

Abstract
A structure-activity relationship (SAR) of the c-Myc (Myc) inhibitor 10074-G5 (N-([1,1′-biphenyl]-2-yl)-7-nitrobenzo[c][1,2,5]oxadiazol-4-amine, 1), which targets a hydrophobic domain of Myc that is flanked by arginine residues, was executed in order to determine its pharmacophore. Whilst the 7-nitrobenzofurazan was found to be critical for inhibitory activity, the ortho-biphenyl could be replaced with a para-carboxyphenyl group to furnish the new inhibitor JY-3-094 (3q). Around five times as potent as the lead with a IC50 of 33 μM for disruption of the Myc–Max heterodimer, JY-3-094 demonstrated excellent selectivity over Max–Max homodimers, with no apparent effect at 100 μM. Importantly, the carboxylic acid of JY-3-094 improves the physicochemical properties of the lead compound, which will facilitate the incorporation of additional hydrophobicity that might enhance Myc inhibitory activity further still.
Keywords: Myc, Max, 10074-G5, pharmacophore, cancer
c-Myc (Myc) is a short-lived (t1/2 = 20 – 30 min) nuclear oncoprotein and is a member of the basic-helix-loop-helix leucine zipper (bHLH-LZ) protein family of transcription factors that includes its obligate binding partner, Max, and its antagonist proteins of the Mad family.1 Myc is responsible for the transcription of multiple target genes involved in cell cycle progression, cell proliferation, differentiation, metabolism and survival.1–4 Though Myc is essential for normal cell function, its over-expression and/or dys-regulation is associated with a wide variety of human cancers, including lung, prostate, pancreatic, breast, gynecological and colon cancers, B-cell lymphomas and leukaemias.5,6 In 2012, it is estimated there will be 577,190 cancer deaths in the United States alone,7 perhaps as many as one-seventh of which will be associated with tumours exhibiting changes in the c-myc proto-oncogene or its expression.1 Inactivation of Myc results in cell-cycle arrest, apoptosis and tumour regression.8–10 Thus, the inhibition of Myc function is an appealing tactic to expand the arsenal of anticancer therapeutics in a highly targeted manner. Currently, there is no such “Myc drug” in the clinic, and the most potent Myc inhibitors to date exhibit only low micromolar IC50 values.5 Given the significant role of Myc in the development and progression of a wide variety of cancers, there is an urgent need for more potent and diverse Myc inhibitors.
In its monomeric form, the bHLH-LZ domain of Myc (and Max) is intrinsically disordered, presenting no obvious binding sites for the development of inhibitors.11 Myc becomes transcriptionally functional only upon its heterodimerization with Max, an event in which the two proteins act as each other’s chaperone to generate a parallel, left-handed four-helix bundle structure that recognizes the obligate E-box DNA sequence CAC/TGTG. The Myc–Max heterodimer thus serves as an excellent example of the coupled folding and binding of two intrinsically disorder proteins. Indeed, most biological activities of Myc require this interaction.12–14 The basic regions of Myc and Max bind the DNA, while the HLH and LZ domains form the dimerization interface. Unlike Myc, Max is constitutively expressed, is stable (t1/2 > 24 h) and can also homodimerize. Max–Max homodimers bind the same DNA sequence as Myc–Max heterodimers but rather inhibit transcription, which is manifested through the direct competition for Max and for the common DNA site. Small-molecules