

Endotoxin-induced neutrophil recruitment in humans and its potential regulation by CXCL8 clearance.
Keywords: Toll-like receptor, endotoxin, neutrophil elastase, Interleukin-8, intradermal
Abstract
This study examined the establishment of neutrophilic inflammation in humans. We tested the hypotheses that neutrophil recruitment was associated with local CXCL8 production and that neutrophils themselves might contribute to the regulation of the size of the inflammatory response. Humans were challenged i.d. with endotoxin. Biopsies of these sites were examined for cytokine production and leukocyte recruitment by qPCR and IHC. Additional in vitro models of inflammation examined the ability of neutrophils to produce and sequester cytokines relevant to neutrophilic inflammation. i.d. challenge with 15 ng of a TLR4-selective endotoxin caused a local inflammatory response, in which 1% of the total biopsy area stained positive for neutrophils at 6 h, correlating with 100-fold up-regulation in local CXCL8 mRNA generation. Neutrophils themselves were the major source of the early cytokine IL-1β. In vitro, neutrophils mediated CXCL8 but not IL-1β clearance (>90% clearance of ≤2 nM CXCL8 over 24 h). CXCL8 clearance was at least partially receptor-dependent and modified by inflammatory context, preserved in models of viral infection but reduced in models of bacterial infection. In conclusion, in a human inflammatory model, neutrophils are rapidly recruited and may regulate the size and outcome of the inflammatory response through the uptake and release of cytokines and chemokines in patterns dependent on the underlying inflammatory stimulus.
Introduction
Neutrophilic inflammation is usually essential in the control of established bacterial infection but is also an important component of numerous disease processes in many organs. Neutrophil trafficking to inflamed tissues is generally held to be mediated through the local production of chemokines of the CXC family, whose production is directly induced by stimuli such as LPS or lies downstream of early proinflammatory cytokines such as IL-1. However, other patterns of mediator generation causing neutrophil recruitment have been described in animals, such as complement activation preceding chemokine generation [1] or leukotriene production causing tissue neutrophilia [2].
Neutrophil recruitment is tightly regulated and is rapidly terminated before the inflammatory response has resolved and even before maximal activation of neutrophils has been achieved [3, 4]. Mechanisms causing defervescence of the inflammatory site are relatively well explored and include induction of apoptosis and production of anti-inflammatory resolvins and cytokines. These key components of inflammatory resolution are later events in the life of an inflammatory focus and do not immediately explain how the rapid termination of neutrophil recruitment occurs. The mechanisms matching levels of recruitment to the size of the insult remain poorly understood.
Using in vitro models, we demonstrated previously that induction of CXCL8 in response to bacterial stimuli was best mediated when tissue cells were cocultured with monocytes in a network where monocytes initiated responses through IL-1 production [5–7]. Here, we sought to establish a human model to investigate the relationship between IL-1 and CXCL8 production and neutrophil recruitment in response to endotoxin and used in vitro models to study how the magnitude of neutrophil recruitment may be determined.
MATERIALS AND METHODS
Ethics
Studies were approved by the Sheffield Research Ethics Committee (UK) and required informed written consent in accordance with the Declaration of Helsinki. Inclusion criteria for the i.d. study were healthy volunteers of either sex and any race, aged 18–50. The i.d. endotoxin study was overseen by a data-monitoring group, independent of the principal investigators. Experiments using mice were approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore (USA). Mice were killed prior to carrying out peritoneal lavage to collect primary macrophages.
Reagents
poly(I:C) was from Invivogen (San Diego, CA, USA). General lab reagents were from Sigma-Aldrich (Poole, Dorset, UK). Cell culture reagents were from Invitrogen (Paisley, Renfrewshire, UK).
i.d. endotoxin challenge
A total of 13 healthy volunteers received four to six i.d. injections (one of saline; the remainder of endotoxin) in the volar surfaces of their forearms at the start of the study. The endotoxin was Escherichia coli strain O:113 {NIH reference stock for clinical trials (Bethesda, MD, USA); a kind gift of Dr. Anthony Suffredini [8]}. The study was divided into an ascending dose phase in eight individuals studied sequentially to safely establish optimal dosing and a second phase using a standardized protocol in five individuals to achieve statistically testable data. Skin sites were measured for area of redness, capturing the outline with sticky tape and quantifying the area using NIH ImageJ. Relevant physiological parameters (blood pressure, temperature, pulse rate) were measured regularly and blood sampled for CRP, hematological parameters, and cytokine analysis. At indicated time-points, the skin sites were anesthetized by the s.c. injection of 1% lignocaine containing adrenaline and 4 mm punch biopsies taken. Biopsies were divided and one-half of each placed into formaldehyde or RNAlater (Ambion, Austin, TX, USA).
IHC
Sections (5 μm) were stained with antibodies to neutrophil elastase (Dako, Glostrup, Denmark) and IL-1β (Abcam, Cambridge, UK). Binding was detected using a Vectastain Elite ABC kit (Vector Laboratories, Peterborough, UK) and a Vector NovaRed substrate kit. Sections were counterstained with hematoxylin, dehydrated to xylene, and mounted in DPX. Staining was analyzed using the Dako Automated Cellular Imaging System (ACIS) III automated image acquisition and analysis system. Thresholds of detection were chosen for each antibody after examination of initial samples, such that all dense staining cells were identified without identification of nonstaining cells (judged by investigators A.B. and L.B.).
qPCR
RNA was extracted from skin biopsies using a TissueRuptor (Qiagen, Crawley, UK) and the RNeasy Lipid Tissue Mini Kit (Qiagen). cDNA was synthesized using the high-capacity cDNA RT kit (Applied Biosystems, Paisley, UK).
qPCR was carried out using assay probe sets specific for IL-1α, IL-1β, and CXCL8 (Applied Biosystems), and reaction mixtures were prepared as described previously [9]. Reactions were carried out using an ABI7900 Automated TaqMan (Applied Biosystems), and absolute copy number was quantified against standards of plasmids (SourceBioscience, Nottingham, UK) containing the target sequences to be amplified.
Leukocyte preparation
Highly purified human neutrophils were prepared from the venous blood of healthy volunteers by density gradient centrifugation and negative magnetic selection using a custom cocktail from Stem Cell Technologies (Vancouver, Canada) as described [10]. Monocytes were prepared from PBMC fractions by negative magnetic selection using the Monocyte Isolation Kit II (Miltenyi Biotec, Audburn, CA, USA) as described [9]. Neutrophils and monocytes were cocultured with BEAS-2B epithelial cells or cultured in isolation in tissue-culture plates, with or without the addition of indicated cytokines (PeproTech EC, London, UK).
To obtain primary murine peritoneal macrophages, WT C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME, USA) or TLR4−/− mice (kindly provided by Dr. S. Akira, Osaka, Japan; bred at the University of Maryland, Baltimore) were injected i.p. with thioglycollate broth as described [11]. Four days later, peritoneal exudates were collected by lavage, washed, and resuspended in RPMI 1640/2% FBS as described [11]. Cells were plated at 2 × 106 cells/ml in 12-well plates and incubated overnight at 37°C. Wells were washed to remove nonadherent cell types and the medium replaced with medium only, NIH endotoxin, or the TLR4-specific positive control, E. coli K235 LPS [12, 13]. Macrophage cultures were incubated for 2 h and RNA prepared for analysis of specific cytokine mRNA by qPCR as described [11].
