

Abstract
The type 1 cannabinoid receptor (CB1) is an integral component of the endocannabinoid system that modulates several functions in the CNS and periphery. The majority of our knowledge of the endocannabinoid system involves ligand–receptor binding, mechanisms of signal transduction, and protein–protein interactions. In contrast, comparatively little is known about regulation of CB1 gene expression. The levels and anatomical distribution of CB1 mRNA and protein are developmental stage-specific and are dysregulated in several pathological conditions. Moreover, exposure to a variety of drugs, including cannabinoids themselves, alters CB1 gene expression and mRNA levels. As such, alterations in CB1 gene expression are likely to affect the optimal response to cannabinoid-based therapies, which are being developed to treat a growing number of conditions. Here, we will examine the regulation of CB1 mRNA levels and the therapeutic potential inherent in manipulating expression of this gene.
Linked Articles
This article is part of a themed section on Cannabinoids. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.167.issue-8
Keywords: cannabinoids, development, Huntington's disease, inflammation, mRNA, obesity, Parkinson's disease, schizophrenia, type 1 cannabinoid receptor (CB1)
Introduction
In the past decade, evidence has accumulated indicating that the endocannabinoid system (ECS) plays a critical role in the regulation of numerous biological processes including embryonic development, metabolism and neurotransmission (Mechoulam and Hanu, 2001; Howlett et al., 2002; Pertwee et al., 2010). The ECS consists of endogenously synthesized endocannabinoids [eCBs, anandamide (AEA) and 2-arachidonoylglycerol (2-AG)], their receptors (the type 1 and type 2 cannabinoid receptors) and their anabolic and catabolic enzymes (Figure 1; Matsuda et al., 1990; Munro et al., 1993; Di Marzo et al., 1994; Cravatt et al., 1996; Martin et al., 1999). In addition to eCBs, phytocannabinoids and synthetic cannabinoids act as cannabinoid receptor ligands. The type 1 cannabinoid receptor (CB1) mediates cannabinoid-dependent signal transduction in the CNS and periphery (Howlett et al., 2002; Basavarajappa et al., 2009), while the type 2 cannabinoid receptor (CB2) is localized to, and highly inducible in, peripheral haemopoietic cells and glial cells in specific areas of the CNS during the inflammatory response (Basavarajappa et al., 2009; Atwood et al., ). To date, the majority of CB1 research has focused on ligand–receptor binding, signal transduction and protein–protein interactions. In contrast, knowledge of CB1 gene regulation is limited. CB1 receptor abundance and the function of the ECS may change in response to altered CB1 gene expression in different developmental or disease conditions or in response to drug exposure. While other reviews have explored the general factors that regulate CB1 and CB2 levels during disease pathogenesis (Miller and Devi, 2011), this review will focus on regulation of CB1 mRNA expression during development and in several pathological conditions where specific pharmacologically tractable regulators of CB1 transcription have been elucidated. The data reviewed here demonstrate that CB1 mRNA transcription is malleable and may be exploited for therapeutic benefit.
Figure 1.
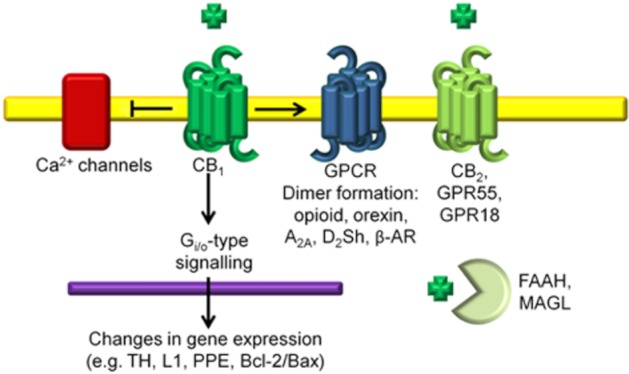
The ECS and CB1. The GPCR CB1 is activated by endocannabinoid ligands such as AEA and exogenous ligands such as THC. In the CNS, activation of CB1, which is typically coupled to Gi/o-proteins, inhibits AC and causes changes in gene expression (e.g. TH, L1 adhesion molecule, PPE and the Bcl-2/Bax regulators of apoptosis). Activation of CB1 also causes inhibition of L-, N- and P/Q-type Ca2+ channels. CB1 can couple to several other GPCRs, which influences receptor trafficking and ligand affinity. Other components of the ECS include CB2, the putative cannabinoid receptors, GPR55 and GPR18, and the catabolic enzymes of cannabinoids fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL).
Architecture, splice variants and isoforms of the CNR1 gene
The human CB1 gene (CNR1) spans 26.1 kb of chromosome 6 (6q14–q15). CNR1 contains four exons (Figure 2A), and the protein coding region of CB1 is contained entirely within exon 4 (Zhang et al., 2004). Outside of the coding region, alternative splicing of CB1 mRNA produces six 5′ untranslated region (5′-UTR) splice variants. The precise transcription start sites within exon 1 included in 5′ UTR variants 1, 3, 4 and 5 have not been defined; although it appears that multiple transcription start sites may exist within the first 60 bp of exon 1 (Shire et al., 1996). Transcription of variant 6 begins within intron 2, and thus the 5′ most exon of variant 6 has been redefined as exon 3a. Transcription of variant 2 begins at the 5′ end of exon 4. Transcript variants 1, 3, 4, 5 and 6 encode full-length CB1 that is 472 amino acids in length encoded without interruption by a single region in exon 4. Exon 4, however, can be differentially spliced to remove 102 nts separating the 5′ end of exon 4 and a new exon identified as exon 4a. This splicing occurs in transcript variant 2 that encodes the truncated, 439 amino acid and CB1b protein (Figure 2D). In CB1a, different intra-exon 4 splice sites result in the loss of 167 nts. Furthermore, two translation start sites are present at the 5′ end of exon 4. Translation from the first produces CB1 and CB1b. Translation from the second is thought to produce the amino-terminal variant CB1a, also known as CB1short (Ryberg et al., 2012). The macaque monkey (Macaca mulatta) CB1 gene is located on chromosome 4. Although the number of exons is not known, the protein coding region of the gene is contained entirely within one contiguous coding region (Figure 2B) (National Centre for Biotechnology Information (NCBI), 2011). The mouse and rat CB1 genes are located on chromosomes 4 and 5, respectively; both genes contain 2 exons with the protein coding regions existing entirely within the second exon in both species (Figure 2C; Miller and Devi, 2011).
Figure 2.