that interfere with this protein–protein dimerization process would be expected to inhibit the oncogenic activity of Myc. Accordingly, several groups have adopted this approach to disrupt the Myc–Max dimer.5,6 Owing to a lack of recognizable binding clefts or motifs, coupled with an inability to predict the structures of potential Myc–Max disruptors, the majority of inhibitors reported to date have been identified by screening large libraries of compounds, albeit with minimal success rates.15–21 Moreover, most compounds identified to date have been hampered by low-affinity binding and/or poor in vivo activity against Myc-dependent tumors.6,22,23
Of particular interest to us are two such-identified Myc–Max dimerization inhibitors: the rhodanine derivative (Z)-5-(4-ethylbenzylidene)-2-thioxothiazolidin-4-one (10058-F4) and the benzofurazan N-([1,1′-biphenyl]-2-yl)-7-nitrobenzo[c][1,2,5]oxadiazol-4-amine (10074-G5, 1).16 Upon further characterization, it has recently been revealed that the mode of Myc–Max dimerization inhibition by these two small-molecules is through the direct binding of the Myc monomer (10058-F4 binds Myc402-412, 10074-G5 binds Myc363-381), thereby limiting its ability to interact with Max.24,25,26 Previously, the activity of 10058-F4 has been improved through the development of a variety of congeners.27 10074-G5 also represents a promising candidate for optimization for several reasons. First, it is accessible in a single chemical reaction from commercially available starting materials, thus facilitating the development of analogues. Second, 10074-G5 comprises three distinct moieties – a 2-aminobiphenyl, a bicyclic benzofurazan and a nitro group – each of which can be studied in a step-wise manner. Third, through employing routine chemical reactions, modification of these moieties is expected to be relatively straightforward and efficient. We have, thus, conducted a structure–activity relationship (SAR) of each of these components of 10074-G5, and our results are disclosed in this Letter.
It has recently been demonstrated that 10074-G5 binds the Myc peptide Myc353-437 with a Kd value of 2.8 μM, more specifically in the region Arg363-Ile381 (Figure 2).24,25 More particularly, NMR NOESY analysis and docking studies independently suggested that 10074-G5 binds in a cavity that is created by a kink (Asp379–Ile381) in the N-terminus of an induced helical domain (Leu370–Arg378), with the biphenyl moiety observed in the hydrophobic domain formed by Phe375, Ile381 and the side chain methylenes of Arg378, and the electron rich 1,2,5-oxadiazole ring and nitro group found near the positively-charged residues Arg366 and Arg367. In order to determine the pharmacophore of 10074-G5, we conducted an SAR study that explored the significance of the biphenyl, the aniline NH, the benzofurazan and the nitro group. As anticipated, the requisite chemical reactions were straightforward and are illustrated in Scheme 1. Variation of the biphenyl moiety was accomplished by reacting 4-chloro-7-nitrobenzofurazan (2) with a range of anilines and aliphatic amines (RNH2) to furnish target molecules 3a – 3q in moderate to excellent yields. Hydrogenation of the nitro group of 10074-G5 provided the corresponding aniline 4 that was subsequently reacted with various acylating agents to deliver compounds 5a – 5c. Finally, N-arylation of 2-aminobiphenyl (6) with 4-fluoronitrobenzene delivered secondary aniline 7 (Supporting Information).
Figure 2.

