

Abstract
Light-controlled modification of the fluorescence emission properties of proteins of the GFP family is of crucial importance for many imaging applications including superresolution microscopy. Here, we have studied the reversibly photoswitchable fluorescent protein mIrisGFP using optical spectroscopy. By analyzing the pH dependence of isomerization and protonation equilibria and the isomerization kinetics, we have obtained insight into the coupling of the chromophore to the surrounding protein moiety and a better understanding of the photoswitching mechanism. A different acid-base environment of the chromophore’s protonating group in its two isomeric forms, which can be inferred from the x-ray structures of IrisFP, is key to the photoswitching function and ensures that isomerization and protonation are correlated. Amino acids near the chromophore, especially Glu212, rearrange upon isomerization, and Glu212 protonation modulates the chromophore pKa. In mIrisGFP, the cis chromophore protonates in two steps, with pKcis of 5.3 and 6, which is much lower than pKtrans (>10). Based on these results, we have put forward a mechanistic scheme that explains how the combination of isomeric and acid-base properties of the chromophore in its protein environment can produce negative and positive photoswitching modes.
Introduction
Fluorescent proteins (FPs) of the green fluorescent protein (GFP) family have become key research tools in a wide range of life science applications. These small proteins of ∼250 amino acids autocatalytically form a fluorescent chromophore in the interior of their 11-stranded β-barrel fold (1–4). When fused to a protein of interest on the DNA level, FPs can serve as genetically encoded, highly specific fluorescence markers in cellular imaging (5–13). In recent years, photoactivatable or optical-highlighter FPs have emerged as especially powerful marker tools, notably for pulse-chase imaging and a variety of superresolution microscopy techniques (7,14–17). Their fluorescence properties can be modified by light irradiation at specific wavelengths. Two different modes of photoactivation are presently known. Irreversible photoactivation, also known as photoconversion, involves a permanent photochemical modification of the FP chromophore. As a consequence, a nonfluorescent (dark) state may become permanently activated to a fluorescent (bright) state, or a bright state may be turned into another bright state with a different emission wavelength (18–23).
Reversible photoactivation, also known as photoswitching, typically arises from chromophore isomerization between two configurations, only one of which emits fluorescence with high yield (24–30). We note in passing that Dreiklang, a recently introduced photoswitching GFP variant, is a notable exception to this rule, featuring a reversible hydration/dehydration reaction of the chromophore (31). Chromophore isomerization is intimately coupled to a chromophore protonation reaction (20,24,32). In most reversibly photoswitchable FPs, the cis configuration is the thermodynamically stable form; its anionic form constitutes the bright fluorophore (24,33,34). Electronic excitation triggers a cis-trans isomerization accompanied by a change of protonation state, resulting in an essentially nonfluorescent chromophore. In a few photoswitchers, including KFP1 (22) and asFP595 (25,35,36), the nonfluorescent trans state rather than the cis state is the thermodynamically stable form, and light irradiation induces a trans-cis isomerization to the fluorescent cis state. In the dark, these FPs spontaneously relax to their thermodynamically stable isomeric state on timescales ranging from minutes to hours. This process can be markedly accelerated by irradiation with light of a suitable wavelength. In the so-called positive photoswitchers, fluorescence excitation enhances the emission intensity; in negative switchers, the emission is quenched by the excitation light (37). The body of acquired knowledge on photoconversion and photoswitching has been presented in several reviews (14,16,38). There is general agreement on the basic principles of photoactivation; the mechanistic details, however, are still controversial. For Dronpa, for example, it was proposed that on-switching is initiated by excited-state proton transfer (ESPT) from the neutral trans chromophore (39), and that off-switching involves an intersystem crossing from the excited anionic cis form to the triplet state and its subsequent protonation (40,41). For asFP595, results from molecular-dynamics simulations led to the suggestion that trans-cis isomerization of the chromophore occurs via a hula-twist mechanism, with a concerted rotation of both bonds of the methine bridge connecting the phenoxy and the imidazolinone rings, possibly accompanied by proton transfer (25). Because the various photoswitching FPs that exist all have monomethine chromophores, Olsen et al. (42) proposed that photoswitching is an intrinsic property of the chromophore. For efficient photoswitching, the protein environment should 1), restrict torsion around the phenoxy bond (P-bond) to enhance the radiative decay channel, and 2), provide a suitable acid-base chemistry so that protonation-state change and isomerization by torsion around the imidazolinone bond (I-bond) are correlated.
We recently introduced FP variants that combine reversible and irreversible photoactivation, tetrameric IrisFP (20) and its monomeric variant, mIrisFP (43). In their green fluorescent form, they display reversible photoswitching between a fluorescent and a nonfluorescent state. In addition, they can be photoconverted irreversibly to red-emitting FPs by ∼400 nm light. In their red form, they can again be toggled between a bright fluorescent and a dark state. Due to their multiple photoactivation modes, these FPs enable entirely novel experiments, e.g., pulse-chase imaging with superresolution in living cells (43).
To understand the competition between photoswitching and photoconversion, we have, as a first step, performed a detailed study on the Phe173Ser variant of the advanced EosFP variant mEosFPthermo (43,44). This protein can be photoswitched efficiently and reversibly between a green fluorescent and a dark state; its irreversible photoconversion from a green fluorescent to a red fluorescent form, however, is largely suppressed and only observable under extremely high laser powers. We refer to this green variant as mIrisGFP because it shares the crucial Phe173Ser mutation of IrisFP, rendering it photoswitchable.
Here, we present an in-depth spectroscopic characterization of the cis and the trans chromophore species of mIrisGFP and analyze the thermally and light-activated chromophore isomerization reactions over a wide pH range. We provide evidence that both chromophore isomerization and protonation occur during reversible photoswitching. Therefore, even a simple model of this process comprises a minimum of four chromophore species in the ground state (cis neutral, cis anionic, trans neutral, trans anionic) and, upon light exposure, the corresponding excited-state species (Fig. 1). In the following, C and T will denote protein subpopulations with cis and trans chromophore isomers, respectively, with superscript minus signs for anionic and superscript H for neutral.
Figure 1.
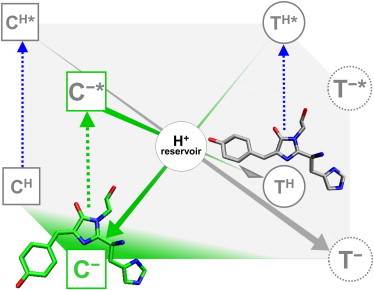
General scheme capturing reversible photoactivation of mIrisGFP and other photoswitchable FPs. It contains eight chromophore species that are potentially involved. The states are denoted by C and T for cis and trans chromophore isomers, respectively, with superscript minus sign for anionic and superscript H for neutral; an asterisk denotes the electronically excited states.
Materials and Methods
Protein mutagenesis, expression, and purification
Taking the mEosFPthermo vector as a template, we introduced an additional mutation, Phe173Ser, by site-directed mutagenesis, yielding mIrisGFP (QuikChange II Mutagenesis Kit, Stratagene, La Jolla, CA). The variant Glu212Gln was produced by further site-directed mutagenesis of mIrisGFP. DNA primers were purchased from Biomers (Ulm, Germany); the resulting clones were sequenced by a commercial provider (GATC, Konstanz, Germany). Proteins were expressed in E. coli strain XL1-blue (Stratagene) and purified as described (43).
Spectroscopic characterization
Absorption spectra of mIrisGFP were recorded on a Cary 50 Bio UV/Vis spectrophotometer (Varian, Darmstadt, Germany). Fluorescence spectra were measured with a SPEX Fluorolog II spectrofluorometer (Spex Industries, Edison, NJ). The excitation line width was set to 0.85 nm; emission spectra were taken at 2.2-nm resolution and corrected for the wavelength dependence of the detector efficiency. Circular dichroism (CD) spectra were recorded on a J-810 spectropolarimeter (Jasco Europe, Cremella, Italy).
