

Abstract
A review of what is presently known about the G protein coupled receptor GPR18 in terms of its expression and distribution, pharmacology and potential implications for central nervous system and endocannabinoid system signalling.
LINKED ARTICLES
This article is part of a themed section on Cannabinoids. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.167.issue-8
Keywords: GPR18, NAGly, abnormal cannabidiol receptor, endocannabinoid, microglia
Introduction
In normal brain, microglia possess a characteristic ramified morphology that facilitates continuous CNS surveillance. Following CNS injury or inflammation, receptor-initiated signalling cascades recruit microglia to sites of damage, where the local microenvironment governs the bespoke morphological and phenotypic characteristics they manifest (Wake et al., 2009). The molecular mechanisms that recruit and instruct microglia to adapt their behaviour in this fashion are poorly understood. Neurons are known to use the endogenous cannabinoid system (eCBs) to communicate with each other (Schlicker and Kathmann, 2001; Pertwee and Ross, 2002), and Walter et al. (2003) reported the involvement of the eCB signalling in recruiting microglia towards dying neurons in vitro. The majority of cannabinoid effects on microglia have been conducted using the immortalized primary microglial cell line (BV-2). While this cell line is not always fully representative of in situ microglia, it has been shown to retain most of the morphological, phenotypical and functional properties described for freshly isolated active microglial cells (Blasi et al., 1990). Indeed, Walter and colleagues demonstrated that pathological stimulation of both neurons and microglia dramatically and selectively increased the production of 2-arachidonoyl glycerol (2-AG), which, together with other structurally related lipids, triggered microglial cell migration by engaging CB2 and abnormal cannabidiol (Abn-CBD) receptors (Walter et al., 2003). N-arachidonoyl glycine (NAGly)-GPR18 signalling has been recently introduced as an important ‘new player’ in microglial–neuronal communication, providing a novel mechanism (both receptor and ligand) for directed migration and phenotypic switches in microglia (McHugh et al., 2010). The published data strongly support a significant role for NAGly and GPR18 in regulating microglia in the CNS and, together with subsequent work, have wider implications for our understanding of the eCB system. This review will outline what is presently known about GPR18, its pharmacology and the potential impact of NAGly-GPR18 signalling on the field.
GPR18 expression and distribution
Mammalian GPCRs constitute a superfamily of diverse proteins with hundreds of members, each having seven transmembrane domains (Bockaert and Pin, 1999). On the basis of shared sequence motifs, they are grouped into four classes: A, B, C and F/S (Horn et al., 1998). The established cannabinoid receptors, CB1 and CB2, belong to class A (i.e. rhodopsin-like) and share considerable structural and phylogenetic homology (see Pertwee et al., 2010 for review). Another GPCR that is activated by the phytocannabinoid, Δ9-tetrahydrocannabinol (Δ9-THC), and by an oxygenated metabolite of the endogenous cannabinoid anandamide (NAGly) has been recently identified (Bradshaw et al., 2009; McHugh et al., 2010; 2012). This GPCR, GPR18, likewise belongs to class A; however, its structural and phylogenic background are dissimilar to CB1 and CB2 (see Pertwee et al., 2010 for review).
The first reports to describe GPR18 arose unanticipated from various broad expression studies of GPCRs. In 1997, while exploiting relaxed stringency PCR to identify a receptor for gastrin-releasing hormone, Gantz et al. inadvertently isolated fragments of a novel 7TM GPCR, 331 amino acids long, from canine gastric mucosa and the human colonic cancer Colo 320DM cell line. Subsequent cloning and genomic library screening identified this gene as GPR18 (designated according to human gene mapping workshop nomenclature) and indicated that the human and canine clones were highly conserved. Fluorescence in situ hybridization was used to localize GPR18 to human chromosome 13q32, where it clusters with Epstein-Barr virus-induced receptor 2 (EBI2) and the lipid receptors, cysteinyl leukotriene receptor 1 and 2 (CysLT1 and CysLT2) (Gantz et al., 1997; Rosenkilde et al., 2006). Northern blot analysis conducted by Gantz et al. in multiple human tissues reported GPR18 transcripts in spleen, thymus, peripheral blood leucocytes, small intestine, appendix and lymph node – suggesting a possible role for GPR18 in immune system regulation. However, the most abundant expression observed was in the testis, where transcripts were found in several cell types. GPR18 mRNA was detected in gametes of all levels of differentiation, with the highest in the most terminally differentiated cells. The tissues Gantz et al. reported to lack any apparent GPR18 mRNA included brain, heart, lung, liver, kidney, pancreas, colon, skeletal muscle, ovary, placenta, prostate, adrenal medulla and adrenal cortex.
Five years later, Vassilatis et al. (2003) published a study that had examined the GPCR repertoires of humans and mice via real-time (RT)-PCR tissue profiling. They reported the following four different expression levels for GPR18: no expression– amygdala, frontal cortex, hippocampus, liver and muscle; low expression– cortex, thalamus, adrenal tissue, colon, intestine, kidney, prostate, skin, spleen, stomach and uterus; moderate expression– lung, ovary, testis, thymus and striatum; strong expression– hypothalamus, thyroid, peripheral blood leucocytes, cerebellum and brain stem (Vassilatis et al., 2003).