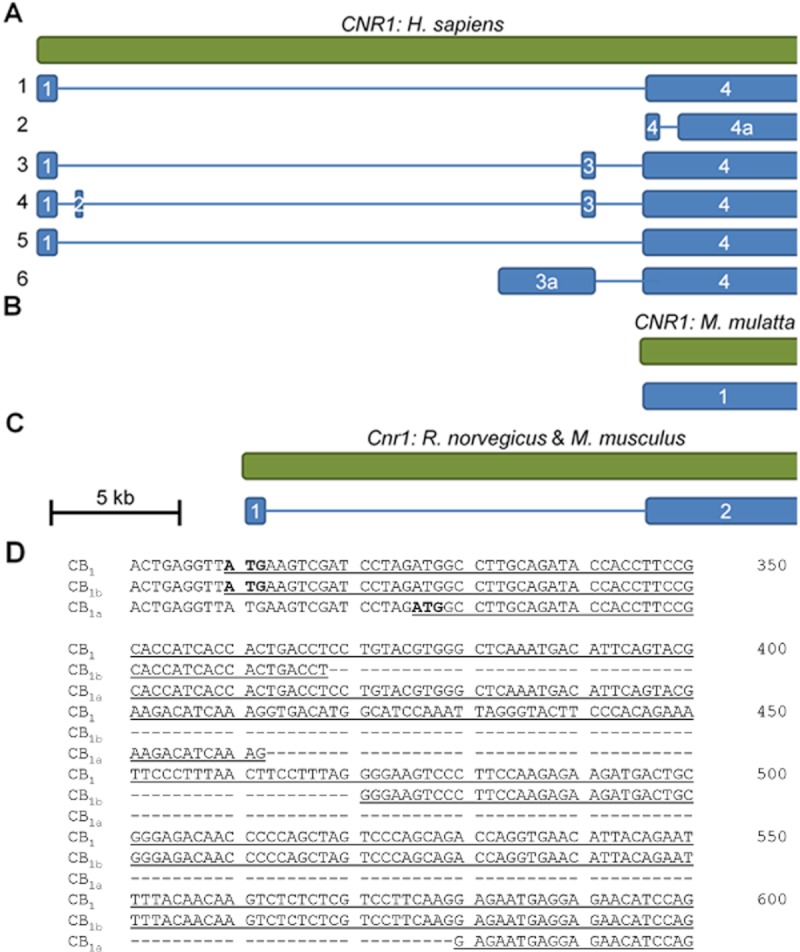
(A) The human CB1 gene, CNR1, spans 26.1 kb on chromosome 6. Six splice variants of the 5′ UTR have been identified by sequencing cDNA ESTs. Splice variants are illustrated in blue and numbered on the left. Exons are numbered within the blue boxes. Each splice variant is aligned with respect to its nucleotide sequence in the CNR1 gene (at top). The scale bar represents 5 kb of nucleotides. (B) The non-human primate (M. mulatta) CB1 gene is poorly characterized, yet it is known that the entire protein coding region is contained within 1 exon (NCBI, 2011). The protein isoforms CB1a and CB1b have been described in non-human primates (Gustafsson et al., 2008). (C) The rat and mouse CB1 genes span approximately 20 kb on chromosome 4 and contain two exons. The second exon contains the entire protein coding region. (D) Three CNR1 coding region variants for protein isoforms of CB1 have been described in humans and non-human primates: the 472 amino acid, intron-less CB1, the 439 amino acid CB1b and the 411 amino acid CB1a. In this figure, position 1 is 300 bp downstream of the 5′ end of exon 4 in CNR1. Translation of CB1 and CB1b begins at the same ATG codon located 309 bp downstream of the first nucleotide in exon 4. Translation of CB1a begins 326 bp downstream of the first nucleotide in exon 4. Fifty-nine basepairs downstream of the CB1b translation start site, CB1b contains a 102 bp intron that is spliced from the pre-mRNA at an atypical intron–exon splice junction (s, CT/cc and ag/GG). Eighty-eight basepairs downstream of the CB1a translation start site, CB1a contains a 167 bp intron that is spliced from the pre-mRNA at a typical 5′ intron-exon boundary (AG/gt) and an atypical ag/GA 3′ splice junction. Downstream of the CB1a intron–exon junction the coding sequences of the three CB1 isoforms are identical.
To date, CB1a and CB1b isoforms have only been identified in humans and higher primates (Ryberg et al., 2012; Gustafsson et al., 2008; Palermo et al., 2009), and some evidence suggests CB1a may be expressed in the rat (Shire et al., 1996). Several authors have demonstrated that CB1, CB1a and CB1b receptors signal via Gi/o-type G-proteins and that the amino-terminal variants CB1a and CB1b have reduced affinity for cannabinoid agonists and antagonists (Rinaldi-Carmona et al., 1996; Ryberg et al., 2012). However, Xiao et al. (2008) did not observe differences in the ligand affinity or localization of the three CB1 protein isoforms. Moreover, the signalling properties of CB1 receptor variants may be altered depending on the model system they are being studied in (Straiker et al., 2012), which complicates our ability to understand receptor differences. In the majority of reports, steady-state CB1 mRNA levels were measured via amplification of the 3′ end of the CB1 coding region outside of the 5′ region in exon 4 involved in differential splicing. The cell-specific relative abundance of CB1 versus CB1a or CB1b is, therefore, poorly characterized (Gustafsson et al., 2008). Early research suggested that CB1a mRNA accounted for approximately 20% of the CB1 transcript population (Shire et al., 1996), yet more recent evidence suggests that less than 5% of the total population of CB1 transcripts obtained from human fetal and adult brain tissue are CB1a or CB1b (Xiao et al., 2008). Studies to define the relative abundance and distribution of the 5′ UTR variants 1–6 have measured the levels of expressed sequence tags. The 5′ UTR transcript variants 1 (5732 bp), 3 (5863 bp), 4 (5901 bp) and 5 (5776 bp) are most abundant in the brain, lymphocytes, testes and liver, relative to other tissues (NCBI, 2011). Transcript variant 2 (5387 bp mRNA) is expressed at highest levels in the brain and testes (NCBI, 2011). Transcript variant 6 (8974 bp mRNA) has only been isolated from brain tissue (NCBI, 2011). Regulation of the transcription of 5′ UTR variants and how 5′ UTR differences relate to CB1 mRNA stability and translation to different CB1 isoforms has not been characterized. The abundance and activity of the different amino-terminal CB1 isoforms may be regulated by different physiological conditions, isoform-specific ligand–receptor affinity and the CB1 isoform complement expressed in a given cell type (Ryberg et al., 2012).
The ‘when and where’ of CB1 mRNA expression
In mammals, steady-state levels of CB1 mRNA vary in different tissues and during different developmental periods. In humans, CB1 is detected in neocortical progenitor cells and in the subventricular zone during the early cortical plate stages of development (9 to 17 weeks gestation; Zurolo et al., 2010). CB1 mRNA is also abundant at 19 weeks gestation in humans in white matter, which is nearly devoid of CB1 expression in adulthood. In the human visual cortex, CB1 mRNA levels rise during early development and plateau approximately 1 year after birth (Romero et al., 1997; Pinto et al., 2010). Following the steady-state CB1 mRNA plateau achieved 1 year after birth, CB1 mRNA levels increase further in the visual cortex to reach a new steady-state level during adolescence, after which CB1 mRNA abundance declines throughout adulthood (Pinto et al., 2010). In the non-human primate, M. mulatta, high levels of CB1 mRNA have been observed in the prefrontal cortex during neonatal development (Eggan et al., 2010); CB1 mRNA abundance increases in the prefrontal cortex until reaching a steady-state at postnatal day 5 (Eggan et al., 2010). In the same manner as is observed in the human visual cortex, a higher steady-state level of CB1 mRNA is observed in the M. mulatta prefrontal cortex during adolescence, and steady-state CB1 mRNA levels decline in the prefrontal cortex following adolescence (Eggan et al., 2010). In mice, CB1 mRNA is detectable during embryonic development as early as four-cell and eight-cell/morula stages (Paria et al., 1995), and can still be detected at embryonic day 12 in glutamatergic neurons of the cerebral cortex and hippocampus (Vitalis et al., 2008). CB1 mRNA is abundant in the adult mouse thalamus, amygdala, dorso-lateral prefrontal cortex, hypothalamus and pituitary (NCBI, 2011). Furthermore, CB1 expression is enriched in the striatum, relative to other brain regions, within the adult mouse CNS (Fernandez-Ruiz et al., 2012; McCaw et al., 2004). It is within the striatum that a high steady-state level of CB1 expression are dysregulated in Parkinson's and Huntington's diseases (Zeng et al., 1999; Denovan-Wright and Robertson, 2000). The temporal and anatomical distribution of CB1 expression during early development is similar in mice and rats (NCBI, 2011). Six to 8-week-old rats, which are sexually mature, have lower levels of CB1 mRNA in the limbic/associative brain areas compared with adolescents (Heng et al., 2011). Following periods of peak neurodevelopment associated with high CB1 levels, CB1 mRNA abundance declines in these brain regions (Heng et al., 2011). Taken together, these data demonstrate that, in mammals, CB1 mRNA levels peak during adolescence within the prefrontal cortex, limbic/associative areas and visual cortex and subsequently with age. Early development and adolescence represent critical developmental windows where the regulation of CB1 expression changes in order for higher levels of expression to be achieved. It is likely that developmental stage-specific transcription factors or modifiers regulate the different steady states of CB1 expression. A representative illustration of the temporal–spatial expression of CB1 mRNA, in the CNS, based on data obtained from mouse, rat, monkey and human is presented in Figure 3.