For spectroscopic experiments, concentrated stock solutions of mIrisGFP were diluted to protein concentrations of ∼10 μM (absorption), ∼1 μM (fluorescence), and ∼30 μM (CD) in 20 mM buffer solutions (sodium citrate/phosphate, sodium phosphate, sodium carbonate, and glycine buffer systems, depending on the desired pH in the range 3–11). The ionic strength was adjusted to 150 mM with sodium chloride.
Photoswitching quantum yields
To measure the kinetics of photoswitching, we illuminated mIrisGFP solutions with 473-nm light at 5–50 mW cm−2 (off-switching) and 405-nm light at 5–25 mW cm−2 (on-switching) from solid-state lasers and recorded the resulting light-induced changes of the fluorescence emission intensity at 520 nm as a function of time. The power density of the activating light was adjusted as appropriate for the specific experiment. The measured kinetic traces were fitted with exponential decays, yielding the sum, kon + koff, as an apparent rate coefficient. Consequently, a single, microscopic rate coefficient results directly from the kinetics only if one of the two predominates. The rate coefficient, k, can be converted to a quantum yield of the different photo-induced processes, ϕ, by using the equation
| (1) |
with wavelength, λ, power density, P, Avogadro’s number, NA, Planck’s constant, h, and the velocity of light, c, and the extinction coefficient, ε, of the reacting species at the wavelength chosen for triggering the process (20).
Thermal relaxation to the fluorescent form
mIrisGFP samples were illuminated with 473-nm light (25 mW cm−2) until the absorption band at ∼490 nm was suppressed to <5% of its original area. Subsequently, thermal relaxation was monitored as a function of time via the absorption increase at 490 nm. The absorption at 490 nm was plotted as a function of time and fitted with a single exponential. Because of the rather slow relaxation kinetics, these experiments were carried out at 310 K (37°C) unless stated otherwise.
Action spectrum of light-induced off-switching
mIrisGFP samples were illuminated for 300 s with light at 20 different wavelengths, λexc, between 330 and 530 nm. The wavelengths were selected from the 450-W xenon lamp of the Fluorolog II spectrometer by using its grating monochromator. The relative fluorescence decrease upon light exposure was measured at 520 nm (λexc = 330–490 nm) and 540 nm (λexc = 490–530 nm). The data were adjusted for intensity variations of the source spectrum and photon energy and plotted against the excitation wavelength to yield the action spectrum.
Results
Spectroscopic characterization of mIrisGFP in the cis isomeric form
In its dark-adapted form, the green chromophore of IrisFP adopts the cis configuration, as shown in the structural model in Fig. 2 A (20). The very similar optical spectra of IrisFP and mIrisGFP suggest that the chromophore of dark-adapted mIrisGFP also assumes the cis configuration. Its absorption spectra show a strong pH dependence (Fig. 3 A), which is often—but not always (45,46)—observed in GFPs and can be described by an exchange between an A (∼400 nm) and a B (∼500 nm) band associated with a neutral and an anionic chromophore, respectively. To assign an absorption band to a protein with a particular chromophore isomer, we use either C (cis) or T (trans) as a subscript. At low pH, the spectrum is dominated by the AC band, peaking at 389 nm (εmax = 24,000 ± 2000 M−1 cm−1). With increasing pH, the BC band at ∼488 nm (εmax = 47,000 ± 1700 M−1 cm−1) gains intensity at the expense of AC (Fig. 3 A). The peak amplitudes of the AC and BC bands are plotted as a function of pH in Fig. 3 B.
Figure 2.
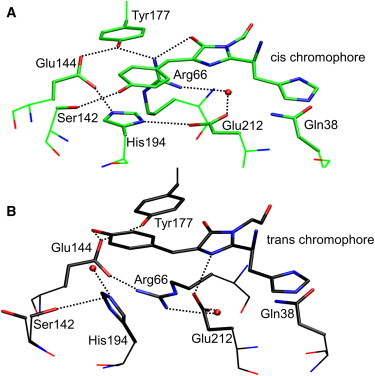
Structural environment of the green chromophore in IrisFP. Hydrogen bonds are shown as dotted lines. (A) Cis conformer (PDB accession code 2VVH). (B) Trans conformer (PDB accession code 2VVI).
Figure 3.
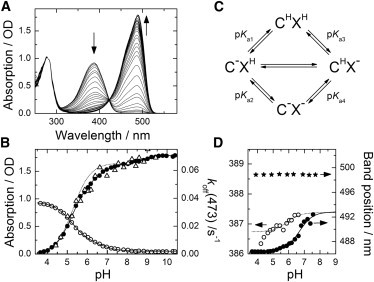
pH variation of the optical absorption spectra of mIrisGFP in the dark-adapted state (cis chromophore). (A) Absorption spectra in the pH range 3.6–10.4. Arrows point in the direction of increasing pH. (B) Peak absorption of the AC (open circles) and BC (solid circles) bands. Solid lines show the fit of the model depicted in panel c in the pH range 3.6–8. An additional protonation step described by the Henderson-Hasselbach equation was added to fit the BC band amplitudes at pH >8. Dotted line shows one-site protonation with pK = 5.3. Open triangles indicate pH dependence of the reaction rate coefficient, koff(473), for off-switching of the fluorescence with 473-nm light. (C) Four-state model describing the protonation equilibrium of the chromophore, C, interacting with a titratable amino acid, X. (D) pH dependence of the peak positions of the AC (open circles) and BC (solid circles) absorption bands of mIrisGFP, as well as the peak position of BC of the Glu212Gln mutant (stars).
For a simple protonation equilibrium between a neutral CH and an anionic C− chromophore species, the fraction of C− varies with pH according to
| (2) |
with a Hill coefficient n = 1; n ≠ 1 indicates that a simple one-site protonation is inadequate. This function, plotted in Fig. 3 B (dotted line) for pKa = 5.3 and n = 1, fails to describe the pH dependence of the C− population. Apparently, a more complex model is needed to fit the observed two-step behavior, which includes (at least) one other protonating group, which we shall denote by X in the following. Located near the chromophore, X modulates the chromophore’s proton affinity via electrostatic interactions. Such a model thus contains four discrete species, CH/XH, CH/X−, C−/XH and C−/X−, as shown in the scheme in Fig. 3 C (47–51). The equations governing the fractional populations of all four species are provided in the Supporting Material. However, those are not directly accessible via the pH-dependent absorption spectra of mIrisGFP, because we can only distinguish between the overall AC and BC bands and, thus, the fractional populations of neutral and anionic chromophores,
| (3) |
Although the protonation state of X is not observable from the relative weights of the AC and BC bands, the presence or absence of a charge in the close vicinity of the neutral or anionic chromophore may affect its delocalized π-electron system. Therefore, the protonation state of X may be inferred from Stark shifts of the AC or BC bands, as discussed below.
A global fit to the data (Fig. 3 B) in the range 3.5 < pH < 7.5, based on the four-state model (Eq. 3 and Eq. S3 in the Supporting Material), yields pKa1 = 5.3 ± 0.1 and pKa4 = 6.0 ± 0.1 for the two-step protonation reaction of the cis chromophore. The amplitude of the BC band at 489 nm displays yet another step with pKa = 8.9 ± 0.2 (Fig. 3 B) that is accompanied by a marked red shift. This effect is likely associated with the appearance of a chromophore species with a more red-shifted absorption band at 496 nm, for which the negative charge of the hydroxybenzyl anion is no longer stabilized by a hydrogen bond to Ser142 (46).