In 2006, while searching for chemokine receptors and GPCRs expressed in adult T-cell leukaemia (ATL) cells, Kohno et al. found GPR18 amongst the genes of relevance. After cloning and further analysis, the study reported that GPR18 was more highly expressed in lymphocytes (CD4+, CD4+CD45RA+, CD4+CD45RO+, CD8+ and CD19+) in comparison with monocytes. Stably transfected GPR18-expressing cell lines were created and used in conjunction with the Bioactive Lipid Library, Ca2+ mobilization and cAMP assays to subsequently identify NAGly as an endogenous ligand for GPR18. Kohno et al. observed concentration-dependent inhibition of forskolin-stimulated cAMP production in GPR18-transfected CHO cells from 1 nM–10 µM NAGly, with an IC50 value of 20 ± 8 nM. The NAGly-mediated inhibition was completely abolished by pertussis toxin (PTX), indicating GPR18 coupling to Gαi (Kohno et al., 2006). Studies by McHugh et al. have shown a similar potency and PTX sensitivity (McHugh et al., 2010; 2012).
Yin et al. (2009) screened a large number of newly ‘deorphaned’ receptors against a collection of ∼400 receptors using the β-arrestin PathHunter™ assay system (DiscoveRx, Fremont, CA, USA) and HEK293 cells stably expressing β-arrestin2-β-gal-EA fusion protein. NAGly was not part of their original lipid collection but was later tested on GPR18. The authors reported that they found NAGly to be inactive at GPR18 but did not present their data or details regarding the concentration(s) of NAGly employed in their GPR18 experiment. A number of possible explanations may account for this negative result: firstly, GPCRs are promiscuous. Their coupling partners vary a lot and include both β-arrestin-dependent and β-arrestin-independent pathways. GPR18 mediated effects of NAGly occurring, although a β-arrestin-independent pathway would be missed by the screening system used in the Yin et al. study. Secondly, it is the experience of multiple investigators that GPR18 is difficult to successfully transfect (unpublished observations); low or no expression of GPR18 would also result in a lack of response to NAGly. Thirdly, the concentration of NAGly may have been inappropriately high or low. Without further information to provide a context, a final interpretation of this piece of data is problematic.
The most current of the PCR-based GPCR screening investigations to involve GPR18 was published in 2010 by Qin et al., who performed a comprehensive array-based, quantitative PCR analysis of the expression profile of 130 genes in three typical sites of melanoma metastases. A comparison between metastases and benign nevi revealed 16 genes that were significantly differentially expressed. Of these, GPR18 and the chemokine ligand CCL4 had the greatest changes in expression levels, which were 24.1- and 27.4-fold higher, respectively. Subsequently, functional experiments in yeast and melanoma were designed to test the hypothesis that GPR18 was able to mediate proliferative or anti-apoptotic signalling. They found that the GPR18 sequence deviated from other GPCRs at position 3.35, where an alanine is present in place of a normally highly conserved asparagine. Asparagine to alanine mutations at 3.35 have been previously shown to result in constitutive activity in CXCR3 and CXCR4 chemokine receptors, precluding the requirement of an agonist ligand to activate them (Ballesteros and Weistein, 1995). Qin et al. reported that mutating the alanine back to asparagine at 3.35 resulted in the loss of constitutive activity of GPR18. This is of interest given that malignant cells are dependent on the constitutive or overexpression of driver genes for maintenance of cell survival or inhibition of apoptosis. Qin et al. (2010) found that in vitro siRNA-mediated knockdown of GPR18 in human melanoma cells enhanced death via apoptosis in further support of this hypothesis.
Using comparative gene microarray analysis, a recent study by Juknat et al. (2012) characterized differential transcriptional profiles in BV-2 microglia when exposed to 10 µM concentrations of CBD and Δ9-THC for 6 h. Relative levels of CB1, CB2, GPR18, TRPV2 and fatty acid amide hydrolase (FAAH) were not significantly altered; however, Juknat and colleagues did find 1298 transcripts that were differentially regulated by the treatments (Juknat et al., 2012). CBD affected many more genes than Δ9-THC, emphasizing that Δ9-THC and CBD can signal through different pathways in BV-2 microglia. This study lays the groundwork for potential insights regarding the signalling mechanisms of GPR18 in BV-2 microglia by identifying transcriptional targets of CBD and Δ9-THC in BV-2.
In summary, on the basis of mRNA transcripts, there is evidence of GPR18 expression in gastrointestinal, immune and testicular tissues, as well as various brain structures and metastatic melanoma. Using GPR18-targetting antibodies, kindly donated by Dr Ken Mackie (Indiana University), McHugh et al. provided evidence of GPR18 receptors in BV-2 microglia. Further immunohisto- and immunocytochemistry in the field will help solidify the pattern of GPR18 expression in humans and the commonly used animal models. This important next step is currently awaiting the availability of GPR18−/− animals in order to definitively verify selectivity of custom or commercially available GPR18 antibodies.