Figure 3.
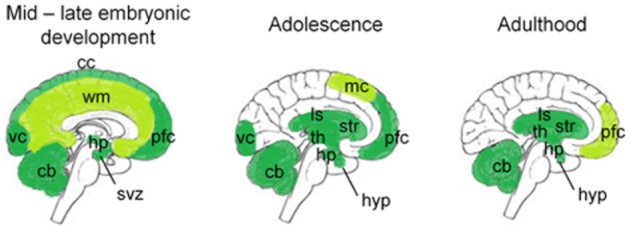
CB1 mRNA abundance and distribution shift throughout development in the CNS. This simplified schematic illustrates the areas of the brain where CB1 mRNA levels are moderate and high relative to other regions of the CNS during mid – late embryonic development, adolescence and adulthood. The images were created using data obtained in humans, non-human primates, mice and rats. cb, cerebellum; cc, cerebral cortex; hp, hippocampus; hyp, hypothalamus; ls, limbic system; mc, motor cortex; pfc, prefrontal cortex; svz, sub-ventricular zone; th, thalamus; str, striatum; vc, visual cortex; wm, white matter (Denovan-Wright and Robertson, 2000; Vitalis et al., 2008; Eggan et al., 2010; Gensat, 2010; Zurolo et al., 2010; Heng et al., 2011; NCBI, 2011).
High levels of CB1 expression are related to the establishment of neuronal circuitry. During critical development periods, such as late gestation and early postnatal life, areas associated with neurogenesis and synapse formation, such as the subventricular zone and white matter, are transiently enriched for CB1 and subsequently depleted of CB1 expression in adulthood (Romero et al., 1997). The activity or abundance of the factors that enabled high steady-state CB1 levels during development and adolescence may decrease in concentration or activity as part of the aging process (Eggan et al., 2010; Heng et al., 2011). Greater expression and subsequent activation of CB1 receptors facilitates higher expression of several genes required for brain development, including tyrosine hydroxylase (TH), preproenkephalin (PPE), the neural adhesion molecule L1 and Bcl-2/Bax genes involved in apoptotic regulation of development (reviewed in Fernandez-Ruiz et al., 2012). Mice lacking CB1 exhibit transcriptional dysregulation of PPE and substance P (Steiner et al., 1999), altered dendritic morphology and lower synapse density in the prefrontal cortex (Fitzgerald et al., 2012), impaired locomotor activity (Zimmer et al., 1999) and increased anxiety (Hill et al., 2011) compared to wild-type littermates. Thus, the developmental stage-specific expression of CB1 facilitates the proper establishment of neuronal circuitry and the consequent normalization of behaviour (Fernandez-Ruiz et al., 2012).
Expression of CB1 is cell-specific within the CNS. Striatal medium spiny projection neurons and interneurons are enriched for CB1 mRNA expression, relative to other cell populations, within the basal ganglia (Marsicano and Lutz, 1999; Fernandez-Ruiz et al., 2012). Consistent with CNS anatomical distribution, CB1 appears to be involved with aspects of motor coordination, mechanisms of reward and motivation, emotion and central endocrine regulation during adulthood (Fernandez-Ruiz et al., 2012). CB1 is co-localized, co-expressed and can dimerize with the pre-synaptic type 2 dopamine short (D2Sh), μ-and δ-opioid, adenosine A2a, and orexin, receptors (Navarro et al., 2008; Pacheco et al., 2009; Uriguen et al., 2009; Bortolato et al., 2010; Rozenfeld et al., ). Thus, in addition to alterations in CB1-mediated signalling, changing CB1 mRNA and protein abundance could cause dysregulated signalling via other GPCRs, such as D2Sh, opioid and orexin receptors; by affecting dimer formation, receptor trafficking and localization, and signal transduction (Hudson et al., 2010; reviewed in Smith et al., 2010).
In non-neuronal tissue, CB1 expression is associated with immune and endocrine homeostasis as well as reproductive system development and maturation. CB1 mRNA is abundant in helper (CD4) and cytotoxic (CD8) T cells, hepatocytes, beta-islet cells and adipose tissue, during adulthood where these receptors regulate inflammatory and metabolic processes through autocrine and paracrine signalling mechanisms (Borner et al., 2008; Mukhopadhyay et al., 2010). CB1 mRNA levels increase in primary cultured rat leydig cells (testosterone-producing testis cells) from postnatal day 14 onwards, spermatids from postnatal days 31 through 61 and sertoli cells from postnatal day 41 onwards (Cacciola et al., 2008). Cells within the testes exhibit a biphasic pattern of CB1 expression, in which CB1 levels are elevated at 1 week post partum, decline to a minimum by 2 weeks post partum and rise again to highest levels as mice reach sexual maturity at 4 weeks post partum (Cacciola et al., 2008). Certain fish species, for example the gilthead seabream (Sparus aurata) and the puffer fish (Fugu rubripes), are capable of undergoing sexual reversal, which is the process of shifting the reproductive organs from being functionally male to functionally female, or vice versa. Sexual reversal, therefore, represents a period of altered gene expression leading to changes in cellular phenotype within teleost reproductive organs. CB1 mRNA expression increases in the testes of these fish during the process of sexual reversal (Cottone et al., 2008). Increased CB1 expression could contribute to sexual reversal by altering the complement of genes expressed and thus the phenotype of the testes (Cottone et al., 2008). In the developing CNS, changes in CB1 expression facilitate downstream changes in the expression of many other genes (Fernandez-Ruiz et al., 2012). Therefore, up-regulation of CB1 expression may also sufficiently alter the gene expression profile in reproductive tissue to produce changes in phenotype and facilitate developmental processes. In reproductive systems, as in the CNS, CB1 expression appears to be coordinated in order to facilitate development and maturation during early development and adolescence.
Understanding changes in CB1 mRNA expression that occur in diverse pathological conditions
CB1 mRNA expression is induced by inflammation in non-neuronal tissue
Although CB2 receptors are considered the major eCB receptor in the periphery, particularly as regulators of inflammation (Rajesh et al., 2008; reviewed in Atwood and Mackie, 2010), CB1 receptors also contribute to regulation of the inflammatory response. Pro-inflammatory molecules induce CB1 and CB2 mRNA expression in cells that mediate the inflammatory responses (Gutierrez et al., 2006; Borner et al., 2008). The involvement of CB1 in the inflammatory response was first examined in rat dorsal root ganglia (DRG), where complete Freund's adjuvant increased CB1 mRNA abundance in glial cells of the DRG 4 h post-treatment, relative to untreated controls (Amaya et al., 2006). Freund's adjuvant produces an inflammatory response and activates such transcription factors as nuclear factor of activated T cells (NFAT) and NF-κB in glial cells (Amaya et al., 2006; Borner et al., 2007a). Activation of NFAT and NF-κB is dependent on the endogenous pro-inflammatory cytokines CD3/28 and IL-4 (Borner et al., 2007a). CD3/28 and IL-4 induce CB1 mRNA expression in human peripheral T cells and immortalized Jurkat cells (Borner et al., 2007a; 2008). Borner et al. (2007a) examined CD3/28- or IL-4-mediated induction of CB1 via a promoter–reporter plasmid in which chloramphenicol acetyl transferase activity was driven by a 3 kb fragment of the CNR1 promoter. Short, double-stranded, decoy oligonucleotides containing the consensus sequences normally bound by NFAT or NF-κB were used to titrate NFAT or NF-κB enhancers of transcription away from their endogenous promoters (Borner et al., 2007a). NFAT and NF-κB facilitate a CD3/28- or IL-4-dependent increase in CB1 expression (Borner et al., 2007a). Using the same techniques, it was found that activator protein 1 (AP-1) and the signal transducers and activators of transcription 5 and 6 (STAT5 and STAT6) are also recruited to the CNR1 promoter to mediate increased mRNA expression in Jurkat cells (Borner et al., 2007a,2007b; 2008). Together, these data demonstrate that pro-inflammatory cues mediate an increase in CB1 mRNA levels from an initial steady-state to a second, higher state through common mechanisms.