Cell lines
BEAS-2B epithelial cells were maintained in RPMI/10% FCS. Cocultures of leukocytes and tissue cells were carried out as described previously [6, 9, 14]. Media were harvested for cytokine analysis.
Cytokine, leukocyte, and serological analysis
Cytokines in supernatants were measured by matched antibody pair ELISAs (R&D Systems, Abingdon, UK) at optimized concentrations and previously noted LDs [9]. Data were analyzed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). Samples whose cytokine levels were below the LD were assigned the LD value for analysis. Cytokine levels in serum from volunteers in the i.d. endotoxin study were measured by sensitive multiplex immunoassays (Meso Scale Discovery, Gaithersburg, MD, USA). Cortisol levels were measured using routine clinical analyzers and protocols in a NHS clinical laboratory, as were full blood counts.
Western blotting
Cell lysates were prepared from 0.5 × 106 neutrophils/point and analyzed as described [15]. Blots were probed with anti-IL-1β (R&D Systems) and anti-p38 (Cell Signaling Technology, Hitchin, UK) and an appropriate secondary antibody. Amersham ECL Western blotting detection reagents (Amersham Pharmacia Biotech, Little Chalfont, UK) were used. Images were scanned using a Bio-Rad ChemiDoc XRS+ (Hemel Hempstead, UK) and quantified using ImageLab V3.0.
Statistics
Data were analyzed using GraphPad Prism v5, with the tests indicated in each figure legend, assuming P < 0.05 as demonstrating statistical significance.
RESULTS
Human i.d. endotoxin challenge
A preparation of E. coli O:113 endotoxic LPS for use in clinical studies was obtained [8]. Its preparation had been prior to the realization that low amounts of bacterial lipopeptide commonly present in endotoxin preparations deliver a TLR2-activating signal in addition to the TLR4-activating LPS signal [12]. We examined whether this NIH preparation to be used in the clinical study activated TLR4 or TLR2 and TLR4 and compared it with a highly purified preparation of E. coli K235 LPS that lacks TLR2 stimulatory activity [12]. Macrophage cultures were prepared from WT mice or mice with a targeted mutation in TLR4 (TLR4−/−). Both LPS species induced TNF-α (MyD88-dependent), IFN-β, and CCL5 (MyD88-independent) mRNA in WT macrophages, but no cytokine mRNA expression was detected in TLR4−/− macrophages (Fig. 1), showing that the NIH clinical endotoxin preparation specifically activated TLR4.
Figure 1. TLR dependence of endotoxin.

Macrophages from WT or TLR4−/− mice were pooled from peritoneal lavages and treated with the indicated concentrations of NIH LPS (N LPS) used in the human challenge studies or a purified laboratory endotoxin known to activate TLR4 alone (C LPS). Samples were cultured for 2 h, and generation of mRNAs for the indicated cytokines was measured by qPCR. (A) TNF-α; (B) IFN-β; (C) CCL5. Cells were pooled from multiple mice (typically five/group), and data shown were derived from a representative experiment.
The experimental examination of human skin responses to endotoxin has only been performed rarely and lastly, as far as we could determine, prior to the discovery of cytokines and TLRs [16]. Thus, we initially studied eight volunteers in an ascending dose-phase protocol with intervals for assessment of cellular responses, safety, and tolerability. Alexander Basran was trained in i.d. injections and skin biopsies. During this phase, volunteers received endotoxin doses from 10 pg–15 ng/site, with total doses ranging from 1.1 ng to 45 ng/volunteer. Biopsies were taken at various predetermined time-points to observe recruitment kinetics, mostly within 6 h of endotoxin administration, although in two subjects, biopsies were taken at 24 h. Blood pressure, pulse, and temperature were monitored routinely at regular intervals during the study. Sutures were removed by the study team at 8–10 days. Subjects were encouraged to report any adverse events. A monitoring group independent of the investigators oversaw progress. No adverse effects deemed to be related to systemic effects of endotoxin were observed in any volunteer. CRP did not rise in any volunteer but was only monitored until the last biopsy. CRP has a relative lag in its response. The two volunteers who had biopsies and blood sampling at 24 h and received a total dose of 45 ng endotoxin each also showed no rise in CRP at this later time-point. The highest endotoxin dose tested, 15 ng/site, caused painless erythema after 1–2 h with mild edema. Based on these studies, five further volunteers received a standardized regime of one saline injection and two injections each of 5 ng and 15 ng endotoxin. For reasons of clarity and brevity, data are henceforward shown for the n = 5 only, with data held on file for all of those in the ascending dose phase. Biopsies (one of each endotoxin dose) were taken at 2 and 6 h. The saline site was biopsied at 6 h. Table 1 shows the physiological parameters monitored. No significant adverse effects of endotoxin challenge were observed. Figure 2 shows representative photographs of the inflammatory lesions in one subject and mean data for erythema areas obtained from the standardized n = 5.
Table 1. Individual Monitoring Parameters.
| Volunteer code | Time (h) | 0 | 1 | 6 |
|---|---|---|---|---|
| S1 | Leukocytes (109/l) | 5.8 | 6.3 | 8.0 |
| Neutrophils (109/l) | 3.5 | 4.4 | 5.7 | |
| CRP (mg/l) | 3.6 | NA | 4.4 | |
| Cortisol (nM) | 617 | NA | 320 | |
| Blood pressure (mmHg, systolic/diastolic) | 136/84 | 137/77 | 136/82 | |
| Heart rate (per min) | 68 | 67 | 69 | |
| Temperature (°C) | 36.9 | 36.7 | 36.9 | |
| J1 | Leukocytes (109/l) | 7.7 | 8.7 | 11.2 |
| Neutrophils (109/l) | 5.1 | 5.6 | 7.8 | |
| CRP (mg/l) | 7.4 | 7.2 | 6.3 | |
| Cortisol (nM) | 387 | NA | 184 | |
| Blood pressure (mmHg, systolic/diastolic) | 138/86 | 129/75 | 126/70 | |
| Heart rate (per min) | 90 | 74 | 72 | |
| Temperature (°C) | 36.7 | 36.8 | 36.6 | |
| A2 | Leukocytes (109/l) | 3.8 | 4.4 | 9.5 |
| Neutrophils (109/l) | 1.9 | 2.4 | 7.8 | |
| CRP (mg/l) | 5 | 5.1 | 4 | |
| Cortisol (nM) | 372 | NA | 259 | |
| Blood pressure (mmHg, systolic/diastolic) | 129/92 | 125/69 | 113/69 | |
| Heart rate (per min) | 67 | 62 | 61 | |
| Temperature (°C) | 35.8 | 35.9 | 36.5 | |
| Q1 | Leukocytes (109/l) | 6.1 | 7.7 | 8.3 |
| Neutrophils (109/l) | 5.1 | 6.4 | 6.3 | |
| CRP (mg/l) | 1.9 | 2.1 | 2 | |
| Cortisol (nM) | NA | NA | NA | |
| Blood pressure (mmHg, systolic/diastolic) | 115/69 | 114/65 | 118/69 | |
| Heart rate (per min) | 57 | 55 | 61 | |
| Temperature (°C) | 36.8 | 36.7 | 36.9 | |
| L4 | Leukocytes (109/l) | 3.6 | 3.7 | NA |
| Neutrophils (109/l) | 2.1 | 2.1 | NA | |
| CRP (mg/l) | 0.3 | 0.3 | 0.3 | |
| Cortisol (nM) | 311 | NA | 202 | |
| Blood pressure (mmHg, systolic/diastolic) | 121/65 | 117/69 | 116/66 | |
| Heart rate (per min) | 67 | 56 | 57 | |
| Temperature (°C) | 35.7 | 36.4 | 36.9 |
Individual monitoring parameters for five volunteers (each recorded by an anonymized case identificiation number) receiving i.d. endotoxin in the standardized endotoxin model. 0, Baseline observations prior to study start; 1, 1 h after endotoxin administration; 6, 6 h after endotoxin administration; NA, data not available at that point.