Pharmacology of GPR18
Two recently published papers have described the pharmacology of NAGly, various cannabinoids and other signalling ligands at GPR18. The first investigated the relationship between GPR18 and Abn-CBD receptor in BV-2 microglia (McHugh et al., 2010). The Abn-CBD receptor is a prominent non-CB1/non-CB2 cannabinoid receptor, discriminated by means of various pharmacological and genetic tools and implicated in the modulation of microglial, endothelial and glioma cell migration, and various cardiovascular responses (Járai et al., 1999; Franklin and Stella, 2003; Offertáler et al., 2003; Walter et al., 2003; Mo et al., 2004; Begg et al., 2005; Vaccani et al., 2005; Mackie and Stella, 2006). Using Boyden chamber migration experiments, yellow tetrazolium (MTT) conversion, in-cell Western, qPCR and immunocytochemistry, it was reported that NAGly at sub-nanomolar, and Abn-CBD and O-1602 at low nanomolar, concentrations potently induced directed cell migration in both BV-2 microglia and HEK293-GPR18-transfected cells, but not in non-transfected HEK293 wild-type cells; the migration effects were blocked or attenuated in both systems by the Abn-CBD receptor antagonist O-1918, and its low efficacy agonist, cannabidiol; NAGly promoted proliferation and activation of MAP kinases in BV-2 microglia and HEK293-GPR18 cells at low nanomolar concentrations – cellular responses correlated with microglial migration; and BV-2 microglia displayed GPR18 immunocytochemical staining and abundant GPR18 mRNA, while qPCR demonstrated that primary microglia, likewise, express abundant amounts of GPR18 mRNA (McHugh et al., 2010). Further work to fully characterize GPR18 expression and function in primary microglia and validate the data arising from BV-2 microglia will be a welcome next step for the field. At the 2011 symposium of the International Cannabinoid Research Society (ICRS), data were presented from siRNA GPR18 knockdown studies showing that migration induced by NAGly, O-1602 and Abn-CBD was significantly attenuated in GPR18 knockdown BV-2 cells compared with the control, whereas migration to vehicle and fMLP (a formylated tripeptide chemoattractant ligand known to stimulate migration through its own distinct GPCRs) remained unchanged (McHugh et al., 2011). Collectively, these data provide definitive evidence that these compounds, characteristic of Abn-CBD receptor pharmacology, are acting via GPR18 in BV-2 microglia.
The second recent publication presented evidence that the endocannabinoid system plays a regulatory role in human endometrial HEC-1B cell migration. Endogenous and phytocannabinoid effects implied a signalling mechanism mediated through CB2 receptors and, to a greater extent, GPR18, which echoes the CB2/Abn-CBD dual expression first described in microglia by Walter et al. (2003). The most effective activator of endometrial cell migration was NAGly, and RT-qPCR revealed that HEC-1B endometrial cells express GPR18 mRNA (McHugh et al., 2012). In addition, the specificity of cannabinoid ligands including traditional CB1 and CB2 receptor agonists and antagonists at GPR18 was screened via p44/42 MAPK activation in stably transfected HEK293-GPR18 cells. In order of potency, NAGly, O-1602, Abn-CBD, Δ9-THC, N-arachidonoyl ethanolamide (AEA) and arachydonylcyclopropylamide (ACPA) are full agonists at GPR18; CBD and AM251 weak GPR18 partial agonists/antagonists; and WIN55212-2, CP55940, R1-methAEA, JWH-133 and JWH-015 had no effect (for EC50 values, refer to McHugh et al., 2012). Figure 1 shows a Venn diagram demonstrating the overlap of AEA, 2-AG and Δ9-THC for all three receptors as well as the specificity of agonists and antagonists at each receptor. Table 1 summarizes the pharmacology of cannabinoids and related compounds at GPR18 known at present.
Figure 1.
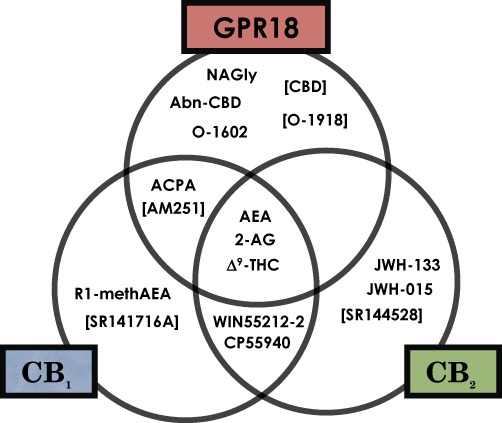
Venn diagram indicating the agonist and antagonist activity that is selective for or shared between CB1, CB2 and GPR18 receptors with regard to various cannabinoid or related compounds. Compounds in square parentheses act as antagonists. CB1, CB2 and GPR18 receptors were stably transfected in HEK293 cells (see McHugh et al., 2010 and McHugh et al., 2012 for further details).
Table 1.