CB1 mRNA expression is changed in various cancers
The ECS regulates cell fate and division during oncogenesis (Malfitano et al., 2012). For example, in breast cancer, CB2 agonism inhibits cell cycle progression via down-regulation of Cdc2 (Caffarel et al., 2006), whereas in fibrosarcoma cells, CB1 antagonism up-regulates the cell cycle inhibitor p21WAF1 and down-regulates cyclins E and D (Malfitano et al., 2012). Up- and down-regulation of CB1 levels influences cell growth, just as cannabinoid treatment affects cell growth. Treatment of transformed cells with anti-neoplastic agents has been shown to both increase and decrease CB1 mRNA expression depending on drug and cell line (Larrinaga et al., 2010b; Proto et al., 2011). CB1 mRNA levels are reduced in the DLD-1 and SW620 cell culture models of colorectal cancer, relative to primary colorectal cell cultures (Proto et al., 2011). Furthermore, the CB1 promoter is highly methylated in human colorectal carcinoma cells, compared with healthy tissue (Wang et al., 2008), suggesting that elevated CB1 expression consequently alters the capacity for cell division in colorectal carcinoma cells. This may be the result of CB1-mediated up-regulation of p21WAF1 or down-regulation of Cdc2 and cyclins, or both (Caffarel et al., 2006; Malfitano et al., 2012). Treatment of DLD-1 and SW620 cells with 17β-estradiol or the synthetic cannabinoid meth-anandamide (mAEA) increases CB1 mRNA levels (Proto et al., 2011). Following treatment with 17β-estradiol or mAEA, the rate of division of these colorectal cancer cells is significantly reduced (Proto et al., 2011). CB1 mRNA levels are lower in primary adrenocarcinoma tumour cells than in healthy tissue (Larrinaga et al., 2010b). In healthy tissue, CB1 activation decreases cell division and proliferation (Larrinaga et al., 2010a,b). The chemotherapeutic agent gemcitabine arrests adrenocarcinoma tumour growth (Larrinaga et al., 2010a). Gemcitabine has also been reported to induce CB1 mRNA expression via NF-κB, as demonstrated by chromatin immunoprecipitation of the CNR1 promoter (Larrinaga et al., 2010a). These data demonstrate that CB1 mRNA expression can be manipulated pharmacologically, and that the level of CB1 expression and activity negatively correlates with cell division. Consequently, pharmacological manipulation of CB1 expression may represent a therapeutic option for the treatment of adrenocarcinomas (Larrinaga et al., 2010a,b).
In contrast to observations of decreased CB1 mRNA levels in adrenocarcinoma, CB1 mRNA abundance is increased in biopsied human tissue taken from patients with prostate cancer or benign prostate hyperplasia relative to healthy tissue (reviewed in Gustafsson et al., 2008). Similarly, CB1 mRNA levels are increased in non-Hodgkin lymphoma tissues, relative to healthy tissues (Gustafsson et al., 2008). Despite the increase in CB1 mRNA levels, Gustafsson et al. (2008) found that the relative proportions of CB1a and CB1b mRNA levels were lower in biopsied lymphoma tissue compared with normal lymphocytes (Gustafsson et al., 2008). If CB1, CB1a and CB1b are not equally abundant, then this suggests that expression of each isoform is differentially regulated (Gustafsson et al., 2008), which is likely the result of differences in mRNA processing and stability (Shire et al., 1996; Ryberg et al., 2012). Conversely, the hypothesis that each CB1 isoform is regulated by different promoter elements and upstream differences in cell signalling (Gustafsson et al., 2008) is unlikely because all three isoforms share a common, highly active promoter (Borner et al., 2007a,2007b; 2008; reviewed in Miller and Devi, 2011). CB1 levels are also elevated in alveolar rhabdosarcoma, which is caused by expression of a chimeric PAX3/7-FOX01 transcription factor (Marshall et al., 2011). Up-regulation of CB1 in alveolar rhabdosarcoma does not affect the rate or capacity of cells to divide but rather increases the metastatic ability of the cells (Marshall et al., 2011). Consequently, elevated CB1 expression in certain cancers, such as prostate cancer, lymphoma or rhabdosarcoma may impact metastasis, but not cell division.
CB1 mRNA abundance fluctuates in obesity and diabetes
The hormone 17β-estradiol and mediators of the inflammatory response, such as CD3/28, increase CB1 mRNA levels. It is not surprising, therefore, that obesity and diabetes – two pathologies associated with hormonal dysregulation and inflammation – are also associated with changes in CB1 mRNA levels (Howlett et al., 2002; Kempf et al., 2007). Whether obesity correlates with higher or lower CB1 mRNA levels remains controversial. CB1 mRNA abundance has been measured in primary cultured adipocytes of lean and obese individuals. In one study, CB1 mRNA was shown to be less abundant in primary cultured adipocytes derived from white adipose tissue of obese children compared with that from lean children (Karvela et al., 2010). Similarly, CB1 levels were lower in adipocytes derived from the visceral adipose tissue of obese adults compared with healthy adults (Kempf et al., 2007). In another study, CB1 mRNA was more abundant in adipocytes derived from the visceral adipose tissue of obese adults, relative to non-obese individuals (Sarzani et al., 2009). Different cell culture conditions may account for the discrepancies in CB1 mRNA levels reported by these groups. Karvela et al. (2010) and Kempf et al. (2007) cultured adipocytes in the presence of adiponectin, while Sarzani et al. (2009) measured CB1 mRNA abundance in tissue samples without culturing the adipocytes. Therefore, the primary culturing and treatment of adipocytes may alter CB1 expression. However, the functional consequence of altered CB1 levels in adipocytes may be altered by cell survival and proliferation because CB1 activation is often associated with pro-survival signalling (Kempf et al., 2007; Sarzani et al., 2009; Karvela et al., 2010). In obese individuals, enhanced survival of adipocytes may exacerbate their condition (Sarzani et al., 2009).
Diabetes is associated with a decreased production of, or response to, insulin. Insulin can penetrate the blood–brain barrier, act on insulin receptors and stimulate glucose uptake in the central nervous system (Bingham et al., 2002). Streptozotocin-treated rats lack insulin-producing beta-islet cells and are used to model diabetes (Zhang et al., 2007). CB1 levels are increased in the striatum and hypothalamus of streptozotocin-treated rats compared to untreated controls (Diaz-Asensio et al., 2008). In the rat pancreas, β-, α, and δ-islet cells express CB1 mRNA (Zhang et al., 2007). Treatment of rats with glucose (20–50 mM) is associated with an increase in plasma insulin concentration and a decrease in CB1 mRNA levels in the pancreas, white adipose tissue and DRG, relative to untreated rats (Zhang et al., 2007). Thus, increased glucose, leading to increased plasma insulin is associated with CB1 down-regulation. Therefore, insulin appears to inhibit CB1 expression in the CNS. Conversely, up- or down-regulation of CB1 may lead to altered insulin receptor expression (Zhang et al., 2007), glucose uptake (Diaz-Asensio et al., 2008) or both.