Figure 2. Inflammatory lesions induced by endotoxin.

The figure shows representative images from one volunteer of inflammation induced by endotoxin. Saline or endotoxin (5 ng) sites are shown at 2 or 6 h as indicated. (A) Saline-injected site at 2 h; (B) 5 ng LPS-injected site at 2 h; (C) LPS 5 ng-injected site at 6 h. Ballpoint lines indicate erythema as demarcated by the operator. Injection sites are marked by ballpoint arrows on the skin. (D) Mean areas of erythema (expressed as mm2) at 2 and 6 h after endotoxin injection are shown on the accompanying graph. In this graph, areas of erythema at 2 h were averaged within each volunteer from two sites at each endotoxin dose, whereas at 6 h, data from a single site remained as the other had been biopsied. Data displayed are n = 5; mean ± sem.
i.d. endotoxin induces tissue and blood neutrophilia
Endotoxin, but not saline, induced rapid recruitment of cells staining positively for the neutrophil marker, NE, with the typical PMN appearance of neutrophils (Fig. 3). As neutrophil recruitment occurred focally across the biopsies, sections were quantified by automated analysis, where the staining of NE was quantified on the entire section of each of two separate sections/biopsy and averaged within and then between donors. We observed significant time-dependent neutrophil recruitment in response to the 15 ng/site dose (Fig. 3). The only detectable systemic response to i.d. endotoxin was a variable blood neutrophilia (Table 1). Serum cortisol declined in each volunteer over the course of the day (Table 1), in keeping with natural diurnal variation in cortisol levels (injections were performed in the morning) and providing further evidence of a lack of a generalized systemic reaction.
Figure 3. Neutrophil recruitment in response to i.d. endotoxin.

Volunteers received i.d. injections of saline or endotoxin (5 or 15 ng/site), and sites were biopsied at 2 or 6 h postinjection (6 h postinjection for the single-saline site). Biopsies were stained for NE expression to measure neutrophil recruitment. Illustrative staining of skin sites from a single volunteer are shown. Left panels show representative photographs of staining at ×200 magnification; right panels show representative photographs taken at ×600 magnification. The identity of positive-staining cells as neutrophils is verified by their PMN morphology on the cropped, enlarged inset image (Panel L). Original scale bars indicate 100 μm distances (×200) and 50 μm distances (×600). Two sections from each biopsy were quantified by automated image analysis, calculating the percentage of NE staining across the whole section. Data from the two sections were averaged for each subject and then the mean of n = 5 calculated and displayed in K as mean ± sem. The existence of a significant dose-dependent response was tested by analysis using ANOVA with Bonferroni's post-test for selected comparisons, with significant difference between the indicated pair shown by *P < 0.05.
Cytokine generation associated with endotoxin challenge
As i.d. endotoxin challenge was associated with tissue and blood neutrophilia, we determined the patterns of cytokine generation associated with these responses. Sera from the five donors who received 40 ng total endotoxin each in the standardized protocol were analyzed by sensitive multiplex immunoassays. No significant changes in IL-1β, TNF-α, IL-6, CXCL8, or G-CSF were seen in serum (Fig. 4). In contrast, analysis of gene expression in skin biopsies by qPCR revealed a rapid local generation of CXCL8 mRNA, which peaked at 2 h (P<0.01 for 5 ng dose and<0.001 for 15 ng dose) and had diminished, although not resolved, by 6 h. Expression of IL-1α and IL-1β (classical early cytokines associated with induction of inflammation) was also evident (Fig. 5). IL-1α expression was only increased significantly at the 6-h time-point for the 15-ng dose. IL-1β expression was increased significantly at 2 h (5 ng, P<0.05; 15 ng, P<0.01) and 6 h (15 ng, P<0.05). Biopsies were stained for production of IL-1β by IHC (Fig. 6). IL-1β protein expression appeared over a similar time course to its mRNA expression, coinciding with the arrival of neutrophils in the tissue. IL-1β-producing cells showed the clear nuclear features of neutrophils, although analysis of the IHC by visual inspection and automated analysis demonstrates that IL-1β protein production by neutrophils was variable. Changes measured by this method failed to reach statistical significance (Fig. 6), likely as a consequence of the smaller proportion of cells staining positively in these sections. Thus, although these data suggested clearly that neutrophils themselves are an important source of IL-1 during human inflammation, further studies were required to test this further.
Figure 4. Peripheral blood cytokines after endotoxin challenge.
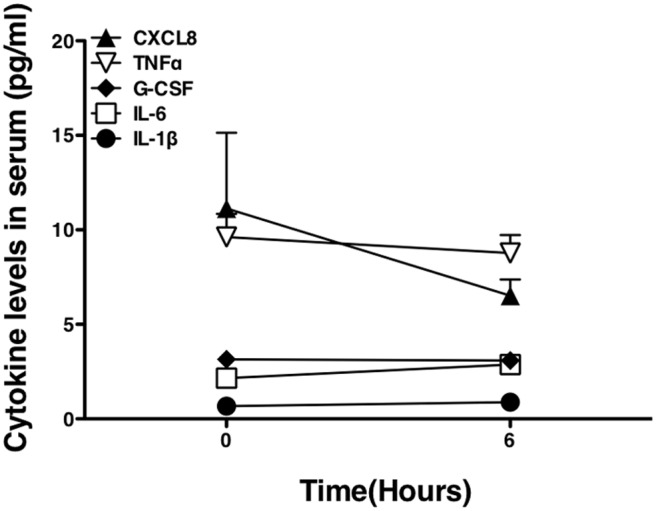
Volunteers who had received i.d. endotoxin (total 40 ng/volunteer) had serum cytokine levels measured by Meso Scale Discovery immunoassay at baseline and 6 h after endotoxin challenge. Data shown are from four of the five volunteers in the standardized component of the challenge protocol, displayed as mean ± sem; samples from one volunteer were not available for analysis.
Figure 5. i.d. cytokine generation after endotoxin challenge.

Volunteers received i.d. injections of saline or endotoxin (5 or 15 ng/site), and sites were biopsied at 2 or 6 h postinjection (6 h postinjection for the single-saline site). Cytokine generation in the skin sites was measured by qPCR and presented as copies/μg total RNA isolated from biopsies. (A) CXCL8; (B) IL-1α; (C) IL-1β. Data are from four of the five volunteers in the standardized component of the challenge protocol. Data displayed are mean ± sem and were analyzed by ANOVA with Tukey's post-test, *P < 0.05; **P < 0.01; ***P < 0.001, displaying significant differences in expression versus the Time 0 control.