Activation of GPR18, CB1 or CB2 receptors stably transfected into HEK293 cells induced p44/42 MAP kinase phosphorylation by cannabinoids and related lipids
| Compound | Activity at GPR18 | EC50 values (95% confidence limits) | Emax values (95% confidence limits) |
|---|---|---|---|
| NAGly | Full agonist | 44.5 nM (32.4–61.0) | 101.6% (98.4–104.9) |
| O-1602 | Full agonist | 65.3 nM (49.3–86.5) | 102.1% (98.9–105.3) |
| Abn-CBD | Full agonist | 835.7 nM (579.6–1205) | 98.6% (92.6–104.6) |
| Δ9-THC | Full agonist | 0.96 µM (0.43–2.12) | 104.9% (88.7–121.2) |
| AEA | Full agonist | 3.83 µM (2.11–6.90) | 109.0% (97.3–120.8) |
| ACPA | Full agonist | 13.5 µM (9.03–20.4) | 105.5% (95.8–115.3) |
| CBD | Partial agonist/antagonist | 51.1 µM (35.8–73.2) | 61.9% (46.8–77.0) |
| AM251 | Partial agonist/antagonist | 96.4 µM (n/a) | 50.3% (n/a) |
| WIN55212-2 | None | – | – |
| CP55,940 | None | – | – |
| R1-methAEA | None | – | – |
| JWH-133 | None | – | – |
| JWH-015 | None | – | – |
| SR141716A | None | – | – |
| SR144528 | None | – | – |
| Compound | Activity at CB1 | EC50 values (95% confidence limits) | Emax values (95% confidence limits) |
|---|---|---|---|
| CP55,940 | Full agonist | 4.56 nM (3.60–5.79) | 97.3% (94.8–98.5) |
| ACPA | Full agonist | 29.5 nM (25.1–34.8) | 98.2% (94.9–100.3) |
| WIN55212-2 | Full agonist | 35.9 nM (30.7–42.0) | 97.1% (95.0–99.2) |
| R1-methAEA | Full agonist | 108.1 nM (97.6–120.0) | 96.7% (94.8–98.9) |
| Compound | Activity at CB2 | EC50 values (95% confidence limits) | Emax values (95% confidence limits) |
|---|---|---|---|
| JWH-133 | Full agonist | 95.7 nM (68.0–115.9) | 98.1% (95.7–100.4) |
| JWH-015 | Full agonist | 207.5 nM (158.7–245.6) | 98.9% (97.3–101.6) |
EC50 and Emax values (percentage of p44/42 MAP kinase activation induced by 10 µM ionomycin) were calculated from sigmoidal concentration–response curves constructed in GraphPad Prism 4 (GraphPad Software, San Diego, CA, USA). The data represent the mean with 95% confidence limits, n= 3 (McHugh et al., 2012).
The background activity of GPR18 receptors in metastatic melanoma reported by Qin et al. (2010), together with the pharmacological profile of CBD and AM251, raises the possibility that low efficacy partial agonist/antagonist or inverse agonist compounds may reduce tonic signalling even in the absence of an agonist. This would be in keeping with the majority of CB1 and CB2 antagonists, including SR141, AM251, SR144 and AM630, which have been reported to behave as inverse agonists. Whether or not this would be expected for GPR18 in a given cell or tissue type depends on the extent of GPR18 receptor expression, the degree to which a subpopulation of GPR18 receptors is constitutively active (i.e. coupled spontaneously to their signalling pathways) and the binding affinity of the ligand in question for the ‘active’ or ‘inactive’ GPR18 receptor state. No binding affinity or quantitative expression data currently exist for GPR18 in microglia.
Takenouchi et al. (2012) have recently examined the effects of NAGly on the mouse macrophage-derived cell line, RAW264.7, and primary mouse peritoneal macrophages. They reported that 30 µM NAGly caused an ∼70% significant reduction in the cell viability of RAW264.7 cells via apoptosis after 24 h. Caspase-3 was activated after the addition of 30 µM NAGly for 12 h, while the Bax/Bcl-2 ratio (an indicator of apoptosis) was increased. Apoptosis was suppressed by pretreatment with PTX and transfection with GPR18-specific siRNA. With regard to the primary mouse cells, 3 µM NAGly significantly reduced the cell viability of M1 and M2 peritoneal macrophages by ∼40 and ∼10%, respectively. Notably, the M1 macrophages exhibited a higher expression of GPR18 mRNA than the M2 cells. The study also demonstrated activation of p44/42 (ERK1/2), p38, and to a lesser extent, JNK MAPKs by NAGly, and that treatment with LPS and interferon-γ augmented GPR18 mRNA levels when compared with treatment with IL-4 or no stimulation (Takenouchi et al., 2012). Collectively, these data confirm the previous findings of Kohno et al. (2006) and McHugh et al. (2012), and suggest that NAGly might function as an anti-inflammatory factor to reduce pro-inflammatory macrophages.