Susceptibility to schizophrenia is associated with changes in CB1 mRNA expression
During adolescence, CB1 mRNA and eCB levels peak in the dorso-lateral prefrontal cortex in mammals (Eggan et al., 2010). Mice exposed to Δ9-tetrahydrocannabinol (THC) during adolescence (postnatal day 40) are more likely to develop schizophrenia modelling behaviours, namely pre-attentional sensorimotor and executive function deficits, than mice exposed to THC later in adulthood (Heng et al., 2011). Eggan et al. (2010) also found that CB1 mRNA abundance peaks in the dorso-lateral prefrontal cortex of macaque monkeys 2 months after puberty. These data suggest that adolescence may be a period of hypersensitivity to cannabinoids (Caspi et al., 2005). Polymorphisms that relate cannabis use to the CNR1 gene have been described. One study reported that a TAG allele in the 5′ region of CNR1 exon 3 and a polymorphic AAT repeat in the 3′ region of exon 4 were more prevalent among European, African American and Japanese substance abusers than individuals that did not have a history of substance abuse from the same region (Zhang et al., 2004). Intriguingly, the presence of the ‘TAG’ allele was associated with less CB1 mRNA compared to other alleles (Zhang et al., 2004). Subsequent analyses support that the length of the polymorphic AAT repeat region may be correlated with substance abuse (Benyamina et al., 2011). Therefore, a strong connection exists between the allelic variability of CNR1 and adolescent exposure cannabis use.
CB1 mRNA abundance differs in individuals with schizophrenia compared with healthy controls. CB1 mRNA levels were higher in postmortem tissue from the dorso-lateral prefrontal cortex of individuals with schizophrenia compared with age-matched healthy subjects (Uriguen et al., 2009). Immunohistochemical analyses of postmortem brains isolated from individuals with schizophrenia revealed that individuals treated with the atypical antipsychotics olanzapine or clozapine expressed significantly less CB1 protein in the dorso-lateral prefrontal cortex than age-matched individuals with schizophrenia who did not receive atypical antipsychotics (Uriguen et al., 2009). Olanzapine and clozapine treatment were not associated with a change in CB1 mRNA levels (Uriguen et al., 2009). Therefore, these atypical antipsychotics that are, among other activities, D2 receptor antagonists, decrease CB1 protein, but not mRNA, levels. The functional implication of this observation is that pharmacological manipulation of the dopaminergic system can impact the cannabinergic system and possibly vice versa (El Khoury et al., 2012). Thus, therapeutics aimed at reducing cannabinergic or dopaminergic tone, such as antipsychotics, may impact both systems simultaneously. Conversely, certain compounds may increase the tone of both systems, which could have undesirable effects on cognition, behaviour and motor control if the ECS is over-activated and beneficial effects on cognition, behaviour and motor control in pathologies where CB1 levels are reduced.
A striatal cell-specific decrease in CB1 mRNA is observed in Parkinson's disease
CB1 mRNA is highly expressed in the caudate and putamen, globus pallidus and substantia nigra of healthy individuals (Fernandez-Ruiz et al., 2012). Hurley et al. (2003) examined CB1 mRNA levels in postmortem tissue from normal controls and individuals with Parkinson's disease. CB1 levels were reduced in the caudate nucleus, anterior dorsal putamen and external segment of the globus pallidus, relative to controls or other brain regions of Parkinson's patients (Hurley et al., 2003). A key feature of Parkinson's disease is decreased dopamine levels. Parkinson's disease can be modelled by selective lesioning of the nigrostriatal pathway by 6-hydroxydopamine or administration of 6-hydroxydopamine to the medial forebrain bundle causes cell loss in the substantia nigra, which in turn depletes the striatum of dopamine (Zeng et al., 1999). Consequently, CB1 mRNA levels are reduced within the dopamine-depleted rat striatum (Zeng et al., 1999). CB1 mRNA expression can be increased in the striatum of 6-hydroxydopamine-lesioned rats by subsequent, chronic, treatment with L-DOPA, which increases dopaminergic signalling (Zeng et al., 1999; Garcia-Arencibia et al., 2009). Reserpine depletes catecholamines, including dopamine, in the CNS (Thrash et al., 2009). Reserpine-treated rats have been used to model Parkinson's disease and depression (Silverdale et al., 2001; Thrash et al., 2009). Similarly to 6-hydroxydopamine-lesioned rats, CB1 mRNA levels were reduced in the caudate and putamen of reserpine-treated rats relative to age-matched, control rats (Silverdale et al., 2001). Overall, CB1 mRNA levels appear to be regulated by dopamine in Parkinson's disease, which provides more evidence for a link between cannabinergic and dopaminergic signalling.
A cell-specific decrease in CB1 mRNA is observed in Huntington's disease and contributes to disease pathogenesis
CB1 mRNA levels are reduced in the caudate and putamen of human subjects with Huntington's disease and the striatum of all Huntington's disease mouse models tested to date relative to age-matched controls (Pazos et al., 2008). Expression and nuclear localization of the amino-terminus of mutant huntingtin protein reduces transcription of CB1 in striatal medium spiny projection neurons early in disease progression (Gafni and Ellerby, 2002; McCaw et al., 2004). CB1 expression is not altered in the presence of mutant huntingtin protein in the hippocampus, or prior to adulthood in mice (Denovan-Wright and Robertson, 2000; McCaw et al., 2004). Also, CB1 mRNA levels are lower in cultured neuronal cell models of Huntington's disease, which lack inter-cellular signalling, compared to cells that do not express mutant huntingtin (Blázquez et al., 2011). Wild-type huntingtin binds to the repressive element 1 silencing transcription factor (REST), which can interact with repressor elements at the CB1 promoter (Blázquez et al., 2011). Blázquez et al. (2011) utilized a CB1 promoter–reporter and decoy oligonucleotide-based assay to demonstrate that REST inhibits transcription of CB1 in cells expressing mutant huntingtin. Therefore, tissue-, cell- and developmental stage-specific factors that normally accommodate high-level CB1 mRNA expression in the adult striatum are affected by the cell-autonomous overexpression of amino-terminal mutant huntingtin. In contrast, CB1 mRNA expression does not appear to change in Alzheimer's disease, a neurodegenerative disease, like Huntington's and Parkinson's diseases, characterized by protein misfolding and aggregation (Kalifa et al., 2011). While CB1 protein levels (Kalifa et al., 2011) and receptor binding (Westlake et al., 1994) may be decreased, in the hippocampus, neocortex and basal ganglia during Alzheimer's disease progression (Westlake et al., 1994; Lee et al., 2010), mRNA abundance is unaffected in humans and mouse models.
Decreased CB1 receptor function may contribute to progressive decline in Huntington's disease. Separate research groups bred two different mouse models of Huntington's disease with homozygous CB1 knock-out mice (CB1−/−; Blázquez et al., 2011; Mievis et al., 2011). Both research groups found that mice over-expressing amino-terminal mutant huntingtin and having reduced CB1 exhibited an earlier Huntington's disease symptom onset, a more rapid disease progression and a greater degree of medium spiny projection neuron degeneration than wild-type mice or mice over-expressing amino-terminal mutant huntingtin with a full complement of CB1 (Blázquez et al., 2011; Mievis et al., 2011). Their findings suggest CB1 normally performs a neuroprotective role in the striatum and loss of this receptor correlates with Huntington's disease pathogenesis. Thus, therapeutic strategies capable of elevating CB1 mRNA abundance may restore or enhance the neuroprotective role of CB1 where decreased expression of this receptor may contribute to disease pathology.
Pharmacological manipulation of CB1 mRNA abundance
CB1 mRNA levels can be modulated by methamphetamine and alcohols
Methamphetamine use is associated with region-specific changes in CB1 mRNA levels in the CNS. Acute treatment of rats with methamphetamine is associated with increases in steady-state CB1 mRNA levels in the prefrontal cortex, caudate and putamen, basolateral amygdala, CA1 hippocampal region and perirhinal cortex, relative to other brain regions and untreated controls (Bortolato et al., 2010). These region-specific increases are detectable up to 3 weeks after a single post-acute treatment (Bortolato et al., 2010). Acute methamphetamine use is associated with increased dopamine neurotransmission. These findings align with findings in schizophrenia and Parkinson's disease that suggest CB1 levels are influenced by dopamine acting on pre- and post-synaptic receptors.