Figure 6. Expression of IL-1β in response to i.d. endotoxin.

Volunteers received i.d. injections of saline or endotoxin (5 or 15 ng/site), and sites were biopsied at 2 or 6 h postinjection (6 h postinjection for the single-saline site). Biopsies were stained for IL-1β expression. Illustrative staining of skin sites from a single volunteer are shown. Left panels show representative photographs of staining at ×200 magnification; right panels show representative photographs taken at ×600 magnification. The identity of positive-staining cells as neutrophils is verified further by their PMN morphology on the cropped, enlarged inset image (Panel L). Original scale bars indicate 100 μm distances (×200) and 50 μm distances (×600). Two sections from each biopsy were quantified by automated image analysis, calculating the percentage of IL-1 staining across the whole section. Data from the two sections were averaged for each subject and then the mean of n = 5, ±sem, calculated and displayed in K. Data are analyzed by ANOVA, but changes observed did not reach statistical significance.
Neutrophils as a source of IL-1β
To confirm the observations that neutrophils may be an important source of IL-1β in inflammation, we prepared highly purified neutrophils by negative magnetic selection and stimulated them with 100 ng/ml LPS for 4 h, a time-point comparable with endotoxin exposure in our skin model. Analysis of the resulting cell lysates revealed that human neutrophils activated with LPS generated pro-IL-1β and processed it to its biologically active, 17-kDa form, in keeping with our observations in the i.d. challenge model (Fig. 7).
Figure 7. Production of IL-1β by neutrophils.

Highly purified human neutrophils from healthy control patients were treated with 100 ng/ml LPS for 4 h. Whole cell lysates were analyzed by Western blot using antibodies specific to IL-1β and total p38 MAPK as a loading control. (A) Data from three independent donors. D1–D3, Days 1–3. The 17-kDa bands, corresponding to cleaved, mature IL-1β, were analyzed by densitometry and displayed in B. Data are presented as mean ± sem of n = 3. Significant differences are indicated by *P < 0.01, as analyzed by paired t test.
Regulation of the intensity of neutrophilic inflammation
Given ongoing production of CXCL8 and IL-1 in response to endotoxin over time courses studied here, we were interested to determine what factors limit neutrophil recruitment so that the infiltration of neutrophils is matched to the size of the initiating inflammatory stimulus.
We examined to what extent neutrophils might, through the well-established, rapid recycling of their principal chemokine receptors CXCR1 and CXCR2 [17, 18], regulate amounts of local CXCL8. Peripheral blood neutrophils were incubated with various concentrations of CXCL8 (chosen so that the minimum was around the Kd of the receptor [17–19].
In Fig. 8A, neutrophils (unprimed or primed with TNF-α) were incubated with varying concentrations of CXCL8 for 24 h. The amount of CXCL8 recoverable in supernatants after this time was measured by ELISA. When CXCL8 was incubated without neutrophils for 24 h in media, it did not show any loss, as measured by ELISA (data not shown). However, where CXCL8 was cultured with neutrophils for 24 h, the amount of CXCL8 remaining in the cultures was reduced markedly, and <25% of even the highest concentration of CXCL8 remained at this time. As tissue neutrophils are exposed to other proinflammatory stimuli that might alter their rates of receptor internalization and recycling, we exposed neutrophils to the priming agents TNF-α or GM-CSF (at doses found in separate experiments to prime respiratory burst; data not shown) for 1 h prior to and continuing during the culture with CXCL8. Exposure to either of these cytokines individually did not have any effect on the rates of CXCL8 uptake by neutrophils over the time course examined (Fig. 8). Neutrophils express CXCL8 receptors, CXCR1 and CXCR2. Clearance of CXCL8 was significantly, although not completely, inhibited by preincubation of neutrophils with blocking mAb to CXCR1 and CXCR2 in combination (Fig. 8). A further hallmark of tissue inflammation is local hypoxia at the site of injury, which is a profound modulator of neutrophil function [20]. Exposure of neutrophils to hypoxia (3 kPa pO2) did not modulate rates of CXCL8 clearance (Fig. 8).
Figure 8. Uptake of CXCL8 by neutrophils.

Highly purified human neutrophils (0.5×106/point) were prepared and incubated with various concentrations of CXCL8 for 24 h, after which, remaining CXCL8 was quantified by ELISA. Incubation of CXCL8 in media alone without neutrophils for 24 h did not reduce amounts of recoverable CXCL8 (data not shown). (A and B) Neutrophils were also primed with 10 ng/ml TNF-α (A) or 50 U/ml GM-CSF (B) for 1 h prior to addition of CXCL8 for 24 h (the priming agents remained present throughout the time course). (C) Neutrophils were preincubated with media, a control IgG, or blocking mAb to CXCR1- or CXCR2-neutralizing antibodies, alone or in combination for 30 min, followed by the addition of CXCL8 (8.3 nM) for 24 h. (D) Neutrophils were cultured with CXCL8 at the indicated concentrations under normoxic or hypoxic (pO2 3 kPa) conditions for 1, 4, and 24 h. In all experiments, at the required time-points, cell-free supernatants were generated and CXCL8 levels measured by ELISA. Data are presented as percentage CXCL8 remaining in the supernatant and are mean ± sem (n=4, A and B; n=3–5, C and D), with each replicate performed with freshly prepared neutrophils from independent donors. (C) Data were analyzed by one-way ANOVA and Tukey's post-test. Significant differences are indicated by *P < 0.05.
We next examined the potential ability of neutrophils to mediate clearance of CXCL8 in well-characterized models of airway inflammation established in our laboratory. In the first model, epithelial cells were stimulated with the viral RNA mimic, poly(I:C), in the presence of increasing numbers of neutrophils. poly(I:C) causes the epithelial cells to generate large amounts of CXCL8 and other cytokines [6]. Increasing numbers of neutrophils added to the culture resulted in a marked decrease in the amount of detectable CXCL8 after 24 h (Fig. 9). These data indicate that CXCL8 uptake by neutrophils arriving at sites of viral infection has the potential to markedly limit cytokine available for ongoing neutrophil recruitment.
Figure 9. Clearance of CXCL8 from models of inflammation.

BEAS-2B epithelial cells were grown to 95% confluence in 24-well plates. In a model of viral inflammation (A), cells were stimulated with poly(I:C) at 10 μg/ml in the absence or presence of neutrophils at the indicated numbers for 24 h. Cell-free supernatants were generated, and CXCL8 in the supernatants was measured by ELISA. Data shown are mean ± sem (n=3), with each replicate performed on separate passages of BEAS-2B cells with freshly prepared neutrophils from independent donors. In a model of bacterial infection (B), BEAS-2B epithelial cells were cultured alone or cocultured with CD14+ monocytes (monos). Cultures were stimulated with LPS (1 ng/ml) in the presence of the indicated numbers of neutrophils for 24 h. Cell-free supernatants were generated, and CXCL8 release was measured by ELISA. Data shown are mean ± sem (n=6), with each replicate performed on separate passages of BEAS-2B cells with freshly prepared monocytes and neutrophils from independent donors. Data were analyzed by one-way ANOVA with Dunnett's post-test, with significant differences indicated by *P < 0.05; **P < 0.01; and ***P < 0.001.