The Takenouchi et al. (2012) study confirms a report by Burstein et al. (2011) regarding the resolution of inflammation by NAGly in a mouse peritonitis model. Here, 0.3 and 1.2 mg·kg−1 oral doses of NAGly significantly reduced the migration of inflammatory leucocytes following injection of the pro-inflammatory agent, thioglycollate, by greater than 50% (Burstein et al., 2011). The authors also report that treatment of GPR18-transfected HEK293 cells with 5 µM NAGly caused a 40-fold increase in the concentration of the prostaglandin, PGJ, in culture media. Addition of polyclonal anti-GPR18 antibody reduced this response to fourfold, suggesting that GPR18 mediates this action. The 3 µM NAGly treatment of HEK293-GPR18 cells caused a 1.78-fold increase in trypan blue staining (an indicator of reduced cell viability), which was blocked by pretreatment with polyclonal anti-GPR18 antibody. FACS analysis following annexin staining of NAGly-treated HEK293-GPR18 supported the occurrence of programmed cell death (i.e. apoptosis) (Burstein et al., 2011).
GPR18 in microglia: implications for neuron–glia interactions
Directed migration, selective phagocytosis and free radical production are critical functions of microglia that have a significant impact on overall CNS stability, both from an acute and a long-term perspective. In both developmental and post-developmental contexts, microglia discriminately engulf and eliminate dead or dying neurons (Ferrer et al., 1990; Wake et al., 2009). These processes must be tightly controlled in order to sustain the least possible collateral damage to adjacent neurons. Indeed, microglial activity is not only orchestrated to exacting standards, but the coupling between the death of neurons and their degradation by microglia is both striking and swift (Peri and Nusslein-Volhard, 2008). This remarkable relationship suggests a fast-acting communication between neurons and microglia, such that the microglia are forewarned of the specific task (i.e. apoptosis, infection or damage). Yet, microglia are also implicated in virtually all CNS neuropathological processes, where they become highly reactive to dying neurons and provoke sustained secondary neurotoxicity (Graeber et al., 1988a,b; Konno et al., 1989; Rao and Lund, 1989; Flaris and Hickey, 1992; Morioka et al., 1992; Berman et al., 1999; Rezaie and Lantos, 2001; Segal, 2003; Games et al., 2006; Gowing et al., 2006; Czigner et al., 2007; Denes et al., 2007; Yasuda et al., 2007). This extreme dichotomy of behaviour underscores the importance of understanding the specific signalling systems that both recruit and instruct microglia to adapt their phenotype selectively in response to damaged/dying/apoptotic neurons, and which are foundational to our ability to monitor and manage dys-regulated microglial activity.
Microglial migration and phenotypic modification are induced by various ligands, including bacterial peptides (e.g. formyl-methionyl-leucyl-phenylalanine, fMLP), lysophospholipids (e.g. lysophosphatidic acid, LPA) and endocannabinoids (Lorton et al., 2000; Schilling et al., 2004). The potential importance of NAGly-GPR18 signalling as a ‘new player’ in microglial–neuronal communication can be appreciated when compared with other typical signalling molecules. Picomolar concentrations of NAGly elicit a response twice that produced by 1 µM fMLP or 1 µM LPA. Additionally, NAGly is 30- and 700-fold more potent than the endocannabinoids, 2-AG and AEA, respectively, and more efficacious than either (McHugh et al., 2010). To date, there are no Food and Drug Administration-approved pharmacological therapies that target microglial–neuronal receptor-ligand interactions. Some promising results have been described regarding the neuroprotective effects of minocycline, a broad spectrum tetracycline antibiotic, against a host of neurodegenerative diseases. Minocycline disrupts T cell-microglia signalling, impairing T-cell-induced TNF-α production by microglia and also blocks activation of microglia via inhibition of NF-κB nuclear translocation (Baptiste et al., 2005; Giuliani et al., 2005; Maier et al., 2007). In line with this, elucidating the NAGly-GPR18 neuronal–microglial communication system has the potential to lead to novel pharmacotherapies centred on enhancing (optimized GPR18 ligands) or suppressing (optimized GPR18 antagonists) microglial activation in the CNS.
GPR18 and N-arachidonoyl glycine: potential impact on the eCB system
A quarter of a century ago, Dr Raphael Mechoulam and Dr Yehiel Gaoni's key breakthrough in isolating Δ9-THC eventually led to the molecular cloning of CB1 and CB2 in the early 1990s. The identification of these two distinct GPCRs, in turn, triggered a search for an endogenous cannabinoid, which culminated in the identification of the lipid molecule N-arachidonoyl ethanolamide or anandamide (AEA; Devane, 1992). Brain-derived NAGly is synthesized primarily from AEA via a fatty acid amide hydrolase (FAAH)-dependent pathway and can be prevented by URB597, an irreversible inhibitor of FAAH (Bradshaw et al., 2009). NAGly has been reported to be ineffective as an agonist at either CB1 or CB2 receptors (Sheskin et al., 1997; Huang et al., 2001), despite the obvious structural overlap with AEA.
As described earlier, AEA and Δ9-THC are full agonists (although neither are especially potent) at GPR18 receptors, while CBD behaves as a low efficacy partial agonist/antagonist. This pharmacology of established cannabinoids at GPR18 receptors, together with the precursor relationship between AEA and NAGly, has raised the prospect that NAGly and GPR18 are either additional pieces of the endocannabinoid system puzzle or components of a distinct signalling mechanism that readily interacts with the eCB system. Further work is necessary before we can discern which hypothesis is correct.