Ethanol use is also associated with region-specific changes in CB1 mRNA levels in the CNS and in cell culture. In humans, CB1 protein levels have been compared in the ventral striatum of individuals that were alcohol-dependent to age-matched, non-alcoholic individuals (Vinod et al., 2005). CB1 protein levels were lower in alcohol-dependent individuals relative to non-alcoholic controls (Vinod et al., 2005). Barbier et al. (2012) found that mice exposed to ethanol in utero have significantly lower levels of CB1 mRNA in the cortex, striatum and hippocampus from postnatal days 14 through 90 relative to age-matched controls. Consequently, exogenous ethanol exposure appears to alter CB1 expression during early development and adulthood and may lead to chronic alterations in neurotransmission and gene expression that are normally facilitated by CB1.
CB1 mRNA levels can be modulated by estradiol and retinoic acid
Estradiol and retinoic acid alter CB1 mRNA levels. 17β-estradiol and retinoic acid act upon their respective nuclear receptors to increase CB1 mRNA levels in cell culture (Mukhopadhyay et al., 2010; Proto et al., 2011). Administration of 17β-estradiol increases CB1 mRNA levels in DLD-1 and SW620 colon cancer cells (Nortarnicola et al., 2008; Proto et al., 2011). The effect of 17β-estradiol requires the oestrogen receptor, retinoic acid receptor (RAR)α and PPARγ to co-localize within the promoter region 1 kb from the human CNR1 transcription start site (Proto et al., 2011). Retinoic acid also increases CB1 mRNA levels in mouse primary hepatic stellate cells (Mukhopadhyay et al., 2010). This induction requires retinaldehyde dehydrogenase and RARγ (Mukhopadhyay et al., 2010). RARγ interacts with a retinoic acid response element approximately 350 bp upstream of the mouse Cnr1 transcription start site. Together, these data demonstrate 17β-estradiol and retinoic acid induce CB1 transcription via oestrogen receptor, RAR- and PPAR-dependent mechanisms from a basal state to a higher steady state. Therefore, CB1 levels can be manipulated by the activation or inhibition of well-known, pharmacologically tractable regulators of transcription.
CB1 mRNA level is modulated by cannabinoids
Cannabinoids modulate steady-state CB1 mRNA abundance. Chronic treatment with THC has been shown to decrease CB1 mRNA levels in the CNS of rodents. Repeated exposure to THC, once daily for 14 days by i.p. injection, decreases CB1 mRNA levels in the caudate and putamen of adult male rats (Corchero et al., 1999). The extent of CB1 mRNA decrease correlates to the number of repeated exposures. In another study, THC treatment increased CB1 mRNA levels in the rat cerebellum and hippocampus over a 3 day period, while simultaneously decreasing CB1 mRNA levels in the rat striatum (Zhuang et al., 1998). Cannabinoids have also been shown to increase CB1 mRNA levels in primary and immortalized cell culture systems. Treatment of primary mouse hepatic, stellate cells with 2-AG induces CB1 mRNA, up to 30-fold relative to basal expression in untreated cells (Mukhopadhyay et al., 2010). 2-AG-mediated CB1 induction is RARγ- and CB1 receptor-dependent in this model system (Mukhopadhyay et al., 2010). AEA has been reported to increase CB1 mRNA levels in DLD-1 and SW620 cells (Proto et al., 2011). This effect was oestrogen receptor- and RARα-dependent. Finally, THC, methanandamide and the CB2-selective agonist JWH-015 induce CB1 mRNA expression in Jurkat cells in a CB2-dependent manner (Borner et al., 2007b). Borner et al. (2007b) observed that CB2 activation leads to phosphorylation of STAT5, transactivation of IL-4 and activation of STAT6, thereby inducing CB1 promoter activity (Borner et al., 2007b). Thus, in some systems, cannabinoid-dependent activation of CB1 and CB2 receptors stimulates the activity of specific transcription factors, such as the oestrogen receptor, RARα and STAT6, and augments steady-state CB1 mRNA levels above basal levels. In other systems, cannabinoid exposure down-regulates CB1 mRNA levels (Corchero et al., 1999). Cannabinoid treatment therefore, as in various pathological conditions, is associated with malleable context-specific regulation of CB1 expression. In vivo, repeated exposure to cannabinoid agonists is associated with tachyphylaxis (Corchero et al., 1999), whereas in cell culture, single acute doses of cannabinoid agonists induce CB1 mRNA expression (Borner et al., 2007b; Mukhopadhyay et al., 2010). From these observations, it is clear that the response of the CB1 mRNA levels to cannabinoid treatment depends on the nature of treatment, chronic versus acute as well as the potency and efficacy of the ligand. For example, CB1 mRNA expression may be inducible in in vivo studies examining acute doses of cannabinoids, indirect cannabinoid agonism via fatty acid amide hydrolase inhibitors (Kim and Alger, 2010), or allosteric modulation of CB1 receptor activity (Navarro et al., 2009; Ahn et al., 2012).
CB1 protein levels are also increased following acute cannabinoid-dependent induction of CB1 mRNA levels (Mukhopadhyay et al., 2010; Proto et al., 2011). This increase is modest (four- to fivefold) compared with the increased mRNA expression observed (29- to 30-fold, Mukhopadhyay et al., 2010; Proto et al., 2011) yet represents an increase in the pool of CB1 receptors. In these studies, CB1 protein abundance was quantified via western blot. Therefore, it is not known whether cannabinoid-mediated CB1 induction affects the localization or functionality of CB1 receptors.
Conclusions
Manipulation of CB1 expression may have wide ranging affects on physiological processes such as embryogenesis (Paria et al., 1995) and neural development (Fitzgerald et al., 2012). Given that CB1 gene expression is highest in many regions of the brain during early development and adolescence, and it is these areas of high expression where expression is often altered during disease progression, the effect of CB1 therapeutics on CB1 expression will depend on the existing level of CB1 expression. CB1 levels can be modulated pharmacologically by pro-inflammatory peptides, oestrogen, insulin, atypical antipsychotics, methamphetamine, ethanol, retinoic acid and, importantly, endogenous and exogenous cannabinoids (Figure 4). The observation that cannabinoids can induce CB1 mRNA expression in cell culture suggests that cannabinoid ligands could regulate CB1 levels. Although long-term treatment with THC in vivo is associated with tachyphylaxis (Corchero et al., 1999), other cannabinoids have not been examined; the extent of tachyphylaxis may vary in a ligand-specific manner (Hudson et al., 2010), as well as with the duration of exposure (Zhuang et al., 1998), half-life, efficacy, and potency of the cannabinoid. For instance, acute doses of less-potent agonists of CB1 (e.g. AEA relative to THC; Pertwee et al., 2010), or allosteric modulators of CB1 (Ahn et al., 2012) may induce CB1 expression in vivo, whereas chronic treatment with more-potent agonists may cause receptor desensitization. If this is the case, then cannabinoid-dependent manipulation of CB1 levels may represent a useful therapeutic strategy for diseases where reduced or elevated CB1 levels correlate with disease progression. Overall, the affect of cannabinoid-based therapies may depend upon their modulation of CB1 gene expression.
Figure 4.