Other data have suggested that neutrophils may take up and degrade IL-1β via expression of the decoy receptor IL-1R2 [21], which could limit inflammatory responses. We found that in vitro, neutrophils did not take up detectable amounts of IL-1β (data not shown). We therefore studied a more complex in vitro inflammatory model with a marked IL-1 dependence, modeling aspects of the initiation of response to bacterial stimuli. BEAS-2B epithelial cells were cocultured with purified human CD14+ monocytes and LPS. In this model of endotoxin-induced inflammation, production of CXCL8 by BEAS-2B cells is dependent on a network in which LPS activates the monocytes, releasing IL-1β, which in turn, induces CXCL8 production from the epithelial cells [22]. Addition of neutrophils to this coculture again resulted in a reduction in CXCL8 levels measured at 24 h, which was dependent on the numbers of neutrophils added. In contrast to the model of viral infection, CXCL8 levels were not reduced to baseline (Fig. 9).
At sites of severe inflammation, e.g., in the ARDS, multiple cytokines are generated and influence disease [23, 24]. Tissue neutrophils might also express other chemokine receptors, such as CCR2 [25]. We therefore exposed neutrophils to a mix of cytokines, CXCL8, CCL2 (the principal ligand for CCR2), and IL-1β, and for some cells, pretreated them with a combination of TNF-α and GM-CSF to mimic the activation stimuli experienced in sites of severe inflammation. In the absence of TNF-α and GM-CSF, peripheral blood neutrophils cleared CXCL8 (Fig. 10) from these cytokine mixes, without reducing levels of CCL2 (Fig. 10) or IL-1β (data not shown). Combined exposure to TNF-α and GM-CSF completely inhibited neutrophil uptake of CXCL8, without activating uptake of CCL2 (Fig. 10) or IL-1β (data not shown), suggesting that in some circumstances, neutrophil-mediated CXCL8 clearance may fail to contain inflammatory responses.
Figure 10. Clearance of cytokines from mixed cultures.

Highly purified human neutrophils (0.5×106/point) were obtained and pretreated with media or TNF-α (10 ng/ml) and GM-CSF (50 U/ml) in combination for 1 h. Following this, treated neutrophils were incubated with increasing concentrations of CXCL8, MCP-1, and IL-1β in combination for 24 h (in the ongoing presence of TNF-α and GM-CSF if added). Cell-free supernatants were generated, and CXCL8 (A), CCL2 (B), and IL-1β (data not shown) levels were measured by ELISA. Data are presented as percentage of CXCL8 or CCL2 remaining in the supernatant and are mean ± sem (n=3), with each replicate performed with freshly prepared neutrophils from independent donors. Data were analyzed by two-way ANOVA and Bonferroni's post-test. Significant differences are indicated by **P < 0.01.
We considered whether release of ROS or chlorinating species might modulate neutrophil uptake of CXCL8 or make CXCL8 inaccessible to the ELISA through modification of the proteins. We therefore incubated neutrophils with the NADPH oxidase inhibitor DPI prior to incubation with CXCL8 or TNF-α and CXCL8. The data in Fig. 11 show that DPI pretreatment of neutrophils resulted in modest increases in detectable remaining CXCL8 at the end of the experimental period; however, this effect of DPI was only significant at one point and was not more evident in the cells treated with TNF-α.
Figure 11. Effects of NADPH oxidase inhibition.

Highly purified human neutrophils (0.5×106/point) were obtained and pretreated with media or TNF-α (10 ng/ml) and/or DPI (10 μM or 20 μM) for 1 h. Following this, treated neutrophils were incubated with CXCL8 at the indicated concentrations for 24 h (in the ongoing presence of TNF-α and/or DPI). Cell-free supernatants were generated, and CXCL8 levels were measured by ELISA. Data are presented as percentage of CXCL8 remaining in the supernatant calculated against the amount of CXCL8 added at the start of the experiment and are mean ± sem (n=3), with each replicate performed with freshly prepared neutrophils from independent donors. Data were analyzed by ANOVA with Bonferroni's post-test for selected comparisons as illustrated on the figure. Significant differences are indicated by *P < 0.05; NS, no significant difference detected.
DISCUSSION
Considerable strides have been made describing the regulation of leukocyte recruitment in animal models, but there are little data examining the time course and mechanisms of neutrophil recruitment in the human. Additionally, whereas the positive stimuli regulating recruitment are becoming increasingly clear, leukocyte recruitment at inflammatory sites is often rapidly terminated [3], and the mechanisms responsible for the termination of recruitment of further cells to an active inflammatory site are poorly understood.
We studied neutrophil recruitment induced in a skin model where dose and time dependence could be investigated. We determined that a preparation of endotoxin made for human studies [8] activates TLR4 exclusively. i.d. injection of endotoxin induced a rapid, limited local inflammatory reaction, with visible erythema preceding infiltration of neutrophils, and CXCL8 mRNA generation was maximal at the earliest time-point examined and diminished but not resolved over a 6-h time course. In total, 13 volunteers received i.d. endotoxin at various doses, with no significant adverse events recorded. The dose chosen for the n = 5 study group of 40 ng/volunteer total endotoxin corresponds to the amount of LPS obtained from ∼16 × 106 E. coli [26].
Although total areas of inflammation were small, a number of volunteers showed a rapid blood neutrophilia, demonstrating a remarkably sensitive ability of the innate immune system to ready the human organism when bacterial infection may be present. A search for mediators responsible for this blood neutrophilia in serum using sensitive immunoassays failed to identify a likely candidate. Neutrophil recruitment was associated with local generation of CXCL8. We were intrigued about the nature of mechanisms matching neutrophil recruitment to the magnitude of the inflammatory stimulus. One mechanism would be a graded induction of chemokines such as CXCL8, with small stimuli producing little chemokine and large stimuli causing the production of large amounts of mediators. However, seminal work in rabbit models of pneumonia indicates that neutrophil recruitment occurs early in the pathology and is terminated before maximal activation is seen at the inflammatory site [3, 4]. Induction of proresolution environments through resolvin expression [27] or neutrophil apoptosis [28] would be unlikely to be active during initiation of recruitment and would be poor pathways to regulate early recruitment, as they would inhibit subsequent effective neutrophil activation. The simplest regulatory system would be if neutrophils themselves could sense the size of the inflammatory response and terminate ongoing cell recruitment by receptor-mediated clearance of CXCL8. We observed that neutrophils were able to mediate a sustained clearance of CXCL8 from their surroundings. Blockade of CXCR1 and CXCR2 inhibited CXCL8 clearance significantly but incompletely. Thus, ligand clearance is at least in part receptor-dependent, despite the well-characterized, rapid process of receptor desensitization, that down-regulates internalization of ligand-exposed CXCR1 and CXCR2 [29–31]. Neutrophils express 20,000–50,000 CXCR1/2 molecules/cell [17], and exposure of 0.5 million neutrophils to 8 nM CXCL8 in the experimental volume studied represented ∼100 molecules of CXCL8/receptor. This scavenging system was selective, with no removal of CCL2 from media by neutrophils. The failure of CXCR1/2 blockade to inhibit CXCL8 clearance completely could suggest that neutrophil protease release is also occurring and potentially breaking down CXCL8, although our experiments were conducted in 10% FCS, which contains many potent antiproteases. We considered whether products of NADPH oxidase might modulate neutrophil CXCL8 uptake through regulation of neutrophil priming [32] or make CXCL8 inaccessible to ELISA through covalent modification. The NADPH oxidase inhibitor DPI, at the top concentration tested at 20 μM, modestly reduced the measured CXCL8 loss seen when CXCL8 was incubated with unprimed neutrophils. However, the ability to clear CXCL8 from our cultures of TNF-α-primed neutrophils was not significantly susceptible to NADPH oxidase inhibition, despite the fact that TNF-α treatment might be expected to up-regulate NADPH oxidase activity. We believe overall that it is most likely that the failure of CXCR1/2 blockade to fully neutralize CXCL8 clearance therefore reflects incomplete blockade of all cell surface receptors, but it remains possible that other mechanisms can contribute to neutrophil-driven CXCL8 clearance.