In the meantime, GPR18 signalling has important and immediate implications with regard to how to interpret past literature and current studies (see Alexander, 2012 for further commentary). The potential role of GPR18 must be brought to the table and considered when interpreting the effects of AEA or Δ9-THC as being due to either or both CB1/CB2; making use of CBD in combination with AEA or Δ9-THC; or employing the CB1-versus-CB2 selective agonist ACPA (also a GPR18 agonist) or CB1-versus-CB2 selective antagonist AM251 (also GPR18 antagonist) to suggest a role for CB1.
Conclusions
We are now in the midst of major advances in biochemistry/physiology associated with therapeutic actions of the endocannabinoids, including fertility, neurodegeneration and neuroprotection, learning and memory, anxiety, pain relief, treatment of cancer, anti-nausea, appetite and obesity, and drug abuse. The discovery of a new endogenous ligand (NAGly) and cannabinoid receptor (GPR18) is a valuable contribution to the field's understanding of the molecular mechanisms responsible for the effects of both endogenous and phytocannabinoids. Indeed, further investigation of NAGly-GPR18 signalling in microglia will greatly enhance our understanding of the extent to which endocannabinoids and related endogenous lipids initiate and maintain neuroimmunological events via microglia.
Acknowledgments
None.
Glossary
- 2-AG
2-arachidonoyl glycerol
- 7TM
seven transmembrane
- Abn-CBD
abnormal cannabidiol
- ACPA
arachidonoyl cyclopropylamide
- AM251
1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidyl)pyrazole-3-carboxamide
- AM630
[6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)-methanone
- AEA
N-arachidonoyl ethanolamine
- BV-2
mouse microglial cell line BV-2
- CBD
cannabidiol
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CP55940
2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl) cyclohexyl]-5-(2-methyloctan-2-yl)phenol
- CysL1
cysteinyl receptor 1
- CysL2
cysteinyl receptor 2
- Δ9-THC
Δ9-tetrahydrocannabinol
- EBI2
Epstein-Barr virus-induced receptor 2
- eCBS
endogenous cannabinoid system
- FAAH
fatty acid amide hydrolase
- FACS
fluorescence activated cell sorting
- FBS
fetal bovine serum
- fMLP
N-formyl-methionine-leucine-phenylalanine
- FPR
formyl peptide receptor 1
- FPRL-1
formyl peptide receptor-like 1
- GFP
green fluorescence protein
- HEC-1B
human endometrial cell line 1-B
- JWH-015
(2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone
- JWH-133
(6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran
- LPA
lysophosphatidic acid
- NAGly
N-arachidonoyl glycine
- O-1602
trans-4- [3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-5-methyl-1,3-benzenediol
- O-1918
1,3-dimethoxy-5-methyl-2- [(1R,6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl)-benzene
- PTX
pertussis toxin
- qPCR
quantitative PCR
- RAW264.7
mouse macrophage-derived cell line RAW264.7
- R1-methAERA
N-(2-hydroxy-1R-methylethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide
- RT-PCR
reverse transcriptase PCR
- simRNA
short interfering messenger RNA
- siRNA
short interfering RNA
- SR141
(SR141716A
- rimonabant)
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride
- SR144
(SR144528), 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N–[(1S,4R,6S)-1,5,5-trimethyl-6bicyclo[2.2.1]heptanyl]pyrazole-3-carboxamide
- WIN55212-2
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanonel
- URB597
(3′-(aminocarbonyl)[1,1′-biphenyl]-3-yl)-cyclohexylcarbamate
Conflicts of interest
The author has no conflicts of interest.
