dichloroethane (20 mL, 0.3 M) at 0 °C. The mixture was stirred at room temperature (3 h). Thionyl chloride and 1,2-DCE were removed by distillation. The product was resuspended in DCM (20 mL) and cooled to 0 °C. To this solution was added 3,5-bis-(trifluoromethyl)aniline (7.6 mL, 48 mmol, 8.0 equiv) and triethylamine (8.4 mL, 60 mmol, 10 equiv), followed by warming to room temperature and stirring for 16 h. The mixture was stirred at room temperature (16 h) and diluted with DCM. The organic layer was washed with water and brine, dried (MgSO4), and concentrated under vacuum. The residue was purified by flash column chromatography (1:10 EtOAc:Hex) to give product 17 (32 mg, 1.4 %). White solid. mp 69–71 °C. 1H NMR (300 MHz, d6-DMSO): δ 6.61 (s, 2H), 6.80 (d, J = 9.0 Hz, 1H), 7.27 (dd, J1 = 9.0 Hz, J2 = 2.4 Hz, 1H), 7.77 (d, J = 2.4 Hz, 1H), 7.79 (s, 1H), 8.45 (s, 2H), 10.76 (s, 1H). HRMS (APCI), m/z calcd for C15H9ClF6N2O-H: 381.0229, found 381.0235.
N-(3,5-Bis(trifluoromethyl)phenyl)2-chlorobenzamide (18)
Prepared by general procedure A, except the reaction was performed in acetonitrile (0.3 M) and pyridine was omitted. White solid. mp 142–143 °C. Yield 77%. 1H NMR (300 MHz, d6-DMSO): δ 7.50–7.70 (m, 4H), 7.87 (s, 1H), 8.40 (s, 2H), 11.20 (s, 1H). IR (neat): 3251, 3087, 1660, 1379, 1276, 11171, 1128, 883 cm−1. HRMS (APCI), m/z calcd for C15H8ClF6NO-H: 366.0120, found 366.0120.
2-[(3,5-Bis-trifluoromethyl-phenylamino)-methyl]-4-chlorophenol (19)
A mixture of 5-chlorosalicylaldehyde (2.5 g, 16 mmol, 1.0 equiv) and 3,5-(trifluoromethyl)aniline (2.5 mL, 16 mmol, 1.0 equiv) in EtOH (80 mL, 0.2 M) was refluxed (16 h). The reaction mixture was cooled to room temperature and the separated crystal was filtered to yield (E)-2-((3,5-bis(trifluoromethyl)phenyl)imino)-methyl)-4-chlorophenol (3.3 g, 56%). This compound (1 g, 2.8 mmol, 1.0 equiv) was dissolved in EtOH (3 mL, 0.9 M). Sodium borohydride (117 mg, 3.1 mmol, 1.1 equiv) was added at 0 °C and the reaction mixture was allowed to warm to room temperature (3 h). After complete reduction of the imine, the pH was adjusted to 8 using 1 N HCl and the reaction mixture was extracted with Et2O. The organic layer was washed with brine, dried (MgSO4), and the product was purified by column chromatography (1:4 EtOAc:Hex) to give product 22 (490 mg, 49%). White solid. mp 120–123 °C. 1H NMR (300 MHz, d6-DMSO): δ 4.26 (d, J = 5.7 Hz, 2H), 6.85 (d, J = 8.6 Hz, 1H), 7.05 (s, 1H), 7.09–7.12 (m, 4H), 7.21 (d, J = 2.6 Hz, 1H), 10.06 (s, 1H). IR (neat): 3292, 1383, 1281, 1125, 1061, 879, 819 cm−1. HRMS (APCI), m/z calcd for C15H10ClF6NO-H: 368.0277, found 368.0277. An alternate synthesis was additionally described previously.(2)
1-(3,5-Bis(trifluoromethyl)phenyl)-3-(5-chloro-2-hydroxyphenyl)-urea (20)24
Prepared by general procedure A. Slightly yellow brown. 192–193 °C. IR (neat): 3361, 1657, 1557, 1381, 1276, 1134, 890 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-2-hydroxy-benzoic acid 3,5-bis-trifluoromethyl-phenyl ester (21)24
Prepared by general procedure A. White solid. mp 98–100 °C. IR (neat): 3258, 1694, 1470, 1276, 1171, 1107, 908 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
N-(5-Chloro-2-hydroxyphenyl)-3,5-bis(trifluoromethyl)benzamide (22)
2-amino-4-chlorophenol (290 mg, 2 mmol, 1.0 equiv) was dissolved in dichloromethane (8 mL, 0.3 M) under argon. A solution of 3,5-bis(trifluoromethyl)benzoyl chloride (0.36 mL, 2 mmol, 1.0 equiv) and pyridine (0.9 mL, 10 mmol, 5 equiv) in DCM (4 mL, 0.5 M) was added dropwise on ice, and the mixture was allowed to warm to room temperature over 3 h. The resultant mixture was concentrated under vacuum and resuspended in EtOAc. This solution was washed successively with water and brine, dried (MgSO4), and concentrated under vacuum. The resultant residue was purified by column chromatography (1:10 EtOAc:Hex) to give product 19 (300 mg, 39%). Red solid. mp 218–221 °C. 1H NMR (300 MHz, d6-DMSO) δ 6.93 (d, J = 8.7 Hz, 1H), 7.12 (d, J = 8.6Hz, 1H), 7.66 (s, 1H), 8.37 (s, 1H), 8.58 (s, 2H), 10.15 (s, 2H). HRMS (APCI), m/z calcd for C15H8ClF6NO2-H: 382.0070, found 382.0056.
N-(3,5-bis(trifluoromethyl)phenyl)-5-chloro-2-hydroxybenzothioamide (23)
Compound 1 (1g, 2.6 mmol, 2.0 eq) was dissolved in pyridine (8 mL, 0.2 M). Phosphorus pentasulfide (591 mg, 1.3 mmol, 1 eq) was added, and the mixture was heated to reflux (3 h). The mixture was concentrated under vacuum and mixed with dilute HCl and toluene. The resulting mixture was refluxed (3 h). The mixture was concentrated under vacuum. Water was added to the residue, and it was extracted with EtOAc. The extract was purified by column chromatography (1:10 EtOAc:Hex) to give product 25 (138 mg, 10%). yellow solid. mp 68–70 °C. 1H NMR (300 MHz, d6-DMSO): δ 6.96 (d, J = 8.8 Hz, 1H), 7.34 (dd, J1 = 8.7 Hz, J2 = 2.7 Hz, 1H), 7.57 (d, J = 2.7 Hz, 1H), 7.99 (s, 1H), 8.73 (s, 2H), 10.56 (s, 1H), 12.40 (s, 1H). HRMS (APCI), m/z calcd for C15H8ClF6NOS-H: 397.9841, found 397.9836.
N-(3,5-Bis(trifluoromethyl)phenyl)-5-chloro-2-hydroxybenzenesulfonamide (24)
Freshly distilled chlorosulfonic acid (13.2 g, 0.1 mol, 1.0 equiv) was cooled to 15 °C in an ice-water bath and 4-chloroanisole (27 mL, 0.2 mol, 2.0 equiv) was carefully added, keeping the temperature of the mixture below 50 °C. The reaction mixture was carefully poured into 250 mL ice water. The resultant white solid (5-chloro-2-methoxybenzene-1-sulfonyl chloride) was filtered and dried.
3,5-Bis(trifluoromethyl)aniline (2.3 g, 10 mmol, 1.0 equiv) and 4-dimethylaminopyridine (50 mg, 0.4 mmol, 0.04 equiv) were dissolved in anhydrous pyridine (20 mL, 0.5 M). 5-chloro-2-methoxybenzene-1-sulfonyl chloride (2.41 g, 10 mmol, 1 equiv) was added. The resulting mixture was stirred at room temperature (10 h). Water (20 mL) was added to quench the reaction. The precipitate formed was filtered and washed with water, cold THF, and Et2O to afford a brown solid (N-(3,5-bis(trifluoromethyl)phenyl)-5-chloro-2-methoxybenzenesulfonamide). 1H NMR (300 MHz, d6-DMSO): δ 3.79 (s, 3H), 7.23 (d, J = 9.0 Hz, 1H), 7.66–7.68 (m, 3H), 7.75 (s, 1H), 7.81 (d, J = 2.6 Hz, 1H), 11.12(s, 1H).