We have also examined the spectra for band shifts that may indicate a change in the protonation state of X. The BC band indeed shows a pronounced red shift by 6 nm, with increasing pH that can be described by pKa2 = 6.6 ± 0.1 (Fig. 3 D). This shift is absent in the mIrisGFP mutant Glu212Gln (compare Fig. 3 D and Fig. S1), strongly suggesting that the protonation state of Glu212 affects the proton affinity of the cis chromophore. Likewise, we have observed a shift of the AC band that signals protonation of X in the presence of a neutral chromophore, CH (Fig. 3 D, dotted line). At pH >5, the shift can be described by pKa3 = 5.9 ± 0.1; the strong deviation below pH 4.5 is due to the onset of acid denaturation (Fig. 3 D). We note that the value of pKa3 agrees quantitatively with that required by the model (Eq. S2) and thus lends strong credence to our two-site protonation model. The coupling free energy between the two sites (Eq. S5) amounts to ∼4 kJ/mol.
The excitation spectra of mIrisGFP, collected with the emission monochromator set to λem = 540 nm, display a single band at 488 nm, showing that C− is the fluorescent species (Fig. 4 A, dotted lines); the emission band is centered on 515 nm (Fig. 4 A, dashed lines). The fluorescence intensity shows the same pH dependence as the BC band absorption at pH <8 and consequently can be modeled with the same pKa values (see Fig. 4 C, triangles). At pH >8, the emission intensity remains constant, whereas the absorption still increases. This discrepancy suggests that the high-pH chromophore species represented by the absorption band at 496 nm is nonfluorescent, as would be expected for a less stabilized chromophore. The fluorescence quantum yield of C− at pH 7 was determined as Φ = 0.63 ± 0.06. The neutral chromophore, CH, is only poorly fluorescent (Φ < 0.03). Its emission peaks at ∼460 nm (λexc = 390 nm); concurrent fluorescence emission at ∼515 nm is not observed, thus excluding excited-state proton transfer as a cause of chromophore deprotonation in the excited state and the subsequent red-shifted emission by the anionic fluorophore seen in a variety of FPs (52–54).
Figure 4.
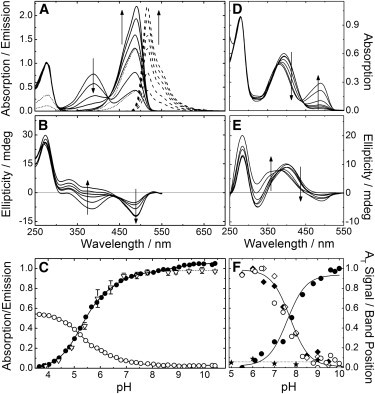
pH dependence of optical spectra of mIrisGFP in the dark-adapted state (cis chromophore) (A–C) and immediately after 473-nm illumination (predominantly trans chromophore) (D–F). (A) Absorption, excitation, and emission spectra (solid, dotted, and dashed lines, respectively). Excitation and emission spectra were measured with emission at 540 nm and excitation at 390 nm. (B) CD spectra. (C) pH dependence of the (normalized) peak amplitudes of the AC (open circles) and BC (solid circles) absorption bands and of the emission intensity (triangles). (D and E) Absorption spectra and CD spectra, respectively. Arrows indicate increasing pH. (F) Normalized peak amplitudes of the A (open circles) and B (solid circles) bands and normalized peak shift of the A band in D (solid diamonds) and E (open diamonds) and of mutant Glu212Gln (stars).
The CD spectra of mIrisGFP display two bands with negative ellipticity corresponding to the AC and BC absorption bands (compare Fig. 4, A and B). Within the experimental error, the CD bands display the same pH-dependent population exchange as the absorption bands. The optical parameters of mIrisGFP (pH 7) with a cis chromophore are compiled in Table 1.
Table 1.
Optical properties of mIrisGFP at pH 7
| Cis chromophore | Anionic (Bc) | Neutral (Ac) |
|---|---|---|
| Wavelength, λmax (Ex/Em)/nm | 488/516 | 391/450 |
| Extinction coefficient, ε (λmax) (M−1 cm−1) | 47,000 ± 1,700 | 24,000 ± 2,000 |
| Fluorescence quantum yield, Φ | 0.63 ± 0.06 | <0.03 |
| pKa | 5.3 ± 0.1 | |
| 6.0 ± 0.1 | ||
| trans chromophore | Anionic (Bt) | Neutral (At) |
| Wavelength, λmax (Abs) (nm) | ND | 390 |
| Extinction coefficient, ε (λmax) (M−1 cm−1) | ND | 18,000 ± 2,300 |
| Quantum yield, Φ | — | — |
| pKa | >10 | |
Spectroscopic characterization of mIrisGFP in the trans state
The protein with the chromophore in trans is a light-induced, metastable conformation that spontaneously reverts back to cis. To measure its absorption spectra as a function of pH, we prepared the trans state by illuminating mIrisGFP solutions with 473-nm light until the color faded completely. Then, the 473-nm light was switched off and spectra were taken. Because of thermal trans-cis relaxation during data acquisition (∼1 min), it was unavoidable that the spectra of the trans form contain minor contributions of the cis species. The absorption spectra (Fig. 4 D) are dominated by an A band at ∼390 nm representing a neutral chromophore species. With increasing pH, a second band emerges at ∼490 nm at the expense of A, which we assign to an anionic chromophore species. Its pH-dependent absorbance change can be modeled with a pKa of 7.8 ± 0.1 (see Fig. 4 F, solid circles). However, even at pH 10, only a minority of proteins are involved in this pH-dependent spectral change, which indicates that it cannot originate from deprotonation of the trans chromophore. Comparison of the spectra in Fig. 4, A and D, rather suggests that the 490-nm band represents anionic cis chromophore species that have reformed during data collection. The pH-dependent variation of the 490-nm band with pKa = 7.8 thus describes the pH dependence of the trans-cis relaxation kinetics rather than a protonation equilibrium. Additional spectral features that would indicate the presence of anionic trans chromophores are absent in the spectra. Consequently, the trans isomer does not deprotonate in the observed pH range, so that pKa > 10 for this isomer.
We have calculated pure absorption spectra of the trans species by removing the cis fraction from the spectra. To this end, the cis spectra in Fig. 4 A (at the respective pH) were scaled to match the 490-nm band of the spectra in Fig. 4 D and subtracted. We note that the apparent pKa of 7.8 is significantly above the pKa values of 5.3 ± 0.1 and 6.0 ± 0.1, which characterize cis chromophore protonation, and that contributions of neutral cis in the spectral region of the A band are therefore small. The pure AT band of the neutral trans species shows a pH-dependent shift from 396.8 to 384.3 nm that can be modeled by proton binding with pKa = 7.8 ± 0.1. The observation that the spectral shift has the same pH dependence as the thermal trans-cis recovery kinetics, shown by plotting the data in a normalized fashion in Fig. 4 F, suggests that the protonation state of the amino acid responsible for this Stark shift also influences the kinetics of isomerization.
Pronounced changes are visible in the CD spectra upon cis-trans isomerization. The bands with negative ellipticity associated with the cis chromophore (see Fig. 4 B) are essentially absent. Instead, there is a strong band with positive ellipticity at ∼390 nm (Fig. 4 E) associated with the nonfluorescent, neutral trans species, TH. Like the AT absorption band, this CD band shifts markedly to the blue, and its pH dependence can also be modeled with pKa = 7.8 ± 0.1 (Fig. 4 F, open diamonds). Because the blue shift is absent for mutant Glu212Gln, it is apparently associated with a change in Glu212 protonation. The 390-nm band dominates the CD spectrum even at pH 10; a band associated with an anionic trans chromophore, T−, could not be discerned (Fig. 4 E). This observation lends further credence to our hypothesis that the trans chromophore protonates with pKa > 10. The optical properties of mIrisGFP with a trans chromophore are also included in Table 1.