References
- Alexander S. So what do we call GPR18 now? Br J Pharmacol. 2012;165:2411–2413. doi: 10.1111/j.1476-5381.2011.01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J, Weistein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G-protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Baptiste DC, Powel KJ, Jollimore CAB, Hamilton C, LeVatte TL, Archibald ML, et al. Effects of minocycline and tetracycline on retinal ganglion cell survival after axotomy. Neuroscience. 2005;134:575–582. doi: 10.1016/j.neuroscience.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Begg M, Pacher P, Bátkai S, Osei-Hyiaman D, Offertáler L, Mo FM, et al. Evidence for novel cannabinoid receptors. Pharmacol Ther. 2005;106:133–145. doi: 10.1016/j.pharmthera.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Berman NE, Marcario JK, Yong C, Raghavan R, Raymond LA, Joag SV, et al. Microglial activation and neurological symptoms in the SIV model of NeuroAIDS: association of MHC-II and MMP-9 expression with behavioral deficits and evoked potential changes. Neurobiol Dis. 1999;6:486–498. doi: 10.1006/nbdi.1999.0261. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzola R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Bockaert J, Pin PJ. Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw HB, Rimmerman N, Hu SS-J, Burstein S, Walker JM. Novel endogenous N-acyl glycines: identification and characterization. Vitam Horm. 2009;81:191–205. doi: 10.1016/S0083-6729(09)81008-X. [DOI] [PubMed] [Google Scholar]
- Burstein SH, McQuian CA, Ross AH, Salmonsen RA, Zurier RE. Resolution of inflammation by N-arachidonoylglycine. J Cell Biochem. 2011;112:3227–3233. doi: 10.1002/jcb.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czigner A, Mihaly A, Farkas O, Buki A, Krisztin-Peva B, Dobo E, et al. Kinetics of the cellular immune response following closed head injury. Acta Neurochir. 2007;149:281–289. doi: 10.1007/s00701-006-1095-8. [DOI] [PubMed] [Google Scholar]
- Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, et al. Proliferating resident microglia after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1941–1953. doi: 10.1038/sj.jcbfm.9600495. [DOI] [PubMed] [Google Scholar]
- Devane WA. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;252:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Bernet E, Soriano E, del Rio T, Fonseca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- Flaris NA, Hickey WF. Development and characterization of an experimental model of brain abscess in the rat. Am J Pathol. 1992;141:1299–1307. [PMC free article] [PubMed] [Google Scholar]
- Franklin A, Stella N. Arachidonylcyclopropylamide increases microglial cell migration through cannabinoid CB2 and abnormal-cannabidiol-sensitive receptors. Eur J Pharmacol. 2003;474:195–198. doi: 10.1016/s0014-2999(03)02074-0. [DOI] [PubMed] [Google Scholar]
- Games D, Buttini M, Kobayashi D, Schenk D, Seubert P. Mice as models: transgenic approaches and Alzheimer's disease. J Alzheimers Dis. 2006;9:133–149. doi: 10.3233/jad-2006-9s316. [DOI] [PubMed] [Google Scholar]
- Gantz I, Muraoka A, Yang Y-K, Samuelson LC, Zimmerman EM, Cook H, et al. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics. 1997;42:462–466. doi: 10.1006/geno.1997.4752. [DOI] [PubMed] [Google Scholar]
- Giuliani F, Hader W, Yong VW. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J Leukoc Biol. 2005;78:135–143. doi: 10.1189/jlb.0804477. [DOI] [PubMed] [Google Scholar]
- Gowing G, Vallieres L, Julien JP. Mouse model for ablation of proliferating microglia in acute CNS injuries. Glia. 2006;53:331–337. doi: 10.1002/glia.20288. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Kreutzberg GW. Axotomy of the rat facial nerve leads to increased CR3 complement receptor expression by activated microglial cells. J Neurosci Res. 1988a;21:18–24. doi: 10.1002/jnr.490210104. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Tetzlaff W, Streit WJ, Kreutzberg GW. Microglial cells but not astrocytes undergo mitosis following rat facial nerve axotomy. Neurosci Lett. 1988b;85:317–321. doi: 10.1016/0304-3940(88)90585-x. [DOI] [PubMed] [Google Scholar]
- Horn F, Weare J, Beukers MW, Hörsch S, Bairoch A, Chen W, et al. GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 1998;26:275–179. doi: 10.1093/nar/26.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Petros TJ, Chang SY, Zavitsanos PA, Zipkin RE, et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J Biol Chem. 2001;276:42639–42644. doi: 10.1074/jbc.M107351200. [DOI] [PubMed] [Google Scholar]
- Járai Z, Wagner JA, Varga K, Lake KD, Compton DR, Martin BR, et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc Natl Acad Sci U S A. 1999;96:14136–14141. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juknat A, Pietr M, Kozela E, Rimmerman N, Levy R, Coppola G, et al. Differential transcriptional profiles mediated by exposure to the cannabinoids cannabidiol and Δ9-tetrahydrocannabinol in BV-2 microglia. Br J Pharmacol. 2012;165:2512–2528. doi: 10.1111/j.1476-5381.2011.01461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno M, Hasegawa H, Inoue A, Muraoka M, Miyazaki T, Oka K, et al. Identification of N-arachidonoylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Commun. 2006;347:827–832. doi: 10.1016/j.bbrc.2006.06.175. [DOI] [PubMed] [Google Scholar]
- Konno H, Yamamoto T, Iwasaki Y, Suzuki H, Saito T, Terunuma H. Wallerian degeneration induces Ia-antigen expression in the rat brain. J Neuroimmunol. 1989;25:151–159. doi: 10.1016/0165-5728(89)90132-x. [DOI] [PubMed] [Google Scholar]
- Lorton D, Schaller J, Lala A, De Nardin E. Chemotactic-like receptors and Aβ peptide induced responses in Alzheimer's disease. Neurobiol Aging. 2000;21:463–473. doi: 10.1016/s0197-4580(00)00092-0. [DOI] [PubMed] [Google Scholar]
- McHugh D, Hu SS-J, Rimmerman N, Juknat A, Vogel Z, Walker JM, et al. N-Arachidonoyl glycine potently drives directed BV-2 microglial migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010;11:44. doi: 10.1186/1471-2202-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Page J, Bradshaw H. 2011. pp. 2–3. siRNA knockdown of GPR18 receptors in BV-2 microglia attenuates the cell migration induced by N-arachidonoyl glycine. The 21st Annual Symposium of the International Cannabinoid Research Society 2011.