Boron tribromide (5 mL, 1.7 mmol, 1.7 equiv) was added to a solution of N-(3,5-bis(trifluoromethyl)phenyl)-5-chloro-2-methoxybenzenesulfonamide (434 mg, 1mmol, 1.0 equiv) in DCM (5 mL, 0.2 M) at −70 °C and allowed to warm to room temperature (12 h). The mixture was added dropwise to 10 mL ice water, with stirring continued for an additional 0.5 h. The mixture was cooled to room temperature and concentrated sodium hydroxide was added dropwise until the pH reached 6–7, followed by extraction with DCM. The organic extracts were combined, dried (MgSO4), concentrated under vacuum, and purified by chromatography (1:2 EtOAc:Hex) (340 mg, 81%). Slightly yellow solid. mp 135–138 °C. 1H NMR (300 MHz, d6-DMSO): δ 6.95 (d, J = 8.8 Hz, 1H), 7.49 (dd, J1 = 8.8 Hz, J2 = 2.3 Hz, 1H), 7.68–7.71 (m, 4H), 11.27 (s, 2H). IR(neat): 3354, 3238, 1413, 1374, 1276, 1125, 966, 826 cm−1. HRMS (APCI), m/z calcd for C14H8ClF6NO3S-H: 417.9766, found 417.9753.
N-(3,5-Bis-trifluoromethyl-benzyl)-5-chloro-2-hydroxy-benzamide (25)
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride was added to a mixture of 5-chlorosalicylic acid (690 mg, 4 mmol, 1.0 equiv), 3,5-bis(trifluoromethyl)benzylamine (973 mg, 4 mmol, 1.0 equiv), 4-dimethylaminopyridine (50 mg, 0.4 mmol, 0.1 equiv) and DCM (12 mL, 0.3 M), and the mixture was stirred at room temperature (3 h). The reaction mixture was poured into dilute HCl (50 mL) and extracted with DCM (70 mL). After the organic layer was washed with water and brine, dried (MgSO4), and concentrated under vacuum, the crude product was purified by column chromatography (1:4 EtOAc:Hex) to give product 23 (245 mg, 55%). White solid. mp 125–127 °C. 1H NMR (300 MHz, d6-DMSO): δ 4.68 (d, J = 5.8 Hz, 2H), 6.97 (d, J = 8.8 Hz, 1H), 7.44 (dd, J1 = 8.8, J2 = 2.6 Hz, 1H), 7.89 (d, J = 2.7 Hz, 1H), 8.01 (s, 1H), 8.05(s, 2H), 9.41(dd, J1 = 5.9 Hz, J2 = 5.9 Hz, 1H), 12.09 (s, 1H). HRMS (APCI), m/z calcd for C16H10ClF6NO2: 396.0226, found 396.0229.
5-Chloro-2-hydroxy-N-phenylbenzamide (26)
Prepared by general procedure A.23 Slightly yellow solid. mp 211–212 °C. Yield 45%. 1H NMR (300 MHz, d6-DMSO): δ 7.00 (d, J = 8.8 Hz, 1H), 7.15 (dd, J1 = 7.2 Hz, J2 = 7.2 Hz, 1H), 7.34–7.39 (m, 2H), 7.46 (dd, J1 = 8.8 Hz, J2 = 2.7 Hz, 1H), 7.69 (d, J = 8.3 Hz, 2H), 7.96 (d, J = 2.6 Hz, 1H), 10.40 (s, 1H), 11.84 (s, 1H). HRMS (APCI), m/z calcd for C13H10ClNO2-H: 246.0322, found 246.0329.
5-Chloro-N-(3-(trifluoromethyl)phenyl)-2-hydroxybenzamide (27)
Prepared by general procedure A.25 White solid, mp 181–182 °C. Yield 51%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (d, J = 8.8 Hz, 1H), 7.45–7.50 (m, 2H), 7.61 (dd, J1 = 8.0 Hz, J2 = 8.0 Hz, 1H), 7.89 (d, J = 2.6 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 8.20 (s, 1H), 10.63 (s, 1H), 11.57 (s, br, 1H). HRMS (APCI), m/z calcd for C14H9ClF3NO2-H: 314.0196, found 314.0202.
5-Chloro-2-hydroxy-N-(2-trifluoromethyl-phenyl)-benzamide (28)20
Prepared by general procedure A. White solid. mp 155–157 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(4-(trifluoromethyl)phenyl)-2-hydroxybenzamide (29)28
Prepared by general procedure A. White solid, mp 222–223 °C. Characterization (NMR, MS) was in accordance with previously reported values.
Aniline substrates for 30 and 31
1,2-Bis(trifluoromethyl)benzene (3.47 mL, 23 mmol, 1 equiv) was added slowly to a mixture of fuming nitric acid (18.3 mL, 440 mmol, 19 equiv) and concentrated sulfuric acid (24.5 mL, 460 mmol, 20 equiv) and the mixture was heated to 100 °C for 4 h. The product was poured onto ice and the aqueous phase was extracted with ether. The organic phase was washed successively with water, saturated sodium carbonate, brine, and then dried (MgSO4). The extract was concentrated under vacuum, yielding a crude oil (4.5 g, 74% combined yield).
The oil containing 2,3-bis(trifluoromethyl)nitrobenzene and 3,4-bis(trifluoromethyl)nitrobenzene) (4 g, 15 mmol) was dissolved in MeOH (160 mL, 0.1 M). 10 wt % Pd/C (220 mg) was added, followed by stirring under hydrogen atmosphere (12 h). The resultant mixture was filtered over Celite and concentrated under vacuum. The obtained residue was purified by column chromatography (1:10 EtOAc:Hex) to isolate the anilines used in the synthesis of compounds 30 (900 mg, 17%, 1H NMR (300 MHz, CDCl3): δ 4.13 (s, 2H), 6.80 (dd, J1 = 8.6 Hz, J2 = 1.9 Hz, 1H), 7.03 (d, J = 2.3 Hz, 1H), 7.58 (d, J = 8.6 Hz, 1H),) and 31 (450 mg, 8%, 1H NMR (300 MHz, CDCl3): δ 4.47 (s, 2H), 6.91 (d, J = 8.3 Hz, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.33 (br t, J = 8.0 Hz, 1H),).
N-(3,4-Bis(trifluoromethyl)phenyl)-5-chloro-2-hydroxybenzamide (30)
Prepared by general procedure A. White solid, mp 215–217 °C. Yield 5%. 1H NMR (300 MHz, CDCl3): δ 6.95 (d, J = 8.8 Hz, 1H), 7.38 (dd, J1 = 8.8 Hz, J2 = 2.3 Hz, 1H), 7.84 (d, J = 8.6 Hz, 1H), 7.97 (d, J = 2.3 Hz, 1H), 8.14–8.16 (m, 2H). HRMS (APCI), m/z calcd for C15H8ClF6NO2-H: 382.0070, found 382.0082.
N-(2,3-Bis(trifluoromethyl)phenyl)-5-chloro-2-hydroxybenzamide (31)
Prepared by general procedure A. White solid. mp 163–164 °C. Yield 7%. 1H NMR (300 MHz, CDCl3): δ 7.00–7.04 (m, 1H), 7.43–7.47 (m, 2H), 7.73–7.80 (m, 2H), 8.20 (d, J = 7.8 Hz, 1H), 8.29 (s, 1H), 11.41 (s, 1H). HRMS (APCI), m/z calcd for C15H8ClF6NO2-H: 382.0070, found 382.0074.