Docking between 10074-G5 and Myc363-381, as described in Reference 24 (adapted from Reference 24, with permission from Steven J. Metallo).
Scheme 1.

a) RNH2, CH3CN, DIPEA, reflux 16 h, 35 – 99%; b) H2, 10% Pd/C, MeOH, RT, 16 h, 76%; c) R2COX, CH2Cl2, Et3N, 0 °C to RT (or 40 °C), 16 h, 38 – 95%.
Our 10074-G5 analogues were initially screened at 100 μM in an electrophoretic mobility shift assay (EMSA) that we have previously described.26 Effective compounds disrupt the interaction of homogeneously purified recombinant Myc bHLH-LZ domain and Max proteins with a fluorescently-tagged (FAM) E-Box-containing double-stranded oligonucleotide. Our results are shown in Tables 1 and 2, and are reported as percentage inhibitions that were determined through comparison of Myc–Max/DNA band intensities in the presence of the candidate inhibitor relative to the control band intensity acquired in its absence. First, as shown by the activity for 3a, an unsubstituted aniline at the 4-position of the 7-nitrobenzofurazan nucleus was insufficient for activity. Furthermore, a small hydrophobic group (3e) or hydrophilic group (3g) at the ortho position afforded compounds with minimal Myc–Max inhibitory activities. On the other hand, bulky, hydrophobic groups at the ortho position (phenyl (10074-G5), bromo (3f)) furnished inhibitors that exhibited about 40% disruption of the Myc–Max dimer. These groups may be directed into the hydrophobic domain composed of Phe375 and Ile381. Shifting the ortho-phenyl ring to the meta and para positions (3l and 3n, respectively) led to a reduction in activity, underscoring the importance of hydrophobicity at the ortho site. Consistent with this is the observation that polar carboxylic acids were poorly tolerated at the ortho and meta positions (3i and 3m, respectively) but afforded excellent inhibition of Myc–Max dimers at the para position (3q, hereafter “JY-3-094”). This trend also suggests that the promising activity of JY-3-094 is not simply a consequence of improved solubility by virtue of the ionizable carboxylic acid since the regioisomers 3i and 3m would be expected to exhibit comparable solubility, yet they demonstrated little to no inhibitory activities.
Table 1.
EMSA-Based SAR analysis of 2-aminobiphenyl moiety of Myc inhibitor 10074-G5.
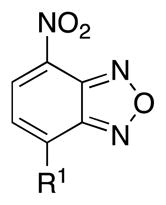
| ||
|---|---|---|
| Compound | R1 | Inhibition at 100 μM (%) |
| 10074-G5 |
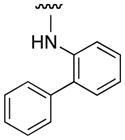
|
38 |
| 3a |

|
2 |
| 3b |
|
11 |
| 3c |

|
0 |
| 3d |

|
10 |
| 3e |

|
11 |
| 3f |

|
39 |
| 3g |
|
13 |
| 3h |
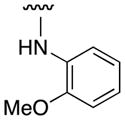
|
0 |
| 3i |

|
8 |
| 3j |

|
36 |
| 3k |
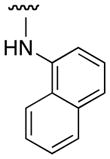
|
0 |
| 3l |

|
20 |
| 3m |
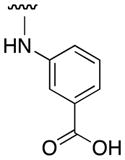
|
0 |
| 3n |

|
28 |
| 3o |

|
0 |
| 3p |

|
0 |
| 3q (“JY-3-094” |
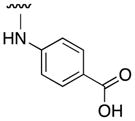
|
93 |
Table 2.
EMSA-Based SAR analysis of 7-nitrobenzofurazan moiety of Myc inhibitor 10074-G5.
| Compound | Structure | Inhibition at 100 μM (%) |
|---|---|---|
| 4 |