- McHugh D, Page J, Dunn E, Bradshaw HB. Δ9-Tetrahydrocannabinol and N-arachidonoyl glycine are full agonists at GPR18 receptors and induce migration in human endometrial HEC-1B cells. Br J Pharmacol. 2012;165:2414–2424. doi: 10.1111/j.1476-5381.2011.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Stella N. Cannabinoid receptors and endocannabinoids: evidence for new players. AAPS J. 2006;8:E298–E306. doi: 10.1007/BF02854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier K, Merkler D, Gerber J, Taheri N, Kuhnert AV, Williams SK, et al. Multiple neuroprotective mechanisms of minocycline in autoimmune CNS inflammation. Neurobiol Dis. 2007;25:514–525. doi: 10.1016/j.nbd.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Mo FM, Offertáler L, Kunos G. Atypical cannabinoid stimulates endothelial cell migration via a Gi/Go-coupled receptor distinct from CB1, CB2 or EDG-1. Eur J Pharmacol. 2004;489:21–27. doi: 10.1016/j.ejphar.2004.02.034. [DOI] [PubMed] [Google Scholar]
- Morioka T, Baba T, Black KL, Streit WJ. Response of microglial cells to experimental rat glioma. Glia. 1992;6:75–79. doi: 10.1002/glia.440060110. [DOI] [PubMed] [Google Scholar]
- Offertáler L, Mo F-M, Batkai S, Liu J, Begg M, Razdan RK, et al. Selective ligands and cellular effectors of a G protein-coupled endothelial cannabinoid receptor. Mol Pharmacol. 2003;63:699–705. doi: 10.1124/mol.63.3.699. [DOI] [PubMed] [Google Scholar]
- Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133:916–927. doi: 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fatty Acids. 2002;66:101–121. doi: 10.1054/plef.2001.0341. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Verdegaal EME, Siderius M, Bebelman JP, Smit MJ, Leurs R, et al. Quantitative expression profiling of G-protein-coupled receptors (GPCRs) in metastatic melanoma: the constitutively active orphan GPCR GPR18 as novel drug target. Pigment Cell Melanoma Res. 2010;24:207–218. doi: 10.1111/j.1755-148X.2010.00781.x. [DOI] [PubMed] [Google Scholar]
- Rao K, Lund RD. Degeneration of optic axons induces the expression of major histocompatibility antigens. Brain Res. 1989;488:332–335. doi: 10.1016/0006-8993(89)90725-7. [DOI] [PubMed] [Google Scholar]
- Rezaie P, Lantos PL. Microglia and the pathogenesis of spongiform encephalopathies. Brain Res Rev. 2001;35:55–72. doi: 10.1016/s0165-0173(01)00042-x. [DOI] [PubMed] [Google Scholar]
- Rosenkilde MM, Benned-Jensen T, Andersen H, Holst PJ, Kledal TN, Lüttichau HR, et al. Molecular phenotyping of EBI2 an orphan seven-transmembrane receptor with constitutive activity. J Biol Chem. 2006;281:13199–13208. doi: 10.1074/jbc.M602245200. [DOI] [PubMed] [Google Scholar]
- Schilling T, Stock C, Schwab A, Eder C. Functional importance of Ca2+-activated K+ channels for lysophosphatidic acid-induced microglial migration. Eur J Neurosci. 2004;19:1469–1474. doi: 10.1111/j.1460-9568.2004.03265.x. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 2001;22:565–572. doi: 10.1016/s0165-6147(00)01805-8. [DOI] [PubMed] [Google Scholar]
- Segal BM. Experimental autoimmune encephalomyelitis: cytokines, effector T cells, and antigen-presenting cells in a prototypical Th1-mediated autoimmune disease. Curr Allergy Asthma Rep. 2003;3:86–93. doi: 10.1007/s11882-003-0017-6. [DOI] [PubMed] [Google Scholar]
- Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J Med Chem. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- Takenouchi R, Inoue K, Kambe Y, Miyata A. N-arachidonoyl glycine induces macrophage apoptosis via GPR18. Biochem Biophys Res Commun. 2012;418:366–371. doi: 10.1016/j.bbrc.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Vaccani A, Massi P, Colombo A, Rubino T, Palolaro D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br J Pharmacol. 2005;144:1032–1036. doi: 10.1038/sj.bjp.0706134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilatis DK, Hohmann JG, Zeng H, Li F, Ranchalis JE, Mortrud MT, et al. The G protein-coupled receptor repertoires of human and mouse. Proc Natl Acad Sci U S A. 2003;100:4903–4908. doi: 10.1073/pnas.0230374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 2009;29:3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23:1398–1405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda Y, Shinagawa R, Yamada M, Mori T, Tateishi N, Fujita S. Long-lasting reactive changes observed in microglia in the striatal and substantia nigral of mice after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Brain Res. 2007;1138:196–202. doi: 10.1016/j.brainres.2006.12.054. [DOI] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. Lipid G protein-coupled receptor ligand identification using β-arrestin PathHunter™ assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]