N-(2,5-Bis(trifluoromethyl)phenyl)-5-chloro-2-hydroxybenzamine (32)20
Prepared by general procedure A. White solid. mp 202–203 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-2-hydroxy-N-((2-phenylsulfonyl)phenyl)benzamide (33)
Prepared by general procedure A. White solid. mp 154–156 °C. Yield 24%. 1H NMR (300 MHz, CDCl3): δ 7.00 (d, J = 8.9 Hz, 1H), 7.32–7.82 (m, 9H), 8.08(d, J = 9.0 Hz, 1H), 8.43(d, J = 8.1 Hz, 1H), 10.62(s, 1H), 11.81 (s, 1H). IR(neat):3340, 1582, 1534, 1331, 1276, 1132, 826 cm−1. HRMS (APCI), m/z calcd for C19H14ClNO4S-H: 386.0254, found 386.0259.
5-Chloro-N-(3-fluorophenyl)-2-hydroxybenzamide (34)
Prepared by general procedure A. White solid. mp >281 °C dec. Yield 38%. 1H NMR (300 MHz, d6-DMSO): δ 6.92–6.98 (m, 2H), 7.35–7.45 (m, 3H), 7.73 (d, J = 5.7 Hz, 1H), 7.85 (s, 1H), 11.06 (s, 1H). HRMS (APCI), m/z calcd for C13H9ClFNO2-H: 264.0228, found 264.0229.
N-(3-Bromophenyl)-5-chloro-2-hydroxybenzamide (35)
Prepared by general procedure A. Yield 10%. White solid. mp 231–232 °C. 1H NMR (300 MHz, d6-DMSO): δ 7.00 (d, J = 8.8 Hz, 1H), 7.33 (d, J = 5.2 Hz, 2H), 7.46 (dd, J1 = 8.8 Hz, J2 = 2.6 Hz, 1H), 7.63–7.66 (m, 1H), 7.88 (d, J = 2.6 Hz, 1H), 8.05 (s, 1H), 10.50 (s, 1H), 11.57 (s, 1H). HRMS (APCI), m/z calcd for C13H9BrClNO2-H: 323.9427, found 323.9433.
5-Chloro-N-(3-cyanophenyl)-2-hydroxybenzamide (36)
Prepared by general procedure A.26 White solid. mp 240–241 °C. Yield 21%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (d, J = 8.8 Hz, 1H), 7.47 (dd, J1 = 8.8 Hz, J2 = 2.6 Hz, 1H), 7.55–7.61 (m, 2H), 7.86 (d, J = 2.6 Hz, 1H), 7.95–7.99 (m, 1H), 8.20 (s, 1H), 10.62 (s, 1H), 11.56 (s, 1H). IR(neat): 3073, 2231, 1632, 1616, 1564, 1438, 1224, 815, 787 cm−1. HRMS (APCI), m/z calcd for C14H9ClN2O2-H: 271.0274, found 271.0283.
3-(5-Chloro-2-hydroxybenzamido)benzoic acid (37)
Sodium hydroxide (130 mg, 3.3 mmol, 5 equiv) in water (1 mL, 3 M) was added to compound 38 (208 mg, 0.65 mmol, 1 equiv) in water (1.5 mL, 0.4 M), and the mixture was stirred at room temperature (5 h). The reaction mixture was diluted with water (10 mL) and washed with EtOAc (20 mL). The water layer was acidified by addition of dilute HCl, and the product was extracted with EtOAc. The EtOAc layer was washed successively with water and brine, then dried (Na2SO4). The resultant solution was concentrated under vacuum to give a residue which was purified by chromatography (MeOH:CHCl3 1:5) to give product 37 (170 mg, 91%). White solid. mp >341 °C dec. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.8 Hz, 1H), 7.46–7.53 (m, 2H), 7.72 (d, J = 7.7 Hz, 1H), 7.92–7.97 (m, 2H), 8.36 (s, 1H), 10.57(s, 1H), 11.91(s, br, 1H), 12.94(s, br, 1H). IR(neat): 3069, 1694, 1591, 1568, 1454, 1294, 1221, 817 ,748 cm−1. HRMS (APCI), m/z calcd for C14H10ClNO4-H: 290.0220, found 290.0216.
Ethyl 3-(5-chloro-2-hydroxybenzamido)benzoate (38)
Prepared by general procedure A. White solid. mp 154–156 °C. Yield 52%. 1H NMR (300 MHz, d6-DMSO): δ 1.31–1.37 (m, 3H), 4.30–4.41 (m, 2H), 7.02 (d, J = 8.8 Hz, 1H), 7.46–7.52 (m, 2H), 7.73 (d, J = 7.8 Hz, 1H), 7.94–7.97 (m, 2H), 8.37 (s, 1H), 10.61 (s, 1H), 11.70 (s, br, 1H). IR(neat): 3322, 3082, 1719, 1561, 1285, 1223, 817, 746 cm−1. HRMS (APCI), m/z calcd for C16H14ClNO4-H: 318.0533, found 318.0532.
N-(3-Carbamoyl-phenyl)-5-chloro-2-hydroxy-benzamide (39)28
Prepared by general procedure A. White solid. mp >272 °C dec. IR(neat): 3359, 3180, 1646, 1561, 1406, 1223, 1119, 828 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-2-hydroxy-N-(3-(phenylcarbamoyl)phenyl)-benzamide (40)
Prepared by general procedure B using 3-amino-N-phenylbenzamide, and the final deprotection step was modified as follows: potassium carbonate (140 mg, 1.02 mmol, 1.2 equiv) was added to a solution of crude product (400 mg, 0.85 mmol, 1 equiv) from the previous step in 1:1 MeOH:1,4-dioxane (8 mL, 0.1 M). The reaction mixture was stirred at room temperature (3 h) and acidified with 1 N HCl, followed by extraction with EtOAc. The organic layer was washed with water and brine, dried (MgSO4), and the solvent was removed under vacuum and recrystallized from Et2O (200 mg, 64%). White solid. mp 269–270 °C. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.7 Hz, 1H), 7.11 (dd, J1 = 7.5 Hz, J2 = 7.2 Hz, 1H), 7.36 (dd, J1 = 7.8 Hz, J2 = 7.8 Hz, 2H), 7.46–7.56 (m, 2H), 7.72–7.79 (m, 3H), 7.94–7.98 (m, 2H), 8.22 (s, 1H), 10.30 (s, 1H), 10.62 (s, 1H). IR(neat): 3281, 1646, 1607, 1525, 1424, 1324, 1221 cm−1. HRMS (APCI), m/z calcd for C20H15ClN2O3-H: 365.0693, found 365.0685.
5-Chloro-N-(4-fluorophenyl)-2-hydroxybenzamide (41)
Prepared by general procedure A.23 White solid. mp 239–240 °C. Yield 8%. 1H NMR (300 MHz, d6-DMSO): δ 7.01 (d, J = 8.8 Hz, 1H), 7.18–7.24 (m, 2H), 7.46 (dd, J1 = 8.8 Hz, J2 = 2.7 Hz, 1H), 7.69–7.74 (m, 2H), 7.93 (d, J = 2.6 Hz, 1H), 10.46 (s, 1H), 11.77 (s, 1H). IR(neat): 3333, 2927, 1621, 1571, 1504, 1422, 1210, 1098, 819 cm−1. HRMS (APCI), m/z calcd for C13H9ClFNO2-H: 264.0228, found 264.0223.
5-Chloro-2-hydroxy-N-(4-trifluoromethoxy-phenyl)-benzamide (42)
Prepared by general procedure A. White solid. mp 200–202 °C. Yield 39%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 8.7 Hz, 2H), 7.47 (dd, J1 = 8.8 Hz, J2 = 2.7 Hz, 1H), 7.82 (dd, J1 = 7.0 Hz, J2 = 2.0 Hz, 2H), 7.91 (d, J = 2.7 Hz, 1H), 10.54 (s, 1H), 11.63 (s, 1H). IR(neat): 3297, 2961, 1614, 1562, 1505, 1160, 1100 cm−1. HRMS (APCI), m/z calcd for C14H9ClF3NO-H: 330.0145, found 330.0156.