Our data showed that CXCL8 clearance is robust and not inhibited by single priming agents or by hypoxia (a potent neutrophil survival factor at inflammatory loci) [20, 33]. These data are striking, given that hypoxia/reperfusion regulates CXCR1 expression [34], and GM-CSF and TNF-α can each regulate CXCR1/2 expression [35–37]. TNF-α- and GM-CSF-induced IL-8R down-regulation is potentiated by endotoxin [35] and for TNF-α, involves mechanisms, including metalloproteinase-mediated receptor shedding [36]. Therefore, despite the potential for CXCL8 receptor down-regulation in inflammatory environments, enough residual receptor function remained in these experiments to clear substantial quantities of CXCL8. Consistent with this, CXCR1 function is maintained in neutrophils from patients with sepsis [38]. However, treatment of neutrophils with combinations of priming agents caused cessation of CXCL8 clearance. It is most likely that this reflects enhanced down-regulation of CXCR1/2 surface expression and function through the mechanisms noted above.
These data generate two further hypotheses. The first is that simple inflammatory networks can regulate the way the neutrophil behaves with respect to modulating further cellular recruitment. Neutrophils have limited efficacy against viruses yet are commonly recruited to the lungs with respiratory virus infection. In our model of airways viral infection, neutrophils rapidly cleared CXCL8 and might thus be expected to limit neutrophil recruitment in a setting in which the neutrophil has an uncertain role. However, in our model of bacterial airways infection, neutrophil-mediated CXCL8 clearance was less effective. This may be important in vivo, as CXCL8 activates bacterial killing by neutrophils [39], and persistent CXCL8 levels are likely to enable a greater total neutrophil recruitment to bacterial infection. Limitations on CXCL8 clearance in bacterial infections may be mediated by direct actions of endotoxin on rates of CXCR1/2 recycling [35], indirect actions of monocyte-derived cytokines [10], or release of excess neutrophil proteases that can cleave CXCR1 [39]. In our skin model, some punctate noncell-associated NE staining was observed in challenged sites. This is consistent with degranulation of neutrophils in response to diffuse local exposure to LPS, and therefore, even in this resolving model of limited inflammation, there is potential for NE to modify local CXCL8 clearance through CXCR1 cleavage. Other mechanisms exist that will also contribute to regulation of cell recruitment. These include the temporal regulation of selectin expression at inflammatory sites [40] and the clearance of chemokines in the circulation and lymphatics, which our in vitro models cannot measure, but our data suggest that neutrophil-mediated clearance of CXCL8 has the potential to be an important regulator of inflammation in vivo. A second hypothesis generated by our observations is that persistent neutrophilic inflammatory disease may at least in part represent a failure of tissue neutrophils to regulate local CXCL8 levels, consequent upon excessive local generation of proinflammatory mediators, which are well-defined features of conditions such as ARDS [23].
We have shown previously that responses of tissue cells to LPS are markedly amplified in the presence of monocytes through their generation of local IL-1β [5, 6, 14, 22], findings that are relevant to animal models of lung [41] and skin inflammation [42, 43]. In our human model, expression of IL-1β correlated with appearance of neutrophils in the skin, IL-1β-expressing cells showed the typical nuclear morphology of neutrophils, and we found highly purified neutrophils could produce and process IL-1β in vitro. Expression of IL-1β mRNA has also been observed in neutrophils recruiting in skin windows to nonmicrobial chemoattractants [44]. A central role for IL-1 in the initiation of inflammation is well established; normally, the presence of neutrophils is taken to indicate that this process is well underway, but these data indicate that in the human, the neutrophil may itself have the potential to amplify inflammation. This is consistent with recent work in a mouse arthritis model, where neutrophils express IL-1 and deliver this to the joint [2], and this may be essential in the later recruitment of monocytes/macrophages to the inflammatory site [45]. Our in vitro assays showed that quiescent and primed neutrophils failed to reduce the local concentration of IL-1β. This observation contradicts one study which showed that neutrophils could take up ∼50% of a local concentration of radiolabeled IL-1 [21], although we studied highly purified neutrophils, having removed contaminating monocytes that modulate neutrophil responses to IL-1β [46]. These data once again emphasize the potential for independent regulation of components of the neutrophilic inflammatory response, as neutrophils appear to be able to limit local CXCL8 but not IL-1β concentrations by receptor-mediated uptake.
In conclusion, we have shown that neutrophils are rapidly recruited to sites of inflammation in the human. Rapid generation of CXCL8 at sites of inflammation is associated with recruitment of neutrophils, which may themselves be an important source of proinflammatory mediators such as IL-1. We have described mechanisms showing how neutrophil recruitment may be regulated at sites of inflammation that may become dysregulated in disease.
ACKNOWLEDGMENTS
This study was supported by Medical Research Council Research grant G0801983, Asthma UK Research grant 07–12, the Sheffield NIHR Cardiovascular Biomedical Research Unit, a Wellcome Trust Senior Clinical Fellowship (to D.H.D.; Reference 076,945), a Wellcome Intermediate Clinical Fellowship (to S.R.W.; Reference 078,244), and U.S. National Institutes of Health AI18797 (to S.N.V.). We thank all of our volunteers who gave us blood or took part in the i.d. endotoxin challenge model. The authors thank Dr. Keith Hunter and Ms. Yvonne Stephenson for their help with histopathological techniques and analyses. The clinical studies would not have been possible without the nursing and administrative support of the Sheffield NIHR Cardiovascular Biomedical Research Unit (Jane Arnold, Sara Walker, John Humphreys, and Sarah Langridge) and the staff of the Sheffield Clinical Research Facility. We thank Professor Rob Read, Dr. Chris Newman, and Professor Simon Heller for providing independent study oversight of the clinical work. Tristan Dyson provided technical support for experiments involving mouse macrophages. I.S. thanks Dr. Paul Collins and Professor Tim Williams for helpful discussions. We thank Dr. Stephen Morley (Sheffield Teaching Hospitals, NHS Foundation Trust) and the staff of the Biochemistry Lab for measurement of serum cortisol levels. The authors are also grateful to Dr. Anthony Suffredini at U.S. National Institutes of Health for his provision of endotoxin and his foresight in creating this reagent.