Thermally activated chromophore isomerization
The neutral trans chromophore of mIrisGFP, TH, relaxes back to cis over time, with a half-life of ∼90 and ∼20 min at 290 and 310 K, respectively (at pH 7). We have confirmed that thermal relaxation goes to completion at all pH values examined here (pH 5–10.4). The recovery kinetics of the cis isomer is pH-dependent (Fig. 5 A). Between pH 5.5 and 10, its rate coefficient, kth, increases ∼40-fold, from 0.167 × 10−3 s−1 to 6.45 × 10−3 s−1, as determined from the increase of the BC band at 488 nm (T = 310 K). The acceleration occurs in two steps. Using a sum of two Henderson-Hasselbalch relations (Eq. 2), we obtain pKth1 = 7.8 ± 0.4 and pKth2 = 9.5 ± 0.6, indicating a destabilizing influence on the trans chromophore due to successive deprotonation of two moieties. The step at lower pH is absent for mutant Glu212Gln (Fig. 5 A).
Figure 5.
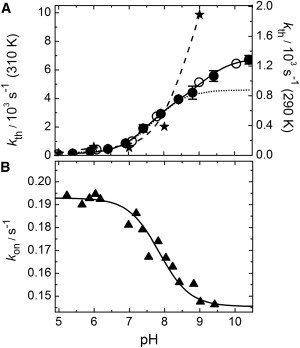
pH dependence of the on-switching kinetics (trans-cis isomerization). (A) Thermally activated recovery of the anionic cis chromophore with rate coefficient, kth, measured at T = 290 K (open circles) and T = 310 K (solid circles). Stars, mIrisGFP Glu212Gln at T = 310 K; solid line, two-site protonation with pKa1 = 7.8 and pKa2 = 9.5; dotted line, one-site protonation with pK = 7.8; dashed line, one-site protonation with pK = 10.3. (B) Photoactivated trans-cis isomerization: kon(405) for P405 = 13 mW cm−2. Solid line shows one-site protonation with pK = 7.8.
Light-activated chromophore isomerization
To study the kinetics of photoactivated cis-trans isomerization of mIrisGFP, we prepared a sample at pH 5.3 with roughly equal fractions of CH and C− chromophores. We illuminated the sample for 300 s with light between 330 and 550 nm in wavelength and measured the relative loss in fluorescence at 520 nm (540 nm for excitation wavelengths >480 nm). These data were normalized for differences in light intensity and photon energy to reveal the wavelength dependence of the photoisomerization efficiency (i.e., the product of extinction coefficient and quantum yield of photoisomerization), which represents the probability of photoisomerization by light irradiation at a particular wavelength.
In Fig. 6, the action spectrum (Fig. 6, symbols) is plotted against the excitation wavelength, together with the absorption spectrum (Fig. 6, solid line). Both spectra were scaled to equal amplitudes at the peak of AC. Remarkably, the action spectrum has a maximum in the AC band and is significantly lower in the region of the BC band. Consequently, light-induced cis-trans isomerization is more efficient when exciting the nonfluorescent CH than the it is for the fluorescent C− species.
Figure 6.
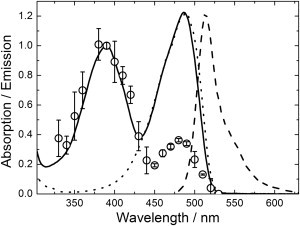
Action spectrum of cis-trans photoisomerization of mIrisGFP. The fluorescence decrease (symbols) at 520/540 nm due to photo-induced off-switching is plotted as a function of the wavelength. Also shown are the absorption (solid line), excitation (dotted line), and emission (dashed line) spectra at pH 5.3. Action spectrum and absorption amplitudes were normalized to 1 at the peak of the AC absorption band at 390 nm; the fluorescence spectra were normalized to match the peak of the BC absorption band.
With our 473-nm laser source (P473 = 24 mW cm−2), light-induced off-switching via excitation of C− occurs within tens of milliseconds; it is about three times faster at pH 9.5 (koff(473) = 0.060 ± 0.001 s−1) than at pH 5 (koff(473) = 0.024 ± 0.004 s−1). Its pH dependence follows that of the C− population (Fig. 3 B). The off-switching quantum yield, ϕoff(473) ≈ koff(473)/εc(473) (Eq. 1), increases from (2.3 ± 0.5) × 10−3 at pH 5 to (6.9 ± 0.3) × 10−3 at pH 9.5.
At the laser powers employed here, off-switching via excitation of the anionic cis chromophore at 473 nm is a simple one-photon process, as shown by the linear dependence of the rate coefficient, koff(473), on the intensity (Fig. 7 A). However, even at the highest intensities (70 mW cm−2), we could not switch off all chromophores: for a sample at pH 7, 4.3 ± 0.1% of the initial fluorescence remained (Fig. 7 A). The residual fluorescence depended on the wavelength used for photoactivation (Fig. 7 B), suggesting that a second light-driven process opposes off-switching. Apparently, the activating light can also excite the neutral trans species, TH, and trigger the back reaction, TH → C−, although with a lower yield. Because of the high pKa of the trans chromophore, light activation of T− is negligible at pH 7.
Figure 7.
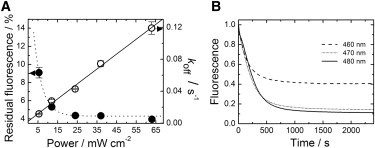
Effect of illumination power and wavelength on photo-induced off-switching of mIrisGFP (cis-trans isomerization). (A) Dependence of the rate coefficient, koff(473), on the 473-nm excitation laser power (open circles, solid line: linear fit) and residual fluorescence upon extended 473-nm laser illumination (solid circles, dotted line: fit) as a function of laser power. (B) Time dependence of the fluorescence decay upon illumination with light at three different wavelengths.
To single out the effect of light on mIrisGFP molecules with a TH chromophore, samples were first illuminated with 473-nm light to populate this state to the largest possible extent. Afterward, the temporal increase of the fluorescence at 520 nm, which is proportional to the amount of C−, was recorded under 405-nm illumination. At pH 7.8, the on-switching rate coefficient, kon(405), was 0.17 s−1 (P405 = 13.3 mW cm−2), so that Eq. 1 yields a quantum yield of on-switching of ϕon(405) = 0.15 ± 0.01. Fig. 5 B shows that kon(405) decreases with increasing pH, with pKa = 7.8 ± 0.1. As the absorption at 405 nm also decreases with pKa = 7.8 ± 0.1 due to the shift of the AT band (Fig. 4 F), ϕon(405) is constant between pH 4.5 and 9.
Because of the large spectral overlap of the A absorption bands of the neutral species, λmax (CH) = 389 nm and λmax (TH) = 391 nm at pH 7, 405-nm light irradiation excites both isomeric forms. Therefore, a light-controlled equilibrium, , is established, with an equilibrium coefficient of
| (4) |
K405 is pH-dependent because koff(405) depends on the protonation equilibrium of the cis chromophore. With 405-nm illumination at pH 5.3 (P = 20 mW/cm2), the fluorescence was reduced to 70% of its initial value, K405 = 0.41 ± 0.05. With the extinction coefficients of the neutral forms, TH and CH, at 405 nm, εT = (12,000 ± 1600) M−1cm−1 and εC = (20,700 ± 2000) M−1 cm−1, respectively, and ϕon(405) = 0.15 (see above), we finally obtain ϕoff(405) = 0.036 ± 0.008. The photophysical parameters of mIrisGFP are compiled in Table 2 together with those of its cousin, mIrisFP (43), for comparison.