5-Chloro-N-(4-cyanophenyl)-2-hydroxybenzamide (43)28
Prepared by general procedure A. Slightly brown solid, mp 246–247 °C [lit28: mp 242–243.5 °C]. IR(neat): 3311, 3075, 2228, 1607, 1548, 1420, 1219, 1103, 831 cm−1. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-2-hydroxy-N-(4-phenoxyphenyl)benzamide (44)
Prepared by general procedure A.31 Grey solid. mp 189–191 °C. Yield 16%. 1H NMR (300 MHz, d6-DMSO): δ 6.99–7.14 (m, 6H), 7.36–7.48(m, 3H), 7.72 (d, J = 8.9 Hz, 2H), 7.96 (d, J = 2.6 Hz, 1H), 10.50 (s, 1H), 11.62(s, br, 1H). HRMS (APCI), m/z calcd for C19H14ClNO3-H: 338.0584, found 338.0575.
N-(4-tert-Butylphenyl)-5-chloro-2-hydroxybenzamide (45)7
Prepared by general procedure A. White solid. mp 219–220 °C [lit7: mp 221–222 °C]. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-2-hydroxy-N-(4-methoxyphenyl)benzamide (46)22
Prepared by general procedure A. Brown solid. mp >207 °C dec. Yield 36%. 1H NMR (300 MHz, d6-DMSO): δ 3.75 (s, 3H), 6.92–7.01 (m, 3H), 7.44–7.58 (m, 1H), 7.57–7.61 (m, 2H), 7.98 (s, 1H), 10.32 (s, 1H), 12.05 (s, 1H). HRMS (APCI), m/z calcd for C14H12ClNO3-H: 276.0427, found 276.0434.
5-Chloro-2-hydroxy-N-(4-morpholin-4-yl-phenyl)-benzamide (47)
Prepared by general procedure A. Grey solid. mp >238 °C dec. Yield 9%. 1H NMR (300 MHz, d6-DMSO): δ 3.05–3.09 (m, 4H), 3.70–3.76 (m, 4H), 6.94–7.01 (m, 3H), 7.45 (dd, J1 = 8.9 Hz, J2 = 2.7 Hz, 1H), 7.56 (d, J = 8.9 Hz, 2H), 8.00 (s, 1H), 10.40 (s, 1H). IR(neat): 3297, 2861, 1612, 1557, 1509, 1217, 1114, 922, 813 cm−1. HRMS (APCI), m/z calcd for C17H17ClN2O3-H: 282.0133, found 282.0137.
N-(4-Carbamoyl-phenyl)-5-chloro-2-hydroxy-benzamide (48)
Compound 43 (50 mg, 0.18 mmol, 1 equiv) was dissolved in EtOH (0.8 mL,) and DMSO (0.4 mL). 1 M NaOH (1.03 mL, 5 equiv) and 30% H2O2 (1.03 mL, 50 equiv) were added and the reaction was stirred at room temperature (16 h). After concentration under vacuum, the residue was purified by column chromatography (1:19 MeOH:CHCl3) to give product 48 (19 mg, 36%). White solid. mp >328 °C dec. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.7 Hz, 1H), 7.30 (s, 1H), 7.46 (dd, J1 = 8.6 Hz, J2 = 2.4 Hz, 1H), 7.78 (d, J = 8.3 Hz, 2H), 7.88–7.91 (m, 4H), 10.55 (s, 1H), 11.69 (s, 1H). HRMS (APCI), m/z calcd for C14H11ClN2O3-H: 289.0380, found 289.0372.
5-Chloro-2-hydroxy-N-(4-(phenylcarbamoyl)phenyl)benzamide (49)
Prepared by general procedure B using 4-amino-N-phenylbenzamide. Red solid. mp 275–277 °C. Yield 11%. 1H NMR (300 MHz, d6-DMSO): δ 7.02–7.12 (m, 2H), 7.35 (dd, J1 = 8.0 Hz, J2 = 8.0 Hz, 2H), 7.48 (dd, J1 = 8.7 Hz, J2 = 2.4 Hz, 1H), 7.78 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.7 Hz, 2H), 7.93 (d, J = 2.1 Hz, 1H), 7.96–8.01 (m, 2H), 10.18 (s, 1H), 10.61 (s, 1H), 11.69 (s, 1H). IR(neat): 3336, 2557, 1635, 1525, 1443, 1221, 810 cm−1. HRMS (APCI), m/z calcd for C20H15ClN2O3-H: 365.0693, found 365.0703.
5-Chloro-2-hydroxy-N-(4-(N-phenylsulfamoyl)phenyl)benzamide (50)
Prepared by general procedure B using 4-amino-N-phenylbenzenesulfonamide. White solid. >291 °C dec. Yield 13%. 1H NMR (300 MHz, d6-DMSO): δ 6.99–7.04 (m, 2H), 7.07–7.10 (m, 2H), 7.20–7.25 (m, 2H), 7.46 (dd, J1 = 9.0 Hz, J2 = 3.0 Hz, 1H), 7.72 (s, 1H), 7.75 (s, 1H), 7.81–7.86 (m, 3H), 10.21 (s, 1H), 10.61 (s, 1H), 11.49 (s, 1H). HRMS (APCI), m/z calcd for C19H15ClN2O4S-H: 401.0363, found 401.0356.
5-Chloro-2-hydroxy-N-(4-(N-(5-methylisoxazol-3-yl)-sulfamoyl)-phenyl)benzamide (51)
Prepared by general procedure B using 4-amino-N-(5-methylisoxazol-3-yl)benzenesulfonamide. Slightly yellow solid. mp >288 °C dec. Yield 71%. 1H NMR (300 MHz, d6-DMSO): δ 2.26 (s, 3H), 6.13 (s, 1H), 7.00(d, J = 8.7 Hz, 1H), 7.45 (dd, J1 = 8.7 Hz, J2 = 2.7 Hz, 1H), 7.82–7.92 (m, 5H), 10.76 (s, 1H). IR(neat): 3187, 1660, 1623, 1506, 1420, 1164, 881 cm−1. HRMS (APCI), m/z calcd for C17H14ClN3O5S-H: 406.0264, found 406.0259.
5-Chloro-N-(3-(trifluoromethyl)-2-methylphenyl)-2-hydroxy-benzamide (52)20
Prepared by general procedure A. White solid. mp 199–200 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(2-fluoro-3-(trifluoromethyl)phenyl)-2-hydroxybenzamide (53)20
Prepared by general procedure A. Slightly yellow solid. mp 250–251 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(2,3-difluorophenyl)-2-hydroxybenzamide (54)
Prepared by general procedure A. White solid. mp 250–251 °C. Yield 58%. 1H NMR (300 MHz, d6-DMSO): δ 7.05 (d, J = 8.7 Hz, 1H), 7.18–7.28 (m, 2H), 7.49 (d, J1 = 8.7 Hz, 1H), 7.95–8.04 (m, 2H), 10.77 (s, 1H), 12.17 (s, 1H). IR(neat): 3315, 3110, 1619, 1557, 1472, 1228, 1004 cm−1. HRMS (APCI), m/z calcd for C13H8ClF2NO2-H: 282.0133, found 282.0135.
5-Chloro-N-(4-fluoro-2-(trifluoromethyl)phenyl)-2-hydroxy-benzamide (55)
Prepared by general procedure A. White solid. mp 173–174 °C. Yield 6%. 1H NMR (300 MHz, d6-DMSO): δ 7.05 (d, J = 8.8 Hz, 1H), 7.52 (dd, J1 = 8.8 Hz, J2 = 2.8 Hz, 1H), 7.59–7.74 (m, 2H), 7.98 (d, J = 2.7 Hz, 1H), 8.13 (dd, J1 = 8.9 Hz, J2 = 5.0 Hz, 1H), 10.79 (s, 1H), 12.20 (s, 1H). HRMS (APCI), m/z calcd for C14H8ClF4NO2-H: 332.0101, found 332.0106.