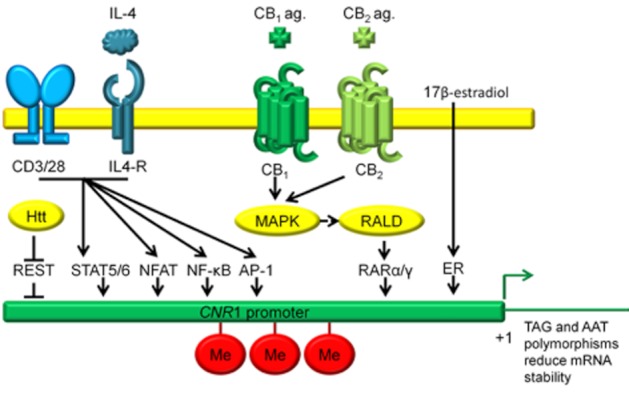
CB1 mRNA expression is regulated by several co-activators of transcription. This schematic illustrates regulation of CB1 transcription at several functional transcription factor binding sites known to enhance CB1 expression within 3 kb of the CNR1 transcription start site at exon 1. STAT5, STAT6 (approximately −2769 bp, Borner et al., 2007a,b), NFAT, NF-κB, AP-1 (within −2490 bp, Borner et al., 2008), REST (approximately −898 bp, Blázquez et al., 2011), RARα/γ (−350 bp, McCaw et al., 2004; Mukhopadhyay et al., 2010; Proto et al., 2011), ERE (−1073 bp and −366 bp, Proto et al., 2011), TAG and AAT polymorphisms 5′ of exon 1 and 3′ of exon 4, respectively (Zhang et al., 2004), Me, DNA methylation leading to promoter repression (Miller and Devi, 2011).
Acknowledgments
We would like to acknowledge the funding support of the Canadian Institutes for Health Research, the Nova Scotia Health Research foundation, the Huntington disease research foundation, the Molly Appeal scholarship for Neuroscience and Dalhousie University. Thanks to Kathleen Murphy and Kimberly Laprairie for help in preparing this article.
Glossary
- 2-AG
2-arachidonyl glycerol
- AEA
anandamide
- AP-1
activator protein 1
- CB1
type 1 cannabinoid receptor
- CB2
type 2 cannabinoid receptor
- CNR1
type 1 cannabinoid receptor gene
- D2Sh
type 2 dopamine receptor – short variant
- DRG
dorsal root ganglion
- eCBs
endocannabinoids
- ECS
endocannabinoid system
- mAEA
meth-anadamide
- NFAT
nuclear factor of activated T cells
- PPE
preproenkephalin
- RAR
retinoic acid receptor
- REST
repressive element 1 silencing transcription factor
- STAT
signal transducers and activators of transcription
- TH
tyrosine hydroxylase
- THC
Δ9-tetrahydrocannabinol
- UTR
untranslated region
Conflict of interest
None declared.
References
- Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem. 2012;287:12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Shimosato G, Kawasaki Y, Hashimoto S, Tanaka Y, Ji RR, et al. Induction of CB1 cannabinoid receptor by inflammation in primary afferent neurons facilitates antihyperalgesic effect of peripheral CB1 agonist. Pain. 2006;124:175–183. doi: 10.1016/j.pain.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160:467–479. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Straiker A, Mackie K. CB2: therapeutic target-in-waiting. Prog Neuropsychopharmacol. 2012;38:16–20. doi: 10.1016/j.pnpbp.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier E, Pierrefiche O, Vaudry D, Vaudry H, Daoust M, Naassila M. Long-term alterations in vulnerability to addiction to drugs of abuse in brain gene expression after early life ethanol exposure. Neuropharmacology. 2008;55:1199–2211. doi: 10.1016/j.neuropharm.2008.07.030. [DOI] [PubMed] [Google Scholar]
- Basavarajappa B, Nixon R, Arancio O. Endocannabinoid system: emerging role from neurodevelopment to neurodegeneration. Mini Rev Med Chem. 2009;9:448–462. doi: 10.2174/138955709787847921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyamina A, Kebir O, Blecha L, Reynaud M, Krebs MO. CNR1 gene polymorphisms in addictive disorders: a systematic review and a meta-analysis. Addict Biol. 2011;16:12–16. doi: 10.1111/j.1369-1600.2009.00198.x. [DOI] [PubMed] [Google Scholar]
- Bingham EM, Hopkins D, Smith D, Pernet A, Hallett W, Reed L, et al. The role of insulin in human brain glucose metabolism: an 18fluoro-deoxyglucose positron emission tomography study. Diabetes. 2002;51:3384–3390. doi: 10.2337/diabetes.51.12.3384. [DOI] [PubMed] [Google Scholar]
- Blázquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR, Resel E, et al. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington's disease. Brain. 2011;134:119–136. doi: 10.1093/brain/awq278. [DOI] [PubMed] [Google Scholar]
- Borner C, Holt V, Kraus J. Activation of human T cells induces upregulation of cannabinoid type 1 transcription. Neuroimmunomodulation. 2007a;14:281–286. doi: 10.1159/000117809. [DOI] [PubMed] [Google Scholar]
- Borner C, Holt V, Kraus J. Transcriptional regulation of the cannabinoid receptor type 1 gene in T cells by cannabinoids. J Leukoc Biol. 2007b;81:336–343. doi: 10.1189/jlb.0306224. [DOI] [PubMed] [Google Scholar]
- Borner C, Bedini A, Holt V, Kraus J. Analysis of promoter regions regulating basal and interleukin-4 inducible expression of the human CB1 receptor gene in T lymphocytes. Mol Pharmacol. 2008;73:1013–1019. doi: 10.1124/mol.107.042945. [DOI] [PubMed] [Google Scholar]
- Bortolato M, Frau R, Bini V, Luesu W, Loriga R, Collu M, et al. Methamphetamine neurotoxicity increase brain expression and alters behavioural functions of CB1 cannabinoid receptors. J Psychiatr Res. 2010;44:944–955. doi: 10.1016/j.jpsychires.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Cacciola G, Chioccarelli T, Mackie K, Meccariello R, Ledent C, Fasano S, et al. Expression of type-1 cannabinoid receptor during rat postnatal testicular development: possible involvement in adult leydig cell differentiation. Biol Reprod. 2008;79:758–765. doi: 10.1095/biolreprod.108.070128. [DOI] [PubMed] [Google Scholar]
- Caffarel MM, Sarrio D, Palacios J, Guzman M, Sanchez C. Delta-9- tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006;66:6615–6621. doi: 10.1158/0008-5472.CAN-05-4566. [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington HL, et al. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-o-methyltrasnferase gene: longitudinal evidence of a gene x environment interaction. Biol Psychiatry. 2005;57:1117–1127. doi: 10.1016/j.biopsych.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Corchero J, Romero J, Berrendero F, Fernandez-Ruiz J, Ramos JA, Fuentes JA, et al. Time-dependent differences of repeated administration with Delta9-tetrahydrocannabinol in proenkephalin and cannabinoid receptor gene expression and G-protein activation by mu-opiod and CB1-cannabinoid receptors in the caudate-putamen. Brain Res Mol Brain Res. 1999;67:148–157. doi: 10.1016/s0169-328x(99)00053-4. [DOI] [PubMed] [Google Scholar]
- Cottone E, Guastalla A, Mackie K, Franzoni MF. Endocannabinoids affect the reproductive functions in teleosts and amphibians. Mol Cell Endocrinol. 2008;286:S41–S45. doi: 10.1016/j.mce.2008.01.025. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Denovan-Wright EM, Robertson HA. Cannabinoid receptor messenger RNA levels decrease in a subset of neurons of the lateral striatum, cortex, and hippocampus of transgenic Huntington's disease mice. Neuroscience. 2000;98:705–713. doi: 10.1016/s0306-4522(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Diaz-Asensio C, Setien R, Echevarna E, Casis L, Casis E, Garrido A, et al. Type 1 diabetes alters brain cannabinoid receptor expression and phosphorylation status in rats. Horm Metab Res. 2008;40:454–458. doi: 10.1055/s-2008-1065323. [DOI] [PubMed] [Google Scholar]