SEE CORRESPONDING EDITORIAL ON PAGE 1
- ARDS
- acute respiratory distress syndrome
- CRP
- C-reactive protein
- DPI
- diphenylene iodonium
- IHC
- immunohistochemistry
- LD
- limit of detection
- NE
- neutrophil elastase
- NHS
- National Health Service
- NIHR
- National Institute of Health Research
- pO2
- oxygen partial pressure
- poly(I:C)
- polyinosinic:polycytidylic acid
- qPCR
- quantitative PCR
- TLR4−/− mice
- TLR-deficient mice
AUTHORSHIP
A.B. performed all of the human experimentation and contributed to protocol development. M.J. performed and helped design the experiments using in vitro models of human inflammation, supervised by L.R.P. and L.C.P., who reviewed the data and helped design the experiments. E.C.P. and C.A.S. purified skin mRNA with A.B. and performed and analyzed experiments using qPCR. K.R.H. undertook the in vitro analysis of neutrophil IL-1 production. H.L.W. contributed to the development of the protocols examining IL-1 production and the interpretation of their data. S.R.W. assisted in the design and execution of experiments examining the effects of hypoxia on neutrophil cytokine uptake. S.N.V. designed and was responsible for the experiments examining effects of endotoxin on mouse leukocytes. L.B. supervised and helped perform the IHC and its analysis. R.A.S. supervised all of the clinical procedures, instructed A.B. in skin surgical techniques, and provided expertise in IHC. The i.d. protocol was devised and supervised by I.S., M.K.B.W., D.H.D., L.B., and R.A.S. with clinical responsibility shared by I.S., M.K.B.W., D.H.D., and R.A.S. All of the authors contributed to the development of the ideas in the manuscript. I.S. was the principal author of the manuscript with input from all of the coauthors.
REFERENCES
- 1. Collins P., Jose P., Williams T. (1991) The sequential generation of neutrophil chemoattractant proteins in acute inflammation in the rabbit in vivo. Relationship between C5a and proteins with the characteristics of IL-8/neutrophil-activating protein 1. J. Immunol. 146, 677–684 [PubMed] [Google Scholar]
- 2. Chou R., Kim N., Sadik C., Seung E., Lan Y., Byrne M., Haribabu B., Iwakura Y., Luster A. (2010) Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33, 266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doherty D., Downey G., Worthen G., Haslett C., Henson P. (1988) Monocyte retention and migration in pulmonary inflammation. Requirement for neutrophils. Lab. Invest. 59, 200–213 [PubMed] [Google Scholar]
- 4. Jones H., Clark R., Rhodes C., Schofield J., Krausz T., Haslett C. (1994) In vivo measurement of neutrophil activity in experimental lung inflammation. Am. J. Respir. Crit. Care Med. 149, 1635–1639 [DOI] [PubMed] [Google Scholar]
- 5. Morris G. E., Whyte M. K. B., Martin G. F., Jose P. J., Dower S. K., Sabroe I. (2005) Agonists of Toll-like receptors 2 and 4 activate airway smooth muscle via mononuclear leukocytes. Am. J. Respir. Crit. Care Med. 171, 814–822 [DOI] [PubMed] [Google Scholar]
- 6. Morris G. E., Parker L. C., Ward J. R., Jones E. C., Whyte M. K., Brightling C. E., Bradding P., Dower S. K., Sabroe I. (2006) Cooperative molecular and cellular networks regulate Toll-like receptor-dependent inflammatory responses. FASEB J. 20, 2153–2155 [DOI] [PubMed] [Google Scholar]
- 7. Ward J. R., Francis S. E., Marsden L., Suddason T., Lord G. M., Dower S. K., Crossman D. C., Sabroe I. (2009) A central role for monocytes in Toll-like receptor-mediated activation of the vasculature. Immunology 128, 58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Suffredini A., Hochstein H., McMahon F. (1999) Dose-related inflammatory effects of intravenous endotoxin in humans: evaluation of a new clinical lot of Escherichia coli O:113 endotoxin. J. Infect. Dis. 179, 1278–1282 [DOI] [PubMed] [Google Scholar]
- 9. Stokes C., Ismail S., Dick E., Bennett J. A., Johnston S., Edwards M., Sabroe I., Parker L. (2011) Role of interleukin-1 and MyD88-dependent signaling in rhinovirus infection. J. Virol. 85, 7912–7921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sabroe I., Prince L. R., Jones E. C., Horsburgh M. J., Foster S. J., Vogel S. N., Dower S. K., Whyte M. K. B. (2003) Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J. Immunol. 170, 5268–5275 [DOI] [PubMed] [Google Scholar]
- 11. Cole L., Elkins K., Michalek S., Qureshi N., Eaton L., Rallabhandi P., Cuesta N., Vogel S. (2006) Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J. Immunol. 176, 6888–6899 [DOI] [PubMed] [Google Scholar]
- 12. Hirschfeld M., Ma Y., Weis J. H., Vogel S. N., Weis J. J. (2000) Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine Toll-like receptor 2. J. Immunol. 165, 618–622 [DOI] [PubMed] [Google Scholar]
- 13. Hogan M., Vogel S. (1987) Lipid A-associated proteins provide an alternate “second signal” in the activation of recombinant interferon-γ-primed, C3H/HeJ macrophages to a fully tumoricidal state. J. Immunol. 139, 3697–3702 [PubMed] [Google Scholar]
- 14. Parker L. C., Prestwich E. C., Ward J. R., Smythe E., Berry A., Triantafilou M., Triantafilou K., Sabroe I. (2008) A phosphatidylserine species inhibits a range of TLR, but not IL-1β, induced inflammatory responses by disruption of membrane microdomains. J. Immunol. 181, 5606–5617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parker L. C., Whyte M. K. B., Vogel S. N., Dower S. K., Sabroe I. (2004) Toll-like receptor (TLR)2 and TLR4 agonists regulate CCR expression in human monocytic cells. J. Immunol. 172, 4977–4986 [DOI] [PubMed] [Google Scholar]
- 16. Sell S., Braude A. (1961) Intradermal reactions in man to autologous erythrocytes sensitized with tuberculin or endotoxin. J. Immunol. 87, 119–125 [PubMed] [Google Scholar]
- 17. Besemer J., Hujber A., Kuhn B. (1989) Specific binding, internalization, and degradation of human neutrophil activating factor by human polymorphonuclear leukocytes. J. Biol. Chem. 264, 17409–17415 [PubMed] [Google Scholar]
- 18. Chuntharapai A., Kim K. J. (1995) Regulation of the expression of IL-8 receptor A/B by IL-8: possible functions of each receptor. J. Immunol. 155, 2587–2594 [PubMed] [Google Scholar]