N-(4-Bromo-2-(trifluoromethyl)phenyl)-5-chloro-2-hydroxy-benzamide (56)
Prepared by general procedure A. Red solid. mp 171–173 °C. Yield 31%. 1H NMR (300 MHz, d6-DMSO): δ 7.05 (d, J = 8.7 Hz, 1H), 7.50 (dd, J1 = 8.7 Hz, J2 = 2.7 Hz, 1H), 7.91–7.95 (m, 3H), 8.20 (d, J = 9.3 Hz, 1H), 10.81 (s, 1H), 12.28 (s, 1H). HRMS (APCI), m/z calcd for C14H8BrClF3NO2-H: 391.9301, found 391.9289.
5-Chloro-N-(2-chloro-4-trifluoromethyl-phenyl)-2-hydroxy-benzamide (57)
Prepared by general procedure A. White solid. mp 195–196 °C. Yield 43%. 1H NMR (300 MHz, d6-DMSO): δ 7.14 (d, J = 8.8 Hz, 1H), 7.58 (dd, J1 = 8.8 Hz, J2 = 2.9 Hz, 1H), 7.84 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 1H), 8.02–8.05 (m, 2H), 8.78 (d, J = 8.6 Hz, 1H), 11.22 (s, 1H), 12.46 (s, 1H). HRMS (APCI), m/z calcd for C14H8Cl2F3NO2-H: 347.9788, found 347.9797.
5-Chloro-N-(2,4-difluorophenyl)-2-hydroxybenzamide (58)
Prepared by general procedure A. Yield 12%. Grey solid. mp >220 °C dec. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.8 Hz, 1H), 7.10–7.17 (m, 1H), 7.37–7.44 (m, 1H), 7.49 (dd, J1 = 8.8 Hz, J2 = 2.8 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 8.07–8.15 (m, 1H), 10.58 (s, 1H), 12.11 (s, 1H). HRMS (APCI), m/z calcd for C13H8ClF2NO2-H: 282.0133, found 282.0134.
5-Chloro-N-(2-chloro-5-(trifluoromethyl)phenyl)-2-hydroxy-benzamide (59)20
Prepared by general procedure A. White solid. mp 180–182 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(2-fluoro-5-(trifluoromethyl)phenyl)-2-hydroxy-benzamide (60)20
Prepared by general procedure A. White solid, mp 199–200 °C. Characterization (NMR, MS) was in accordance with previously reported values.
N-(2-Bromo-5-(trifluoromethyl)phenyl)-5-chloro-2-hydroxy-benzamide (61)24
Prepared by general procedure A. White solid. mp 174–176 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-2-hydroxy-N-(5-methoxy-2-(trifluoromethyl)-phenyl)benzamide (62)
Prepared by general procedure A. White solid. mp 162–163 °C. Yield 35%. 1H NMR (300 MHz, d6-DMSO): δ 3.98 (s, 3H), 7.06 (d, J = 8.7 Hz, 1H), 7.29 (d, J = 8.6 Hz, 1H), 7.49 (dd, J1 = 8.7 Hz, J2 = 3.1 Hz, 2H), 7.96 (d, J = 2.8 Hz, 1H), 8.82 (d, J = 2.1 Hz, 1H), 11.05 (s, 1H), 12.18 (s, 1H). HRMS (APCI), m/z calcd for C15H11ClF3NO3-H: 344.0301, found 344.0310.
5-Chloro-N-(2-chloro-5-fluoro-phenyl)-2-hydroxy-benzamide (63)
Prepared by general procedure A. White solid. mp 218–221 °C. Yield 26%. 1H NMR (300 MHz, d6-DMSO): δ 7.04–7.11 (m, 2H), 7.51–7.66 (m, 2H), 7.98 (d, J = 2.8 Hz, 1H), 8.38 (dd, J1 = 11.4 Hz, J2 = 3.0 Hz, 1H), 11.09 (s, 1H), 12.40 (s, 1H). HRMS (APCI), m/z calcd for C13H8Cl2FNO2-H: 297.9838, found 297.9834.
5-Chloro-N-(2-fluoro-5-nitro-phenyl)-2-hydroxy-benzamide (64)
Prepared by general procedure A. Slightly yellow solid. mp 254–255 °C. Yield 19%. 1H NMR (300 MHz, d6-DMSO): δ 7.07 (d, J = 8.8 Hz, 1H), 7.52 (dd, J1 = 8.8 Hz, J2 = 2.8 Hz, 1H), 7.61–7.68 (m, 1H), 7.95 (d, J = 2.4 Hz, 1H), 8.06–8.12 (m, 1H), 9.25 (dd, J1 = 6.7 Hz, J2 = 2.9 Hz, 1H), 11.00 (s, 1H), 12.28 (s, 1H). HRMS (APCI), m/z calcd for C13H8ClFN2O4-H: 309.0078, found 309.0085.
5-Chloro-N-(2-chloro-6-(trifluoromethyl)phenyl)-2-hydroxybenzamide (65)
Prepared by general procedure A. White solid. mp 167–170 °C. Yield 9%. 1H NMR (300 MHz, CDCl3): δ 7.00 (d, J = 8.9 Hz, 1H), 7.48 (m, 2H), 7.57 (d, J = 2.4 Hz, 1H), 7.68–7.76 (m, 2H), 11.46 (s, 1H). HRMS (APCI), m/z calcd for C14H8Cl2F3NO2-H: 347.9806, found 347.9805.
5-Chloro-N-(2,6-difluorophenyl)-2-hydroxybenzamide (66)
Prepared by general procedure A. Slightly yellow solid. mp >238 °C dec. Yield 7%. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.8 Hz, 1H), 7.19–7.27 (m, 2H), 7.38–7.46 (m, 1H), 7.54 (m, 1H), 7.98 (d, J = 2.6 Hz, 1H), 10.23 (s, 1H), 11.87 (s, 1H). HRMS (APCI), m/z calcd for C13H8ClF2NO2-H: 282.0133, found 282.0129.
5-Chloro-N-(4-fluoro-3-(trifluoromethyl)phenyl)-2-hydroxybenzamide (67)20
Prepared by general procedure A. White solid. mp 203–205 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(4-chloro-3-(trifluoromethyl)phenyl)-2-hydroxy-benzamide (68)30
Prepared by general procedure A. White solid(Slightly yellow), mp 230–232 °C. Yield 38%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 7.5 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.85 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 8.30 (s, 1H), 10.68 (s, 1H). HRMS (APCI), m/z calcd for C14H8Cl2F3NO2-H: 347.9828, found 347.9819.
N-(4-Bromo-3-(trifluoromethyl)phenyl)-5-chloro-2-hydroxy-benzamine (69)
Prepared by general procedure A. White solid, mp 238–240 °C. Yield 26%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (d, J = 8.8 Hz, 1H), 7.46 (dd, J1 = 8.8 Hz, J2 = 2.6 Hz, 1H), 7.85–7.93 (m, 3H), 8.29 (s, 1H), 10.65 (s, 1H), 11.53 (s, br, 1H). HRMS (APCI), m/z calcd for C13H8BrClF3NO-H: 391.9301, found 391.9311.
5-Chloro-N-(4-cyano-3-(trifluoromethyl)phenyl)-2-hydroxy-benzamide (70)
Prepared by general procedure A. White solid. mp 214–216 °C. Yield 16%. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.8 Hz, 1H), 7.48 (dd, J1 = 8.8 Hz, J2 = 2.6 Hz, 1H), 7.79 (d, J = 2.6 Hz, 1H), 8.15 (s, 2H), 8.42 (s, 1H), 10.97 (s, 1H), 11.34 (s,1H). IR(neat): 3197, 2235, 1680, 1534, 1317, 1182, 1116, 1048, 890, 817 cm−1. HRMS (APCI), m/z calcd for C15H8ClF3N2O2-H: 339.0148, found 339.0150.