Table 2.
Photophysical parameters of mIrisFP and mIrisGFP at pH 7
| mIrisFP (green) | mIrisGFP | |
|---|---|---|
| ϕoff(473) | 0.0069 ± 0.0001 | 0.0045 ± 0.0004 |
| ϕoff(405) | ND | 0.036 ± 0.008 |
| ϕon(405) | 0.36 ± 0.02 | 0.15 ± 0.01 |
| Residual fluorescence (%)∗ | 4.8 ± 0.7 | 5.0 ± 0.6 |
| Thermal recovery half-life t1/2 (min)† | 60 ± 4.6 | 64 ± 2 |
All data were taken on samples in potassium phosphate buffer. ND, not determined.
Determined with 473-nm illumination at 25 mW cm−2.
Measured at 290 K.
Discussion
Chromophore protein interactions in mIrisGFP
The photophysical properties of (photoactivatable) FPs strongly depend on the coupling between the chromophore and the surrounding protein moiety. In the cis and trans isomeric forms of mIrisGFP, the chromophore’s protonating hydroxyphenyl group resides in two very different local environments (Fig. 2), as revealed by the x-ray structures of IrisFP (20). In the cis configuration, the hydroxyphenyl group forms a hydrogen bond to the Ser142 hydroxyl group and, therefore, is presumably deprotonated (Fig. 2 A). The anionic state is further stabilized by a hydrogen bond between Arg66 and the imidazolinone carbonyl stabilizing the electron density on the imidazolinone ring. In contrast, the hydroxyphenyl group of the mIrisGFP trans chromophore points toward the Glu144 side chain (Fig. 2 B). As in IrisFP (20), Glu144 is most likely deprotonated. Consequently, the trans chromophore has a much higher proton affinity (pKa > 10) than the cis chromophore (pKa = 5.3/6.0) and is thus protonated up to pH values of ∼9.5.
Chromophore protonation and the kinetics of trans-cis isomerization are coupled to the protonation of a neighboring group (Fig. 3), which we assign to the side chain of Glu212 based on several observations. In the Glu212Gln mutant, the shifts of the absorption bands are absent and, moreover, the kinetics of photoisomerization are altered, strongly suggesting that it is the protonation of Glu212 that affects the chromophore properties. Glu212 is part of a tightly coupled network of residues that also includes His194, Arg66, and Glu144. If, instead, Glu144 were the residue in question, its deprotonation at pH >7.8 would cause a stabilization of the proton on the neutral trans chromophore and, concomitantly, a destabilization of the anionic cis form. Consequently, we would expect a slower thermal trans-cis isomerization but the contrary is observed (Fig. 5 A).
In the presence of anionic and neutral cis chromophores, Glu212 protonation occurs with pKa values of 6.6 and 5.9, respectively. This pKa difference of 0.7 pH units is identical to that between the chromophore’s pKa values of 6.0 and 5.3, and it reflects their mutual coupling free energy of ∼4 kJ/mol. We note that an earlier report assumed a much stronger coupling for GFP (55). With a neutral trans chromophore, Glu212 is more basic (pKa = 7.8). This pronounced pKa difference likely originates from the changed orientation of the Glu212 side chain (Fig. 2). Next to the trans chromophore, it points at the free electron pair on the imidazolinone nitrogen, so it should have a rather high proton affinity. With the chromophore in cis, Glu212 is part of an extended, planar network parallel to the chromophore plane, in which its acidity is governed by its direct-interaction partners, His194 and Arg66.
Thermal versus photoactivated trans-cis isomerization
The metastable, trans form of mIrisGFP is produced by light irradiation and spontaneously relaxes back to cis in the dark. This process is pH-dependent; its rate coefficient, kth, increases in two steps toward higher pH (Fig. 5 A). The first step has a pKa of 7.8 and is, therefore, presumably connected to a change of the Glu212 protonation state. Upon deprotonation, the stabilizing hydrogen bond with the imidazolinone nitrogen is relinquished and, instead, the charged Glu212 carboxylate side chain causes electrostatic repulsion. This structural change is expected to enhance chromophore flexibility and thereby may facilitate twisting of the chromophore and isomerization to cis. Concomitantly, the Glu212 carboxylate reorients to form a salt bridge to the presumably positively charged His194 side chain (56,57) and a hydrogen bond to a water molecule. Furthermore, an extensive network of salt bridges and hydrogen-bonding interactions is established adjacent to the chromophore, which also involves Arg66, Glu144, and Tyr177 (Fig. 2 A). Such a network has also been found for the cis species of Dronpa (58) and mTFP0.7 (59). The trans-cis relaxation kinetics is further accelerated upon deprotonation of the chromophore hydroxyphenyl (pKa > 10, Fig. 5 A). Removal of a stabilizing hydrogen bond to the Glu144 side chain and introduction of electrostatic repulsion between the charged phenolate moiety and the deprotonated Glu144 side chain may cause an enhanced mobility of the chromophore. Thermally activated on-switching upon chromophore deprotonation is markedly faster for mutant Glu212Gln than for mIrisGFP (Fig. 5 A), supporting the idea of a more flexible chromophore. We note that the fluorescence quantum yield of the mutant in decreased, Φ = 0.26 ± 0.01, as expected for a more mobile chromophore.
Photoactivated trans-cis isomerization is also a pH-dependent process (pKa = 7.8). Its rate coefficient, kon, decreases with increasing pH (Fig. 5 B), in contrast to that of thermal relaxation (Fig. 5 A). Apparently, protonation of Glu212 has a different effect on the light-induced transition in the electronically excited state than on thermal recovery. Trans-cis photoisomerization is initiated by excitation of TH with 405-nm light, so its kinetics depends on the probability of absorbing a photon, i.e., on the absorption at 405 nm and on the probability to subsequently isomerize. The latter is an intrinsic property of the particular chromophore species and, therefore, depends only weakly on pH due to coupling to other, more remote groups. However, the absorption of TH at 405 nm decreases with increasing pH because of the AT band shift, with the pH dependence described by pKa = 7.8 (Fig. 4 F). Note that the overall concentration of TH species and, therefore, the concentration of reactants, is essentially constant for 5 < pH < 9 because of the high pKtrans.
In general, one should expect the isomerization probability to depend on the stabilization of the chromophore in its particular isomeric state, which is substantially determined by the numbers and strengths of hydrogen-bonding interactions between the chromophore hydroxyphenyl and the protein moiety. In the C− conformation of mIrisFP, the cis chromophore is engaged in hydrogen-bonding with Ser142 (2.7 Å) and two water molecules (2.7 Å), and by π-stacking with His194 (20). In the TH species, the chromophore hydrogen-bonds to Glu144 (2.9 Å) and a water molecule (20) and, therefore, appears less stabilized. Indeed, the trans-cis photoisomerization (on-switching) quantum yield is much larger than the cis-trans photoisomerization (off-switching) quantum yields via either the CH or the C− states (Table 2).