- 19. Samanta A., Oppenheim J., Matsushima K. (1989) Identification and characterization of specific receptors for monocyte-derived neutrophil chemotactic factor (MDNCF) on human neutrophils. J. Exp. Med. 169, 1185–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walmsley S. R., Chilvers E. R., Thompson A. A., Vaughan K., Marriott H. M., Parker L. C., Shaw G., Parmar S., Schneider M., Sabroe I., Dockrell D. H., Milo M., Taylor C. T., Johnson R. S., Pugh C. W., Ratcliffe P. J., Maxwell P. H., Carmeliet P., Whyte M. K. B. (2011) Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J. Clin. Invest. 121, 1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bourke E., Cassetti A., Villa A., Fadlon E., Colotta F., Mantovani A. (2003) IL-1β scavenging by the type II IL-1 decoy receptor in human neutrophils. J. Immunol. 170, 5999–6005 [DOI] [PubMed] [Google Scholar]
- 22. Chaudhuri N., Paiva C., Donaldson K., Duffin R., Parker L., Sabroe I. (2010) Diesel exhaust particles override natural injury-limiting pathways in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 299, L263–L271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bhatia M., Moochhala S. (2004) Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 202, 145–156 [DOI] [PubMed] [Google Scholar]
- 24. Goodman E., Stricker P., Velavicius M., Fonseca R., Kleinstein E., Lavery R., Deitch E., Hauser C., Simms H. (1999) Role of granulocyte-macrophage colony-stimulating factor and its receptor in the genesis of acute respiratory distress syndrome through an effect on neutrophil apoptosis. Arch. Surg. 134, 1049–1054 [DOI] [PubMed] [Google Scholar]
- 25. Johnston B., Burns A. R., Suematsu M., Issekutz T. B., Woodman R. C., Kubes P. (1999) Chronic inflammation upregulates chemokine receptors and induces neutrophil migration to monocyte chemoattractant protein-1. J. Clin. Invest. 103, 1269–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cross A., Opal S., Sadoff J., Gemski P. (1993) Choice of bacteria in animal models of sepsis. Infect. Immun. 61, 2741–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Serhan C. N., Chiang N., Van Dyke T. E. (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bianchi S., Dockrell D., Renshaw S., Sabroe I., Whyte M. (2006) Granulocyte apoptosis in the pathogenesis and resolution of lung disease. Clin. Sci. 110, 293–304 [DOI] [PubMed] [Google Scholar]
- 29. Sabroe I., Williams T. J., Hébert C. A., Collins P. D. (1997) Chemoattractant cross-desensitization of the human neutrophil IL-8 receptor involves receptor internalization and differential receptor subtype regulation. J. Immunol. 158, 1361–1369 [PubMed] [Google Scholar]
- 30. Ali H., Richardson R. M., Haribabu B., Snyderman R. (1999) Chemoattractant receptor cross-desensitization. J. Biol. Chem. 274, 6027–6030 [DOI] [PubMed] [Google Scholar]
- 31. Nasser M., Raghuwanshi S., Grant D., Jala V., Rajarathnam K., Richardson R. (2009) Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 183, 3425–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moreland J., Davis A., Matsuda J., Hook J., Bailey G., Nauseef W., Lamb F. (2007) Endotoxin priming of neutrophils requires NADPH oxidase-generated oxidants and is regulated by the anion transporter ClC-3. J. Biol. Chem. 282, 33958–33967 [DOI] [PubMed] [Google Scholar]
- 33. Walmsley S. R., Print C., Farahi N., Peyssonnaux C., Johnson R. S., Cramer T., Sobolewski A., Condliffe A. M., Cowburn A. S., Johnson N., Chilvers E. R. (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1α-dependent NF-κB activity. J. Exp. Med. 201, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grutkoski P. S., Graeber C. T., D'Amico R., Keeping H., Simms H. H. (1999) Regulation of IL-8RA (CXCR1) expression in polymorphonuclear leukocytes by hypoxia/reoxygenation. J. Leukoc. Biol. 65, 171–178 [DOI] [PubMed] [Google Scholar]
- 35. Sabroe I., Jones E. C., Whyte M. K. B., Dower S. K. (2005) Regulation of human neutrophil chemokine receptor expression and function by activators of Toll-like receptors (TLR)2 and TLR4. Immunology 115, 90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khandaker M. H., Mitchell G., Xu L., Andrews J. D., Singh R., Leung H., Madrenas J., Ferguson S. S., Feldman R. D., Kelvin D. J. (1999) Metalloproteinases are involved in lipopolysaccharide- and tumor necrosis factor-α-mediated regulation of CXCR1 and CXCR2 chemokine receptor expression. Blood 93, 2173–2185 [PubMed] [Google Scholar]
- 37. Asagoe K., Yamamoto K., Takahashi A., Suzuki K., Maeda A., Nohgawa M., Harakawa N., Takano K., Mukaida N., Matsushima K., Okuma M., Sasada M. (1998) Down-regulation of CXCR2 expression on human polymorphonuclear leukocytes by TNF-α. J. Immunol. 160, 4518–4525 [PubMed] [Google Scholar]
- 38. Cummings C. J., Martin T. R., Frevert C. W., Quan J. M., Wong V. A., Mongovin S. M., Hagen T. R., Steinberg K. P., Goodman R. B. (1999) Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J. Immunol. 162, 2341–2346 [PubMed] [Google Scholar]
- 39. Hartl D., Latzin P., Hordijk P., Marcos V., Rudolph C., Woischnik M., Krauss-Etschmann S., Koller B., Reinhardt D., Roscher A. A., Roos D., Griese M. (2007) Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat. Med. 13, 1423–1430 [DOI] [PubMed] [Google Scholar]
- 40. Harari O. A., McHale J. F., Marshall D., Ahmed S., Brown D., Askenase P. W., Haskard D. O. (1999) Endothelial cell E- and P-selectin up-regulation in murine contact sensitivity is prolonged by distinct mechanisms occurring in sequence. J. Immunol. 163, 6860–6866 [PubMed] [Google Scholar]
- 41. Hollingsworth J. W., Chen B. J., Brass D. M., Berman K., Gunn M. D., Cook D. N., Schwartz D. A. (2005) The critical role of hematopoietic cells in lipopolysaccharide induced airway inflammation. Am. J. Respir. Crit. Care Med. 171, 806–813 [DOI] [PubMed] [Google Scholar]
- 42. Cybulsky M. I. (1988) Neutrophil leukocyte emigration induced by endotoxin. Mediator roles of interleukin 1 and tumor necrosis factor α 1. J. Immunol. 140, 3144–3149 [PubMed] [Google Scholar]
- 43. Reckless J., Tatalick L., Grainger D. (2001) The pan-chemokine inhibitor NR58-3.14.3 abolishes tumour necrosis factor-α accumulation and leucocyte recruitment induced by lipopolysaccharide in vivo. Immunology 103, 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Theilgaard-Mönch K., Knudsen S., Follin P., Borregaard N. (2004) The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J. Immunol. 172, 7684–7693 [DOI] [PubMed] [Google Scholar]
- 45. Rider P., Carmi Y., Guttman O., Braiman A., Cohen I., Voronov E., White M. R., Dinarello C. A., Apte R. N. (2011) IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 187, 4835–4843 [DOI] [PubMed] [Google Scholar]
- 46. Prince L. R., Allen L., Jones E. C., Hellewell P. G., Dower S. K., Whyte M. K., Sabroe I. (2004) The role of interleukin-1β in direct and Toll-like receptor 4-mediated neutrophil activation and survival. Am. J. Pathol. 165, 1819–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]