5-Chloro-N-(3-chloro-4-fluoro-phenyl)-2-hydroxy-benzamide (71)
Prepared by general procedure A. Grey solid. mp 224–225 °C. Yield 30%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (d, J = 8.8 Hz, 1H), 7.40–7.49 (m, 2H), 7.62–7.67 (m, 1H), 7.87 (d, J = 2.6 Hz, 1H), 8.03 (dd, J1 = 6.9 Hz, J2 = 2.6 Hz, 1H), 10.51 (s, 1H), 11.61 (s, 1H). HRMS (APCI), m/z calcd for C13H8Cl2FNO2-H: 297.9833, found 297.9835.
5-Chloro-N-(3,4-difluoro-phenyl)-2-hydroxy-benzamide (72)
Prepared by general procedure A. Slightly yellow solid. mp 250–251 °C. Yield 25%. 1H NMR (300 MHz, d6-DMSO): δ 7.01 (d, J = 8.8 Hz, 1H), 7.39–7.49 (m, 3H), 7.85–8.00 (m, 2H), 10.53 (s, 1H), 11.59 (s, 1H). HRMS (APCI), m/z calcd for C13H8ClF2NO2-H: 282.0133, found 282.0137.
5-Chloro-2-hydroxy-N-(3-iodo-4-methyl-phenyl)-benzamide (73)
Prepared by general procedure A. Yellow brown solid. mp 245–247 °C. Yield 14%. 1H NMR (300 MHz, d6-DMSO): δ 2.35 (s, 3H), 7.00 (d, J = 8.7 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.46 (dd, J1 = 8.8 Hz, J2 = 2.7 Hz, 1H), 7.60 (dd, J1 = 8.3 Hz, J2 = 2.1 Hz, 1H), 7.92 (d, J = 2.7 Hz, 1H), 8.27 (d, J = 2.1 Hz, 1H), 10.44 (s, 1H), 11.74 (s, 1H). HRMS (APCI), m/z calcd for C14H11ClINO2-H: 385.9445, found 385.9459.
N-(3-Bromo-5-(trifluoromethyl)phenyl)-5-chloro-2-hydroxy-benzamide (74)24
Prepared by general procedure A. White solid, mp 200–202 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-2-hydroxy-N-(3-methoxy-5-(trifluoromethyl)-phenyl)benzamide (75)24
Prepared by general procedure A. White solid, mp 191–192 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(3,5-dichlorophenyl)-2-hydroxybenzamide (76)20
Prepared by general procedure A. White solid, mp 247–249 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(3,5-difluorophenyl)-2-hydroxybenzamide (77)21
Prepared by general procedure A. Slightly yellow solid. mp 244–246 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(3,5-di-tert-butylphenyl)-2-hydroxybenzamide (78)19
Prepared by general procedure A. White solid. mp 186–188 °C. Characterization (NMR, MS) was in accordance with previously reported values.
5-Chloro-N-(3,5-dimethylphenyl)2-hydroxybenzamide (79)
Prepared by general procedure A. White solid, mp 183–184 °C. Yield 22%. 1H NMR (300 MHz, d6-DMSO): δ 2.49 (s, 6H), 6.79 (s, 1H), 7.00 (d, J = 8.8 Hz, 1H), 7.32 (s, 2H), 7.46 (dd, J1 = 8.8 Hz, J2 = 2.6 Hz, 1H), 7.97 (d, J = 2.6 Hz, 1H), 10.28 (s, 1H), 11.91 (s, 1H). HRMS (APCI), m/z calcd for C15H14ClNO2-H: 274.0635, found 274.0625.
Substrates for 80 and 81: 4-bromo-3,5-bis-(trifluoromethyl)aniline & 2-bromo-3,5-bis-(trifluoromethyl)aniline
3,5-Bis(trifluoromethyl)aniline (2 g, 8.7 mmol, 1 equiv) was dissolved in DMF (6 mL, 1.5 M). N-bromosuccinimide (1.7 g, 9.6 mmol, 1.1 equiv) was added, and this mixture was stirred at 50 °C for one hour. After cooling to room temperature, water was added (100 mL) followed by extraction with EtOAc. The crude product was purified by column chromatography (1:10 → 1:5 EtOAc:Hex) to give 4-bromo-3,5-bis-(trifluoromethyl)-aniline (340 mg) & 2-bromo-3,5-bis-(trifluoromethyl)aniline (680 mg).
5-Chloro-2-hydroxy-N-(4-methyl-3,5-bis(trifluoromethyl)-phenyl)benzamide (80)
4-Bromo-3,5-bis-(trifluoromethyl)aniline (616 mg, 2 mmol, 1 equiv), potassium carbonate (829 mg, 6 mmol, 3 equiv), Pd(PPh3)4 (231 mg, 0.2 mmol, 0.1 equiv), DMF (5 mL, 0.4 M) and (MeO)3B (0.28 mL, 2 mmol, 1 equiv) were added to a flask and the contents heated to 110 °C overnight. The reaction solution was filtered using silica gel, Celite, and sea sand. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography (1:5 EtOAc:Hex), to give 4-methyl-3,5-bis(trifluoromethyl)aniline (760 mg, 67%). 1H NMR (300 MHz, d6-DMSO): δ 2.42 (s, 3H), 3.88 (s, 2H), 7.09 (s, 2H). This aniline was used for general procedure A (EtOAc:Hex 1:20 → 1:10) (630 mg, 51%). Slightly brown solid, mp 192–193 °C. 1H NMR (300 MHz, d6-DMSO): δ 2.49 (s, 3H), 7.02 (d, J = 8.8 Hz, 1H), 7.47 (dd, J1 = 8.8 Hz, J2 = 2.7 Hz, 1H), 7.87 (d, J = 2.6, 1H), 8.41 (s, 2H), 10.75 (s, 1H), 11.48 (s, br, 1H). HRMS (APCI), m/z calcd for C16H10ClF6NO2-H: 396.0226, found 396.0238.
5-Chloro-2-hydroxy-N-(2-methyl-3,5-bis(trifluoromethyl)-phenyl)benzamide (81)
Synthesis proceeded as for 80 using 2-bromo-3,5-bis-(trifluoromethyl)aniline as starting material (330 mg, 33%). Slightly yellow solid. mp 191–193 °C. 1H NMR (300 MHz, d6-DMSO): δ 2.46 (s, 3H), 7.07 (d, J = 8.8 Hz, 1H), 7.50 (dd, J1 = 8.7 Hz, J2 = 2.4 Hz, 1H), 7.82 (s, 1H), 7.95 (d, J = 2.7 Hz, 1H), 8.60 (s, 1H), 10.64 (s, 1H), 12.11 (s, 1H). HRMS (APCI), m/z calcd for C16H10ClF6NO2-H: 396.0226, found 396.0233.
N-(4-Bromo-3,5-difluorophenyl)-5-chloro-2-hydroxybenzamide (82)
Prepared by general procedure A. White solid. mp >271 °C dec. Yield 32%. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.8 Hz, 1H), 7.47 (d, J = 6.7 Hz, 1H), 7.68 (d, J = 9.5 Hz, 2H), 7.79 (s, 1H), 10.72 (s, 1H), 11.45 (s, br, 1H). HRMS (APCI), m/z calcd for C13H7BrClF2NO2-H: 359.9238, found 359.9234.
5-Chloro-2-hydroxy-N-(3,4,5-trifluoro-phenyl)-benzamide (83)
Prepared by general procedure A. Slightly yellow solid. mp 234–236 °C. Yield 52%. 1H NMR (300 MHz, d6-DMSO): δ 7.03 (d, J = 8.8 Hz, 1H), 7.47 (dd, J1 = 8.8 Hz, J2 = 2.7 Hz, 1H), 7.67–7.72 (m, 2H), 7.80 (d, J = 2.7 Hz, 1H), 10.59 (s, 1H), 11.47 (s, br, 1H). HRMS (APCI), m/z calcd for C13H7ClF3NO2-H: 300.0039, found 300.0055.
5-Chloro-2-hydroxy-N-(3,4,5-trichloro-phenyl)-benzamide (84)29
Prepared by general procedure A. White solid. mp 287–290 °C. Yield 44%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (d, J = 8.8 Hz, 1H), 7.47 (dd, J1 = 8.7 Hz, J2 = 2.5 Hz, 1H), 7.81 (d, J = 2.5 Hz, 1H), 8.05 (s, 2H), 10.68 (s, 1H), 11.41 (s, br, 1H). HRMS (APCI), m/z calcd for C13H7Cl4NO2-H: 347.9153, found 347.9156.