Photoisomerization mechanism
Reversible photoswitching of FPs involves a light-activated change of both isomerization and protonation states of the chromophore (24,25,57,60), but the mechanistic details are not fully understood (41,57,61,62). Especially the order in which isomerization and protonation occur is still under debate (63,64). The results obtained here provide several pieces of evidence in support of the model of Olsen et al. (42). Trans-cis isomerization can be induced by 405-nm excitation of the neutral trans chromophore (Fig. 5 B) with a quantum yield of ϕon(405) = 0.15 ± 0.01. According to the photoswitching model (42), the excited neutral trans chromophore twists around the I-bond of the methylene bridge, deprotonates, and isomerizes to the anionic cis state (Fig. 1). The molecular structure (Fig. 2 B) suggests that Glu144 may accept the proton from the neutral trans chromophore. Irradiation of the anionic cis chromophore with 473-nm light causes the opposite chromophore transition, i.e., cis-trans isomerization (off-switching), with ϕoff(473) = 0.0045 ± 0.0004. Rather unexpectedly, mIrisGFP can be switched off much more efficiently by exciting the neutral, nonfluorescent cis chromophore (ϕoff(405) = 0.036 ± 0.008) (Fig. 6). This observation also agrees with the result of the quantum-chemical calculations by Olsen et al. (42), who found a barrierless energy surface for excited-state isomerization of the neutral chromophore, whereas a substantial barrier was found for the anionic chromophore. In addition, one may argue that the C− species should be especially stabilized by hydrogen-bonding so as to ensure a high fluorescence quantum yield, which makes a decay via isomerization less probable.
Finally, we note that off-switching by exciting the neutral cis chromophore occurs with a lower quantum yield than on-switching via the neutral trans chromophore (Table 2). This difference likely originates from the different proton-accepting properties of the moieties near the neutral hydroxyphenyl group in the cis and trans forms, which contribute to stabilization of the chromophore by hydrogen bonding or electrostatic interactions.
Conclusions for the photoswitching behavior
Reversible photoswitching of FPs may proceed along several pathways (Fig. 1). Depending on the illumination wavelength and the chromophore species present, several pathways may even be activated in parallel. So a whole range of FP properties may arise depending on a number of determinants, including the relative free energies of the protein conformations with a cis or a trans chromophore in the ground and excited states, the isomer-dependent proton affinities of the chromophore’s hydroxyphenyl group in its local environment in the ground and excited states, and the shapes of the energy surfaces connecting the different states, which control the yields of fluorescence and isomerization transitions. An important issue is also that the availability of protons at the internal donor and acceptor sites must be assured. Taking a GFP as an example, blue light (∼400 nm) excites both protonated species, CH and TH, and green light (∼500 nm) excites both anionic species, C− and T−, owing to their spectral overlap. Therefore, the net result of irradiating into the blue and green bands is governed by the relative populations and isomerization yields of the cis and trans species.
If the proton affinities are such that pKcis < pH < pKtrans, only the C− and TH species will be appreciably populated. The low pKcis results from a strong hydrogen bond formed between a serine and the anionic cis chromophore. This bond contributes significantly to the overall stabilization of C−, so it is typically the thermodynamically stable and thus predominant species at neutral pH in the absence of light. Green light excites the fluorescent species, C−, and also switches it to the nonfluorescent TH form; blue light induces the reverse transition from TH to C− and switches the fluorescence back on. Because irradiation into the excitation band of the fluorescent species causes a reduction of fluorescence emission, this scheme has been termed a negative switching mode (37). Dronpa (pKcis = 5 (65)) and mTFP0.7 (pKcis = 4.0 (32,59)) are two examples of such negative photoswitchers. If pKcis is closer to neutral, which is relevant for marker applications, e.g., in mIrisGFP (pKcis = 5.3/6), fastLime (27), the mGeos variants (66), rsTagRFP (pKcis = 6.6 (29)), and rsEGFP (pKcis = 6.6 (26)), a third state (CH) comes into play, resulting in a lower dynamic range between the on and off states, as explained in this work in connection with the data in Fig. 7. In a similar way, for pH ≈ pKtrans, T− as the third state gives rise to more complicated behavior.
For a reverse order of pKs, i.e., pKcis > pH > pKtrans, only the nonfluorescent CH and T− species are appreciably populated. In this case, the low pKtrans results from a strong hydrogen bond formed between the amino acid at position 158 (residue numbering according to mIrisGFP), which is often a serine, and the anionic trans chromophore, so T− is typically the thermodynamically stable species at neutral pH. Photoisomerization can be induced but remains unnoticed in the fluorescence emission due to the lack thereof. If, however, pKcis ≈ pH ≫ pKtrans, the fluorescent C− species is present in addition to CH and T−. This scenario is characteristic of the so-called positive photoswitchers, for which fluorescence excitation enhances the emission (37). One example is Padron, with pKcis = 6 and pKtrans = 4.5 (27,64). Excitation of the ground state T− chromophore with green light generates CH. Subsequently, the protonation equilibrium readjusts to maintain the ratio of the two species, so the net effect is an increase in the fluorescent species, C−. Green-light irradiation excites C− fluorescence, and moreover, photoisomerization may occur as well. However, due to preferential T− excitation and, possibly, different isomerization quantum yields of T− and C−, the net effect is an enhanced C− species. Off-switching is achieved by blue-light excitation of CH, which photoisomerizes to generate nonfluorescent T−. Subsequently, the pH equilibrium between CH and C− readjusts, thereby reducing the fraction of the fluorescent C− species. We note that the fluorescent C− is a minority species in this positive photoswitching mode, so the emission will, in principle, be significantly reduced in comparison to that of negative photoswitchers.
Red FPs can exhibit similar photoswitching properties because the chromophore extension leading to red-shifted optical spectra is practically decoupled from the protonation/isomerization properties of the hydroxyphenyl group (67). The red asFP595 also belongs to the class of positive photoswitchers (25,68). In the dark, the chromophore adopts the anionic trans state. Illumination of T− induces isomerization to the CH state (see Fig. 1 B in Chudakov et al. (69). and Schüttrigkeit et al. (70).). Because of the high pKcis, most cis chromophores are neutral and thus nonfluorescent. The reported fluorescence quantum yield, Φ = 0.001 (71), is extremely low, presumably because it was referenced to the total amount of protein rather than the tiny fraction of C− species responsible for fluorescence.
To conclude, we have studied the coupling of the chromophore to its protein environment in the reversibly photoswitchable mIrisGFP by using optical spectroscopy. By analyzing pH-dependent equilibria and kinetic properties in the native protein and the mutant Glu212Gln, chromophore-protein interactions could be interpreted in structural terms based on the x-ray structure of mIrisFP (20). Our measured quantum yields of photoisomerization are in qualitative agreement with a recently published photoswitching model (42). From these results, we have put forward a general scheme as to how isomeric and acid-base properties of the chromophore in its protein environment can give rise to negative and positive photoswitching modes. This knowledge is required to further analyze the competition between photoconversion and photoswitching in mIrisFP and beneficial for the design of advanced FPs.
Acknowledgments
We gratefully acknowledge our long-standing, fruitful collaboration with Dr. Jörg Wiedenmann (University of Southampton, Southampton, United Kingdom).
G.U.N. was supported by the Deutsche Forschungsgemeinschaft (DFG) and the State of Baden-Württemberg through the Center for Functional Nanostructures (CFN) and by DFG grant Ni 291/9.
Footnotes
Susan Gayda’s present address is Diagnostics Research, Abbott Diagnostics Division, 100 Abbott Park Road, Abbott Park, IL 60064.