|
2 |
| 5a |
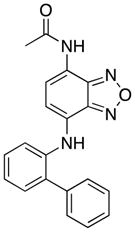
|
2 |
| 5b |

|
25 |
| 5c |

|
0 |
| 5d |

|
17 |
| 5e |

|
33 |
| 7 |

|
0 |
The isosteric replacement of the aniline NH group with oxygen (compound 3j) had no impact on the disruption of the Myc–Max dimer, indicating that a hydrogen bond donor is probably not important here. Reduction of the nitro group to the corresponding amine (4) was not tolerated, and this may be a consequence of the loss of the interaction between the electron rich nitro group and the positively-charged Arg366 and Arg367 residues. Acetylation of the primary amino group of 4 to furnish acetamide 5a did not lead to recovery of Myc inhibitory activity. However, replacement of the acetyl group with the more-electron rich trifluoroacetyl bioisostere delivered inhibitor 5b that effected 25% inhibition of the Myc–Max heterodimer. Moreoever, the coupling of 4 to diglycolic anhydride furnished N-acyl derivative 5e with a carboxylic acid that demonstrated even greater inhibition of the Myc–Max heterodimer (33%). Taken together, these data suggest the nitro group of 10074-G5 does, indeed, make contacts with postiviely-charged residues, such as Arg367. Deletion of the 1,2,5-oxadiazole ring of the bicyclic benzofurazan system to give 7 abolished activity, indicating the importance of the nitrogen and oxygen heteroatoms that may be involved in polar interactions with Arg366 and Arg367 as originally proposed.24,25 The nitro group and benzofurazan ring, therefore, appear critical for Myc inhibitory activity.
On the basis of the EMSA screening data, we can, thus, define the pharmacophore of 10074-G5 as follows. Maximal Myc inhibitory activity requires the electron rich 7-nitrobenzofurazan ring, which is consistent with its predicted binding mode to the positively-charged residues Arg366 and Arg367 that would be expected to contribute considerably to the binding energy. Furthermore, the 7-nitrobenzofurazan should be substituted with a secondary aniline (or phenol) at the 4-position. More particularly, this aniline requires a bulky, hydrophobic substituent (e.g. phenyl) at the ortho position, which may engage in van der Waals interactions with Phe375 and Ile381, or a carboxylic acid at the para position, which may be involved in a salt bridge with Arg378 that is located at the N-terminus of the induced helix. We believe this information may prove useful in the development of future congeners of 10074-G5.
Upon further characterization, JY-3-094 was shown to exhibit a dose-response inhibitory effect against Myc–Max dimerization (Figure 3), with an IC50 value of 33 μM (Figure 4), which is around five-fold as potent as the original lead 10074-G5 (IC50 = 146 μM). Moreover, JY-3-094 showed no activity against Max–Max homodimers at 100 μM (Figures 3 & 4), demonstrating a greater than three-fold selectivity for Myc–Max heterodimers, and suggesting that the mode of inhibition of JY-3-094 is through binding Myc rather than Max or DNA. Indeed, a mixture of JY-3-094 and the ds oligo used in the EMSA assay were incubated together, then subjected to PAGE; no binding of the small-molecule to the DNA was observed. We were next keen to investigate the activity of JY-3-094 in Myc-overexpressing cell lines. Daudi Burkitt lymphoma cells and HL60 promyelocytic leukaemia cells both express high levels of Myc. The lead compound 10074-G5 performs well in these cell lines, with IC50 values of about 30 μM in HL60 cells and about 10 μM in Daudi cells.16 Unfortunately, JY-3-094 demonstrated limited cytotoxicities toward these cell lines, with IC50s of 184 μM (HL60) and 56 μM (Daudi). However, this was to be expected, since a hydrophobic phenyl group had been replaced with a polar and charged carboxylic acid group that is known to impede cell entry.
Figure 3.

JY-3-094 exhibits a dose-dependent inhibition of the Myc–Max heterodimer, with no effect against Max–Max homodimers, as demonstrated by (A) a reduction in the Myc–Max/DNA band intensity and (B) no change in the Max–Max/DNA band intensity in EMSA assays (for further details, see Supporting Information).
Figure 4.

JY-3-094 is selective for Myc–Max heterodimers over Max–Max homodimers, as determined by EMSA assays.
In summary, we have conducted a focused SAR study on the Myc inhibitor 10074-G5 and have determined its pharmacophore. The 7-nitro group and 1,2,5-oxadiazole of the benzofurazan moiety are both crucial for activity. Furthermore, a substituted aniline (or phenol) is necessary at the 4-position of the benzofurazan: ortho-substitution requires a bulky, hydrophobic group; para-substitution requires a carboxylic acid function. Reintroduction of the ortho-phenyl ring into new Myc inhibitor JY-3-094 is anticipated to afford a further improvement in Myc inhibitory activity through restoration of interactions with the hydrophobic pocket created by Phe375 and Ile381. In fact, the incorporation of especially hydrophobic groups to target the aforementioned hydrophobic pocket will likely be facilitated by the ionizable carboxylic acid that promotes the solubility of JY-3-094. This is in contrast to 10074-G5 whose limited solubility restricts its likewise optimization. In order to furnish analogues of JY-3-094 that are more active in cells, we are developing ester pro-drugs of the carboxylic acid function. Our efforts towards the optimization of JY-3-094 shall be reported in due course.
Supplementary Material
Figure 1.