5-Chloro-2-hydroxy-N-(2,4,5-trifluorophenyl)benzamide (85)
Prepared by general procedure A. Slightly yellow solid. mp 250–251°C. Yield 10%. 1H NMR (300 MHz, d6-DMSO): δ 7.04 (d, J = 8.8 Hz, 1H), 7.50 (dd, J1 = 8.8 Hz, J2 = 2.8 Hz, 1H), 7.69–7.79 (m, 1H), 7.93 (d, J = 2.7 Hz, 1H), 8.26–8.36 (m, 1H), 10.88 (s, 1H), 12.19 (s, br, 1H). HRMS (APCI), m/z calcd for C13H7ClF3NO2-H: 300.0026, found 300.0032.
5-Chloro-2-hydroxy-N-(2,3,4-trifluoro-phenyl)-benzamide (86)
Prepared by general procedure A. Slightly yellow solid. mp 244–246 °C. Yield 16%. 1H NMR (300 MHz, d6-DMSO): δ 7.05 (d, J = 8.8 Hz, 1H), 7.32–7.42 (m, 1H), 7.51 (dd, J1 = 8.8 Hz, J2 = 2.7 Hz, 1H), 7.85–7.94 (m, 2H), 10.67 (s, 1H), 12.08 (s, 1H). HRMS (APCI), m/z calcd for C13H7ClF3NO2-H: 300.0026, found 300.0030.
5-Chloro-N-(2,4-dichloro-5-methoxy-phenyl)-2-hydroxy-benzamide (87)
Prepared by general procedure A. White solid. mp >268 °C dec. Yield 43%. 1H NMR (300 MHz, d6-DMSO): δ 3.89 (s, 3H), 7.06 (d, J = 8.7 Hz, 1H), 7.50 (dd, J1 = 8.7 Hz, J2 = 2.7 Hz, 1H), 7.70 (s, 1H), 7.97 (d, J = 2.7 Hz, 1H), 8.38 (s, 1H), 11.20 (s, 1H), 12.49 (s, 1H). HRMS (APCI), m/z calcd for C14H10Cl3NO3-H: 343.9648, found 343.9647..
5-Chloro-N-(2,4-dichloro-5-hydroxy-phenyl)-2-hydroxy-benzamide (88)
To a solution of compound 88 (400 mg, 1.15 mmol, 1 equiv) in DCM (100 mL, 0.01 M) was added a 1 M solution of BBr3 in hexane (4.5 mL, 4 equiv). The suspension was stirred at room temperature (24 h). Water (10 mL) was added and stirring continued for another 15 min. DCM was added and the mixture was washed with 1 N NaOH, and dried and concentrated under vacuum. The residue was purified by column chromatography (1:5 EtOAc:Hex) to give 87 (260 mg, 67%). White solid. mp 224–226 °C. 1H NMR (300 MHz, d6-DMSO): δ 7.08 (d, J = 8.7 Hz, 1H),7.52 (dd, J1 = 8.7 Hz, J2 = 2.7 Hz, 1H), 7.58 (s, 1H), 7.98 (d, J = 2.7 Hz, 1H), 8.31 (s, 1H), 10.68 (s, 1H), 10.88 (s, 1H), 12.31 (s, 1H). IR(neat): 3603, 3194, 1637, 1591, 1534, 1224, 826 cm−1. HRMS (APCI), m/z calcd for C13H8Cl3NO3-H: 329.9492, found 329.9492
N-(3-Bromo-5-trifluoromethyl-phenyl)-5-fluoro-2-hydroxy-benzamide (89)
Prepared by general procedure A. White solid. mp 212–214 °C. Yield 40%. 1H NMR (300 MHz, d6-DMSO): δ 7.00–7.05 (m, 1H), 7.30–7.36 (m, 1H), 7.64 (dd, J1 = 9.4 Hz, J2 = 3.2 Hz, 1H), 7.72 (s, 1H), 8.18 (s, 1H), 8.28 (s, 1H), 10.71 (s, 1H), 11.24 (s, 1H). HRMS (APCI), m/z calcd for C14H8BrF4NO2-H: 375.9596, found 375.9590.
N-(2-Chloro-5-(trifluoromethyl)phenyl)-5-fluoro-2-hydroxy-benzamide (90)
Prepared by general procedure A. White solid. mp 176–180 °C. Yield 10%. 1H NMR (300 MHz, d6-DMSO): δ 7.05–7.10 (m, 1H), 7.34–7.39 (m, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.75 (dd, J1 = 9.4 Hz, J2 = 3.1 Hz, 1H), 7.83 (d, J = 6.7 Hz, 1H), 8.90 (s, 1H), 11.22 (s, 1H), 12.16 (s, br, 1H). HRMS (APCI), m/z calcd for C14H8ClF4NO2-H: 332.0090, found 332.0093.
N-(3,5-Difluorophenyl)-5-fluoro-2-hydroxybenzamide (91)
Prepared by general procedure A. White solid. mp 205–206 °C. Yield 18%. 1H NMR (300 MHz, d6-DMSO): δ 6.96–7.03 (m, 2H), 7.29–7.33 (m, 1H), 7.50 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 9.3 Hz, 1H), 10.61 (s, 1H), 11.26 (s, br, 1H). HRMS (APCI), m/z calcd for C13H8F3NO2-H: 266.0429, found 266.0439.
5-Fluoro-2-hydroxy-N-(4-trifluoromethyl)phenyl)benzamide (92)28
Prepared by general procedure A. White solid. mp 227–230 °C [lit28: mp 222–223 °C]. Characterization (NMR, MS) was in accordance with previously reported values.
5-Fluoro-N-(2-fluoro-5-(trifluoromethyl)phenyl)-2-hydroxy-benzamide (93)
Prepared by general procedure A. Yield 29%. White solid. mp 228–230 °C. 1H NMR (300 MHz, d6-DMSO): δ 7.05 (dd, J1 = 8.9 Hz, J2 = 4.5 Hz, 1H), 7.32–7.39 (m, 1H), 7.59 (d, J = 5.6 Hz, 2H), 7.72 (dd, J1 = 9.6 Hz, J2 = 3.3 Hz, 1H), 8.74 (d, J = 7.3 Hz, 1H), 10.99 (s, 1H), 12.00 (s, 1H). HRMS (APCI), m/z calcd for C14H8F5NO2-H: 316.0397, found 316.0403.
5-Fluoro-2-hydroxy-N-((3-trifluoromethyl)phenyl)benzamide (94)
Prepared by general procedure A. White solid. mp 216–217 °C. Yield 55%. 1H NMR (300 MHz, d6-DMSO): δ 7.02 (dd, J1 = 9.0 Hz, J2 = 4.7 Hz, 1H), 7.28–7.35 (m, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.60 (dd, J1 = 8.1 Hz J2 = 7.9 Hz, 1H), 7.70 (dd, J1 = 9.5 Hz, J2 = 3.2Hz, 1H), 7.91 (d, J = 8.6 Hz, 1H), 8.22 (s, 1H), 10.63 (s, 1H), 11.38 (s, 1H). HRMS (APCI), m/z calcd for C14H9F4NO2-H: 298.0491, found 298.0487.
Biological characterization
Initial minimum inhibitory concentration (MIC) screening was performed using Mtb H37Rv(pMSP12) in the presence of 30 µg/mL kanamycin (to maintain plasmid). Later follow-up was performed using the parental wild-type strain Mtb H37Rv (ATCC27294) since it demonstrated identical MIC values to the original strain. MIC values were determined in 96-well plate format at 37 °C. Mtb was grown to early log-phase (OD650=0.2–0.3) and diluted 1:1000 in Middlebrook 7H9 supplemented with 0.5% BSA, 0.2% glucose, 0.081% NaCl, 0.05% Tyloxapol (with glucose as the sole carbon source). Two-fold dilutions were made across 96-well plates with the final column used as negative control (no compound). DMSO was used at 0.5% (v/v) final concentration and none of the compounds appeared to be sufficiently insoluble to obscure final MIC values although solubility may have limited the maximum value that could be tested in some cases (signified by a greater than sign in the data). Isoniazid was used as positive control. Plates were read at two weeks. MIC was defined as the minimum compound concentration causing complete cessation of visual growth. All assays were performed at least two independent times.