Supporting Material
References
- 1.Tsien R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 2.Ormö M., Cubitt A.B., Remington S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- 3.Zimmer M. Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem. Rev. 2002;102:759–781. doi: 10.1021/cr010142r. [DOI] [PubMed] [Google Scholar]
- 4.Remington S.J. Fluorescent proteins: maturation, photochemistry and photophysics. Curr. Opin. Struct. Biol. 2006;16:714–721. doi: 10.1016/j.sbi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Chudakov D.M., Lukyanov S., Lukyanov K.A. Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol. 2005;23:605–613. doi: 10.1016/j.tibtech.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Nienhaus G.U. The green fluorescent protein: a key tool to study chemical processes in living cells. Angew. Chem. Int. Ed. Engl. 2008;47:8992–8994. doi: 10.1002/anie.200804998. [DOI] [PubMed] [Google Scholar]
- 7.Wiedenmann J., Oswald F., Nienhaus G.U. Fluorescent proteins for live cell imaging: opportunities, limitations, and challenges. IUBMB Life. 2009;61:1029–1042. doi: 10.1002/iub.256. [DOI] [PubMed] [Google Scholar]
- 8.Day R.N., Davidson M.W. The fluorescent protein palette: tools for cellular imaging. Chem. Soc. Rev. 2009;38:2887–2921. doi: 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giepmans B.N., Adams S.R., Tsien R.Y. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 10.Lippincott-Schwartz J., Patterson G.H. Development and use of fluorescent protein markers in living cells. Science. 2003;300:87–91. doi: 10.1126/science.1082520. [DOI] [PubMed] [Google Scholar]
- 11.Shaner N.C., Patterson G.H., Davidson M.W. Advances in fluorescent protein technology. J. Cell Sci. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- 12.Shaner N.C., Steinbach P.A., Tsien R.Y. A guide to choosing fluorescent proteins. Nat. Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y., Shyy J.Y., Chien S. Fluorescence proteins, live-cell imaging, and mechanobiology: seeing is believing. Annu. Rev. Biomed. Eng. 2008;10:1–38. doi: 10.1146/annurev.bioeng.010308.161731. [DOI] [PubMed] [Google Scholar]
- 14.Lukyanov K.A., Chudakov D.M., Verkhusha V.V. Innovation: photoactivatable fluorescent proteins. Nat. Rev. Mol. Cell Biol. 2005;6:885–891. doi: 10.1038/nrm1741. [DOI] [PubMed] [Google Scholar]
- 15.Fernández-Suárez M., Ting A.Y. Fluorescent probes for super-resolution imaging in living cells. Nat. Rev. Mol. Cell Biol. 2008;9:929–943. doi: 10.1038/nrm2531. [DOI] [PubMed] [Google Scholar]
- 16.Lippincott-Schwartz J., Patterson G.H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009;19:555–565. doi: 10.1016/j.tcb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hedde P.N., Nienhaus G.U. Optical imaging of nanoscale cellular structures. Biophys. Rev. 2010;2:147–158. doi: 10.1007/s12551-010-0037-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ando R., Hama H., Miyawaki A. An optical marker based on the UV-induced green-to-red photoconversion of a fluorescent protein. Proc. Natl. Acad. Sci. USA. 2002;99:12651–12656. doi: 10.1073/pnas.202320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurskaya N.G., Verkhusha V.V., Lukyanov K.A. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006;24:461–465. doi: 10.1038/nbt1191. [DOI] [PubMed] [Google Scholar]
- 20.Adam V., Lelimousin M., Bourgeois D. Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations. Proc. Natl. Acad. Sci. USA. 2008;105:18343–18348. doi: 10.1073/pnas.0805949105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kremers G.J., Hazelwood K.L., Piston D.W. Photoconversion in orange and red fluorescent proteins. Nat. Methods. 2009;6:355–358. doi: 10.1038/nmeth.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chudakov D.M., Belousov V.V., Lukyanov K.A. Kindling fluorescent proteins for precise in vivo photolabeling. Nat. Biotechnol. 2003;21:191–194. doi: 10.1038/nbt778. [DOI] [PubMed] [Google Scholar]
- 23.Wiedenmann J., Ivanchenko S., Nienhaus G.U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andresen M., Stiel A.C., Jakobs S. Structural basis for reversible photoswitching in Dronpa. Proc. Natl. Acad. Sci. USA. 2007;104:13005–13009. doi: 10.1073/pnas.0700629104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andresen M., Wahl M.C., Jakobs S. Structure and mechanism of the reversible photoswitch of a fluorescent protein. Proc. Natl. Acad. Sci. USA. 2005;102:13070–13074. doi: 10.1073/pnas.0502772102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grotjohann T., Testa I., Hell S.W. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature. 2011;478:204–208. doi: 10.1038/nature10497. [DOI] [PubMed] [Google Scholar]
- 27.Andresen M., Stiel A.C., Jakobs S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat. Biotechnol. 2008;26:1035–1040. doi: 10.1038/nbt.1493. [DOI] [PubMed] [Google Scholar]
- 28.Stiel A.C., Trowitzsch S., Wahl M.C. 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochem. J. 2007;402:35–42. doi: 10.1042/BJ20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subach F.V., Zhang L., Verkhusha V.V. Red fluorescent protein with reversibly photoswitchable absorbance for photochromic FRET. Chem. Biol. 2010;17:745–755. doi: 10.1016/j.chembiol.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subach F.V., Patterson G.H., Verkhusha V.V. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods. 2009;6:153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brakemann T., Stiel A.C., Jakobs S. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat. Biotechnol. 2011;29:942–947. doi: 10.1038/nbt.1952. [DOI] [PubMed] [Google Scholar]
- 32.Henderson J.N., Ai H.W., Remington S.J. Structural basis for reversible photobleaching of a green fluorescent protein homologue. Proc. Natl. Acad. Sci. USA. 2007;104:6672–6677. doi: 10.1073/pnas.0700059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Habuchi S., Tsutsui H., van Oijen A.M. mKikGR, a monomeric photoswitchable fluorescent protein. PLoS ONE. 2008;3:e3944. doi: 10.1371/journal.pone.0003944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voliani V., Bizzarri R., Beltram F. Cis-trans photoisomerization of fluorescent-protein chromophores. J. Phys. Chem. B. 2008;112:10714–10722. doi: 10.1021/jp802419h. [DOI] [PubMed] [Google Scholar]
- 35.Quillin M.L., Anstrom D.M., Remington S.J. Kindling fluorescent protein from Anemonia sulcata: dark-state structure at 1.38 Å resolution. Biochemistry. 2005;44:5774–5787. doi: 10.1021/bi047644u. [DOI] [PubMed] [Google Scholar]
- 36.Martynov V.I., Savitsky A.P., Lukyanov S.A. Alternative cyclization in GFP-like proteins family. The formation and structure of the chromophore of a purple chromoprotein from Anemonia sulcata. J. Biol. Chem. 2001;276:21012–21016. doi: 10.1074/jbc.M100500200. [DOI] [PubMed] [Google Scholar]
- 37.Stiel A.C., Andresen M., Jakobs S. Generation of monomeric reversibly switchable red fluorescent proteins for far-field fluorescence nanoscopy. Biophys. J. 2008;95:2989–2997. doi: 10.1529/biophysj.108.130146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourgeois D., Adam V. Reversible photoswitching in fluorescent proteins: a mechanistic view. IUBMB Life. 2012;64:482–491. doi: 10.1002/iub.1023. [DOI] [PubMed] [Google Scholar]
- 39.Fron E., Flors C., Hofkens J. Ultrafast excited-state dynamics of the photoswitchable protein Dronpa. J. Am. Chem. Soc. 2007;129:4870–4871. doi: 10.1021/ja069365v. [DOI] [PubMed] [Google Scholar]