The chemical structure of Myc inhibitor 10074-G5 and its three components that may be readily modulated in an SAR research program.
Acknowledgments
Financial support for this work is gratefully acknowledged from the University of Maryland School of Pharmacy (SF), an American Chemical Society Pre-Doctoral Medicinal Chemistry Fellowship (JLY) and NIH grant R01 CA140624 (EVP).
Footnotes
Experimental procedures and characterization data (1H NMR, 13C NMR and LRMS) of compounds 10074-G5, 3a–3q, 4, 5a–5e and 7.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dang CV. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nesbit CE, Tersak JM, Prochownik EV. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 3.Boxer LM, Dang CV. Oncogene. 2001;20:5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- 4.Gardner LB, Lee LA, Dang CV. In: Encyclopedia of Cancer. 2. Bertino J, editor. Elsevier Science; Burlington, MA: pp. 555–561. [Google Scholar]
- 5.Yap JL, Chauhan J, Jung KY, Chen L, Prochownik EV, Fletcher S. Med Chem Commun. 2012;3:541–550. [Google Scholar]
- 6.Prochownik EV, Vogt PK. Genes Cancer. 2010;1:650–659. doi: 10.1177/1947601910377494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegel R, Naishadham D, Jemal A. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 8.Felsher DW, Bishop JM. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 9.Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. Exp Hematol. 2006;34:1480–1489. doi: 10.1016/j.exphem.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 10.Mo H, Henriksson M. Proc Natl Acad Sci USA. 2006;103:6344–6349. doi: 10.1073/pnas.0601418103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nair SK, Burley SK. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 12.Blackwood EM, Eisenman RN. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 13.Prendergast GC, Lawe D, Ziff EB. Cell. 1991;65:395–407. doi: 10.1016/0092-8674(91)90457-a. [DOI] [PubMed] [Google Scholar]
- 14.Kretzner L, Blackwood EM, Eisenman RN. Nature. 1992;359:426–429. doi: 10.1038/359426a0. [DOI] [PubMed] [Google Scholar]
- 15.Berg T, Cohen SB, Desharnis J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, Vogt PK. Proc Natl Acad Sci USA. 2002;99:3830–3835. doi: 10.1073/pnas.062036999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin X, Giap C, Lazo JS, Prochownik EV. Oncogene. 2003;22:6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. Bioorg Med Chem. 2006;14:2660–2673. doi: 10.1016/j.bmc.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 18.Kiessling A, Sperl B, Hollis A, Eick D, Berg T. Chem Biol. 2006;13:745–751. doi: 10.1016/j.chembiol.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Kiessling A, Wiesinger R, Sperl B, Berg T. ChemMedChem. 2007;2:627–630. doi: 10.1002/cmdc.200600294. [DOI] [PubMed] [Google Scholar]
- 20.Boger DL, Lee JK, Goldberg J, Jin Q. J Org Chem. 2000;65:1467–1474. doi: 10.1021/jo9916481. [DOI] [PubMed] [Google Scholar]
- 21.Shi J, Stover JS, Whitby LR, Vogt PK, Boger DL. Bioorg Med Chem Lett. 2009;19:6038–6041. doi: 10.1016/j.bmcl.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. J Pharmacol Exp Ther. 2010;335:715–727. doi: 10.1124/jpet.110.170555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo J, Parise RA, Joseph E, Egorin MJ, Lazo JS, Prochownik EV, Eiseman JL. Cancer Chemother Pharmacol. 2009;63:615–625. doi: 10.1007/s00280-008-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Chem Biol. 2008;15:1149–1155. doi: 10.1016/j.chembiol.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 25.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. J Am Chem Soc. 2009;131:7390–7401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- 26.Michel J, Cuchillo R. PLoS One. 2012;7:e41070. doi: 10.1371/journal.pone.0041070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, Prochownik Mol Cancer Ther. 2007;6:2399–2408. doi: 10.1158/1535-7163.MCT-07-0005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.