Cytotoxicity was determined using the murine macrophage cell line J774.1 (ATCC TIB-67) grown in DMEM supplemented with 10% fetal bovine serum (FBS), 0.25 mM pyruvate, and 0.5× MEM nonessential amino acids (Mediatech). Cells used had fewer than 10 passages. Two-fold dilutions of compound starting at 60 µM were added to cells plated (5×103 cells per well) onto 96-well plates. Plates were incubated at 37 °C for 3 days (5% CO2) and were read by Alamar Blue (Invitrogen) following the manufacturer’s instructions.
Supplementary Material
Acknowledgments
We thank Anton Simeonov, Paul Shinn, Adam Yasgar, Carleen Klumpp, and Ajit Jadhav for construction and help with screening and analysis of the NCGC library. We also thank Gwendolyn Marriner for helpful discussions, Noel Whittaker for HRMS data collection, and Kathryn Soucy for assistance in data collection. This work was funded (in part) by the Intramural Research Program of the National Institute of Allergy and Infectious Disease, National Institutes of Health, the Bill and Melinda Gates Foundation, the Korea Research Institute of Chemical and Technology, and the Ministry of Education, Science and Technology of Korea.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/
References and notes
- 1.World Health Organization. Global tuberculosis control: WHO report. Geneva: WHO Press; 2011. [Google Scholar]
- 2.Ogawa H, Azuma M, Muto S, Nishioka Y, Honjo A, Tezuka T, Uehara H, Izumi K, Itai A, Sone S. Clin. Exp. Allergy. 2011;41:104. doi: 10.1111/j.1365-2222.2010.03564.x. [DOI] [PubMed] [Google Scholar]
- 3.Macielag MJ, Demers JP, Fraga-Spano SA, Hlasta DJ, Johnson SG, Kanojia RM, Russell RK, Sui Z, Weidner-Wells MA, Werblood H, Foleno BD, Goldschmidt RM, Loeloff MJ, Webb GC, Barrett JF. J. Med. Chem. 1998;41:2939. doi: 10.1021/jm9803572. [DOI] [PubMed] [Google Scholar]
- 4.De La Fuente R, Sonawane ND, Arumainayagam D, Verkman AS. Br. J. Pharmacol. 2006;149:551. doi: 10.1038/sj.bjp.0706873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imramovsky A, Vinsova J, Ferriz JM, Buchta V, Jampilek J. Bioorg. Med. Chem. Lett. 2009;19:348. doi: 10.1016/j.bmcl.2008.11.080. [DOI] [PubMed] [Google Scholar]
- 6.Chandrawathani P, Yusoff N, Wan LC, Ham A, Waller PJ. Vet. Res. Commun. 2004;28:479. doi: 10.1023/b:verc.0000040240.69004.dc. [DOI] [PubMed] [Google Scholar]
- 7.Waisser K, Matyk J, Divisova H, Husakova P, Kunes J, Klimesova V, Kaustova J, Mollmann U, Dahse HM, Miko M. Arch. Pharm. (Weinheim) 2006;339:616. doi: 10.1002/ardp.200600093. [DOI] [PubMed] [Google Scholar]
- 8.Ferriz JM, Vavrova K, Kunc F, Imramovsky A, Stolarikova J, Vavrikova E, Vinsova J. Bioorg. Med. Chem. 2010;18:1054. doi: 10.1016/j.bmc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 9.Cheng TJ, Wu YT, Yang ST, Lo KH, Chen SK, Chen YH, Huang WI, Yuan CH, Guo CW, Huang LY, Chen KT, Shih HW, Cheng YS, Cheng WC, Wong CH. Bioorg. Med. Chem. 2010;18:8512. doi: 10.1016/j.bmc.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 10.Dahlgren MK, Kauppi AM, Olsson IM, Linusson A, Elofsson M. J. Med. Chem. 2007;50:6177. doi: 10.1021/jm070741b. [DOI] [PubMed] [Google Scholar]
- 11.Brown ME, Fitzner JN, Stevens T, Chin W, Wright CD, Boyce JP. Bioorg. Med. Chem. 2008;16:8760. doi: 10.1016/j.bmc.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 12.Liechti C, Sequin U, Bold G, Furet P, Meyer T, Traxler P. Eur. J. Med. Chem. 2004;39:11. doi: 10.1016/j.ejmech.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Terada H. Biochim. Biophys. Acta. 1981;639:225. doi: 10.1016/0304-4173(81)90011-2. [DOI] [PubMed] [Google Scholar]
- 14.Terada H, Goto S, Yamamoto K, Takeuchi I, Hamada Y, Miyake K. Biochim. Biophys. Acta. 1988;936:504. doi: 10.1016/0005-2728(88)90027-8. [DOI] [PubMed] [Google Scholar]
- 15.Dolezal R, Van Damme S, Bultinck P, Waisser K. Eur. J. Med. Chem. 2009;44:869. doi: 10.1016/j.ejmech.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 16.Vandal OH, Pierini LM, Schnappinger D, Nathan CF, Ehrt S. Nat. Med. 2008;14:849. doi: 10.1038/nmXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Carvalho LP, Darby CM, Rhee KY, Nathan C. ACS Med. Chem. Lett. 2011;2:849. doi: 10.1021/ml200157f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo L, Wang Q-L, Jiang Q-Q, Jiang Q-J, Jiang Y-B. J. Og. Chem. 2007;72:9947. doi: 10.1021/jo701823d. [DOI] [PubMed] [Google Scholar]
- 19.Garner AL, Gloeckner C, Tricoche N, Zakhari JS, Samje M, Cho-Ngwa F, Lustigman S, Janda KD. J. Med. Chem. 2011;54:3963. doi: 10.1021/jm200364n. [DOI] [PubMed] [Google Scholar]
- 20.Itai A, Muto S, Nagano T, Saotome T. Patent EP1352650. European Patent Office. 2003
- 21.Itai A, Muto S. Patent EP1535609. European Patent Office. 2005
- 22.Imramovsky A, Ferriz JM, Pauk K, Kratky M, Vinsova J. J. Comb. Chem. 2010;12:414. doi: 10.1021/cc900168s. [DOI] [PubMed] [Google Scholar]
- 23.Ciampa G, Grieco C. Accad. Sci. Fis. Naples. 1966;Vol. 33:386. [Chem. Abstracts, vol 68, 95469a] [Google Scholar]
- 24.Itai A, Muto S. Patent EP1510210. European Patent Office. 2005
- 25.Drain DJ, Davy B, Horlington M, Howes JG, Scruton JM, Selway RA. J. Pharm. Pharmacol. 1971;23:857. doi: 10.1111/j.2042-7158.1971.tb10203.x. [DOI] [PubMed] [Google Scholar]
- 26.Wood R, Welsh W, Ekins S, Ai N. WO2010101648. World Intellectual Property Organization Patent. 2010
- 27.Muto S, Itai A. Patent EP1512397. European Patent Office. 2005
- 28.Waisser K, Bures O, Holy P, Kunes J, Oswald R, Jiraskova L, Pour M, Klimesova V, Kubicova L, Kaustova J. Arch. Pharm. (Weinheim) 2003;336:53. doi: 10.1002/ardp.200390004. [DOI] [PubMed] [Google Scholar]
- 29.Itai A, Muto S. Patent EP1512396. European Patent Office. 2005
- 30.Bindler J, Geigy AG. US2703332. United States Patent. 1952
- 31.Moyle C. US3577550. United States Patent. 1971 [Chem. Abstracts, vol 76, 140263]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.