- 40.Habuchi S., Ando R., Hofkens J. Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proc. Natl. Acad. Sci. USA. 2005;102:9511–9516. doi: 10.1073/pnas.0500489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizuno H., Mal T.K., Miyawaki A. Light-dependent regulation of structural flexibility in a photochromic fluorescent protein. Proc. Natl. Acad. Sci. USA. 2008;105:9227–9232. doi: 10.1073/pnas.0709599105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen S., Lamothe K., Martínez T.J. Protonic gating of excited-state twisting and charge localization in GFP chromophores: a mechanistic hypothesis for reversible photoswitching. J. Am. Chem. Soc. 2010;132:1192–1193. doi: 10.1021/ja907447k. [DOI] [PubMed] [Google Scholar]
- 43.Fuchs J., Böhme S., Nienhaus G.U. A photoactivatable marker protein for pulse-chase imaging with superresolution. Nat. Methods. 2010;7:627–630. doi: 10.1038/nmeth.1477. [DOI] [PubMed] [Google Scholar]
- 44.Adam V., Moeyaert B., Hofkens J. Rational design of photoconvertible and biphotochromic fluorescent proteins for advanced microscopy applications. Chem. Biol. 2011;18:1241–1251. doi: 10.1016/j.chembiol.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 45.Ward W.W., Prentice H.J., Reeves S.C. Spectral perturbations of the Aequorea green-fluorescent protein. Photochem. Photobiol. 1982;35:803–808. [Google Scholar]
- 46.Nienhaus K., Renzi F., Nienhaus G.U. Chromophore-protein interactions in the anthozoan green fluorescent protein asFP499. Biophys. J. 2006;91:4210–4220. doi: 10.1529/biophysj.106.087411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elsliger M.A., Wachter R.M., Remington S.J. Structural and spectral response of green fluorescent protein variants to changes in pH. Biochemistry. 1999;38:5296–5301. doi: 10.1021/bi9902182. [DOI] [PubMed] [Google Scholar]
- 48.Scharnagl C., Raupp-Kossmann R., Fischer S.F. Molecular basis for pH sensitivity and proton transfer in green fluorescent protein: protonation and conformational substates from electrostatic calculations. Biophys. J. 1999;77:1839–1857. doi: 10.1016/S0006-3495(99)77028-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nienhaus K., Renzi F., Nienhaus G.U. Exploring chromophore-protein interactions in fluorescent protein cmFP512 from Cerianthus membranaceus: x-ray structure analysis and optical spectroscopy. Biochemistry. 2006;45:12942–12953. doi: 10.1021/bi060885c. [DOI] [PubMed] [Google Scholar]
- 50.Müller J.D., McMahon B.H., Nienhaus G.U. Connection between the taxonomic substates and protonation of histidines 64 and 97 in carbonmonoxy myoglobin. Biophys. J. 1999;77:1036–1051. doi: 10.1016/s0006-3495(99)76954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ullmann G.M. Relations between protonation constants and titration curves in polyprotic acids: a critical view. J. Phys. Chem. B. 2003;107:1263–1271. [Google Scholar]
- 52.Chattoraj M., King B.A., Boxer S.G. Ultra-fast excited state dynamics in green fluorescent protein: multiple states and proton transfer. Proc. Natl. Acad. Sci. USA. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lossau H., Kummer A., Michel-Beyerle M.E. Time-resolved spectroscopy of wild-type and mutant green fluorescent proteins reveals excited state deprotonation consistent with fluorophore-protein interactions. Chem. Phys. 1996;213:1–16. [Google Scholar]
- 54.Nienhaus G.U. The “wiggling and jiggling of atoms” leading to excited-state proton transfer in green fluorescent protein. ChemPhysChem. 2010;11:971–974. doi: 10.1002/cphc.200901016. [DOI] [PubMed] [Google Scholar]
- 55.Bizzarri R., Nifosì R., Beltram F. Green fluorescent protein ground states: the influence of a second protonation site near the chromophore. Biochemistry. 2007;46:5494–5504. doi: 10.1021/bi602646r. [DOI] [PubMed] [Google Scholar]
- 56.Henderson J.N., Remington S.J. Crystal structures and mutational analysis of amFP486, a cyan fluorescent protein from Anemonia majano. Proc. Natl. Acad. Sci. USA. 2005;102:12712–12717. doi: 10.1073/pnas.0502250102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X., Chung L.W., Morokuma K. A theoretical study on the nature of on- and off-states of reversibly photoswitching fluorescent protein Dronpa: absorption, emission, protonation, and Raman. J. Phys. Chem. B. 2010;114:1114–1126. doi: 10.1021/jp909947c. [DOI] [PubMed] [Google Scholar]
- 58.Wilmann P.G., Turcic K., Rossjohn J. The 1.7 Å crystal structure of Dronpa: a photoswitchable green fluorescent protein. J. Mol. Biol. 2006;364:213–224. doi: 10.1016/j.jmb.2006.08.089. [DOI] [PubMed] [Google Scholar]
- 59.Ai H.W., Henderson J.N., Campbell R.E. Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: structural characterization and applications in fluorescence imaging. Biochem. J. 2006;400:531–540. doi: 10.1042/BJ20060874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abbruzzetti S., Bizzarri R., Beltram F. Photoswitching of E222Q GFP mutants: “concerted” mechanism of chromophore isomerization and protonation. Photochem. Photobiol. Sci. 2010;9:1307–1319. doi: 10.1039/c0pp00189a. [DOI] [PubMed] [Google Scholar]
- 61.Luin S., Voliani V., Beltram F. Raman study of chromophore states in photochromic fluorescent proteins. J. Am. Chem. Soc. 2009;131:96–103. doi: 10.1021/ja804504b. [DOI] [PubMed] [Google Scholar]
- 62.Bizzarri R., Serresi M., Beltram F. Single amino acid replacement makes Aequorea victoria fluorescent proteins reversibly photoswitchable. J. Am. Chem. Soc. 2010;132:85–95. doi: 10.1021/ja9014953. [DOI] [PubMed] [Google Scholar]
- 63.Faro A.R., Adam V., de Rosny E. Low-temperature switching by photoinduced protonation in photochromic fluorescent proteins. Photochem. Photobiol. Sci. 2010;9:254–262. doi: 10.1039/b9pp00121b. [DOI] [PubMed] [Google Scholar]
- 64.Brakemann T., Weber G., Jakobs S. Molecular basis of the light-driven switching of the photochromic fluorescent protein Padron. J. Biol. Chem. 2010;285:14603–14609. doi: 10.1074/jbc.M109.086314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ando R., Mizuno H., Miyawaki A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- 66.Chang H., Zhang M., Xu T. A unique series of reversibly switchable fluorescent proteins with beneficial properties for various applications. Proc. Natl. Acad. Sci. USA. 2012;109:4455–4460. doi: 10.1073/pnas.1113770109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nienhaus G.U., Wiedenmann J. Structure, dynamics and optical properties of fluorescent proteins: perspectives for marker development. ChemPhysChem. 2009;10:1369–1379. doi: 10.1002/cphc.200800839. [DOI] [PubMed] [Google Scholar]
- 68.Schäfer L.V., Groenhof G., Grubmüller H. Photoswitching of the fluorescent protein asFP595: mechanism, proton pathways, and absorption spectra. Angew. Chem. Int. Ed. Engl. 2007;46:530–536. doi: 10.1002/anie.200602315. [DOI] [PubMed] [Google Scholar]
- 69.Chudakov D.M., Feofanov A.V., Lukyanov K.A. Chromophore environment provides clue to “kindling fluorescent protein” riddle. J. Biol. Chem. 2003;278:7215–7219. doi: 10.1074/jbc.M211988200. [DOI] [PubMed] [Google Scholar]
- 70.Schüttrigkeit T.A., von Feilitzsch T., Michel-Beyerle M.E. Femtosecond study of light-induced fluorescence increase of the dark chromoprotein asFP595. Chem. Phys. 2006;323:149–160. [Google Scholar]
- 71.Lukyanov K.A., Fradkov A.F., Lukyanov S.A. Natural animal coloration can be determined by a nonfluorescent green fluorescent protein homolog. J. Biol. Chem. 2000;275:25879–25882. doi: 10.1074/jbc.C000338200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.