- Eggan SM, Melchitsky DS, Sesack SR, Fish KN, Lewis DA. Relationship of cannabinoid CB1 receptor and cholecystokinin immunoreactivity in monkey dorsolateral prefrontal cortex. Neuroscience. 2010;169:1651–1661. doi: 10.1016/j.neuroscience.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury MA, Gorgievski V, Moutsimilli L, Giros B, Tzavara ET. Interactions between the cannabinoid and dopaminergic systems: evidence from animal studies. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38:56–60. doi: 10.1016/j.pnpbp.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Gomez M, Hernandez M, de Miguel R, Ramos JA. Cannabinoids and gene expression during brain development. Neurotox Res. 2004;6:389–401. doi: 10.1007/BF03033314. [DOI] [PubMed] [Google Scholar]
- Fitzgerald ML, Chan J, Mackie K, Lupica CR, Pickel VM. Altered dendritic distribution of dopamine D2 receptors and reduction in mitochondrial number in parvalbumin-containing interneurons in medial prefrontal cortex of cannabinoid-1 (CB1) receptor knockout mice. J Comp Neurol. 2012 doi: 10.1002/cne.23141. DOI: 10.1002/cne.23141. E-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Arencibia M, Garcia C, Kurz A, Rodriguez-Navarro JA, Gispert-Sachez S, Mena MA, et al. Cannabinoid CB1 receptors are early downregulated followed by a further upregulation in the basal ganglia of mice with deletion of specific park genes. J Neural Transm Suppl. 2009;73:269–275. doi: 10.1007/978-3-211-92660-4_22. [DOI] [PubMed] [Google Scholar]
- Gene Expression Nervous System Atlas (GENSAT) Project. 2010. Gene name: cannabinoid receptor 1 (brain). NINDS Contracts N01NS02331 & HHSN271200723015 to The Rockefeller University (New York, NY). Available at: http://www.gensat.org/GeneProgressTracker.jsp?gensatGeneID=397. [DOI] [PubMed]
- Gustafsson K, Wang X, Severa D, Eriksson M, Kimby E, Merup M, et al. Expression of cannabinoid receptors type 1 and type 2 in non-Hodgkin lymphoma: growth inhibition by receptor activation. Int J Cancer. 2008;123:1025–1033. doi: 10.1002/ijc.23584. [DOI] [PubMed] [Google Scholar]
- Gutierrez T, Farthing JN, Zvonok AM, Makriyannis A, Hohmann AG. Activation of peripheral CB1 and CB2 receptors suppresses the maintenance of inflammatory nociception: a comparative analysis. Br J Pharmacol. 2006;150:153–163. doi: 10.1038/sj.bjp.0706984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng L, Beverley JA, Steiner H, Tseng KY. Differential developmental trajectories for CB1 cannabinoid receptor expression in limbic/associative and sensorimotor cortical areas. Synapse. 2011;65:278–286. doi: 10.1002/syn.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MN, Hillard CJ, McEwen BS. Alterations in corticolimbic dendritic morphology and emotional behaviour in cannabinoid CB1 receptor-deficient mice parallel the effects of chronic stress. Cereb Cortex. 2011;21:2056–2064. doi: 10.1093/cercor/bhq280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Hudson BD, Hebert TE, Kelly ME. Physical and functional interaction between CB1 cannabinoid receptors and beta2-adrenoceptors. Br J Pharmacol. 2010;160:627–642. doi: 10.1111/j.1476-5381.2010.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley MJ, Mash DC, Jenner P. Expression of cannabinoid CB1 receptor mRNA in basal ganglia of normal and parkinsonian human brain. J Neural Transm. 2003;110:1279–1288. doi: 10.1007/s00702-003-0033-7. [DOI] [PubMed] [Google Scholar]
- Kalifa S, Polston EK, Allard JS, Manaye KF. Distribution patterns of cannabinoid CB1 receptors in the hippocampus of APPswe/PS1ΔE9 double transgenic mice. Brain Res. 2011;1376:94–100. doi: 10.1016/j.brainres.2010.12.061. [DOI] [PubMed] [Google Scholar]
- Karvela A, Rojas-Gil AP, Samkinidou E, Papadaki H, Pappa A, Georgiou G, et al. Endocannabinoid (EC) receptor, CB1, and EC enzymes' expression in primary adipocyte cultures of lean and obese pre-pubertal children in relation to adiponectin and insulin. J Pediatr Endocrinol Metab. 2010;23:1011–1024. doi: 10.1515/jpem.2010.162. [DOI] [PubMed] [Google Scholar]
- Kempf K, Hector J, Strate T, Schwartzloh B, Rose B, Herder C, et al. Immune-mediated activation of the endocannabinoid system in visceral adipose tissue in obesity. Horm Metab Res. 2007;39:596–600. doi: 10.1055/s-2007-984459. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrinaga G, Sanz B, Perez I, Blanco L, Candenas ML, Pinto FM, et al. Cannabinoid CB1 receptor is downregulated in clear cell renal cell carcinoma. J Histochem Cytochem. 2010a;58:1129–1134. doi: 10.1369/jhc.2010.957126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrinaga G, Varona A, Perez I, Sanz B, Ugalde A, Candenas ML, et al. Expression of cannabinoid receptors in human kidney. Histol Histopathol. 2010b;25:1133–1138. doi: 10.14670/HH-25.1133. [DOI] [PubMed] [Google Scholar]
- Lee JH, Agacinski G, Williams JH, Wilcock GK, Esiri MM, Francis PT, et al. Intact cannabinoid CB1 receptors in Alzheimer's disease cortex. Neurochem Int. 2010;57:985–989. doi: 10.1016/j.neuint.2010.10.010. [DOI] [PubMed] [Google Scholar]
- McCaw EA, Hu H, Gomez GT, Hebb AL, Kelly ME, Denovan-Wright EM. Structure, expression, and regulation of the cannabinoid receptor gene (CB1) in Huntington's disease transgenic mice. Eur J Biochem. 2004;271:4909–4920. doi: 10.1111/j.1432-1033.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- Malfitano AM, Laezza C, Galgani M, Matarese G, D'Alessandro A, Gazzerro P, et al. The CB1 receptor antagonist rimonabant controls cell viability and ascitic tumour growth in mice. Pharmacol Res. 2012;65:365–371. doi: 10.1016/j.phrs.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Marshall AD, Lagutina I, Grosveld GC. PAX3-FOX01 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res. 2011;71:7471–7480. doi: 10.1158/0008-5472.CAN-11-0924. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Martin BR, Mechoulam R, Razdan RK. Discovery and characterization of endogenous cannabinoids. Life Sci. 1999;65:573–595. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Hanu L. The cannabinoids: an overview. Therapeutic implications in vomiting and nausea after cancer chemotherapy, in appetite promotion, in multiple sclerosis, and in neuroprotection. Pain Res Manag. 2001;6:67–73. doi: 10.1155/2001/183057. [DOI] [PubMed] [Google Scholar]
- Mievis S, Blum D, Ledent C. Worsening of Huntington's disease phenotype in CB1 receptor knockout mice. Neurobiol Dis. 2011;42:524–529. doi: 10.1016/j.nbd.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Miller LK, Devi LA. The highs and lows of cannabinoid receptor expression in disease: mechanisms and their therapeutic implications. Pharmacol Rev. 2011;63:461–470. doi: 10.1124/pr.110.003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay B, Liu J, Osei-Hyiaman D, Godlewski G, Mukhopadhyay P, Wang L, et al. Transcriptional regulation of cannabinoid receptor-1 expression in the liver by retinoic acid acting via retinoic acid receptor-gamma. J Biol Chem. 2010;285:19002–19011. doi: 10.1074/jbc.M109.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- National Centre for Biotechnology Information (NCBI) 2011. EST Profile Hs.75110-CNR1: cannabinoid receptor 1 (brain). Database of Expressed Sequence Tags (dbEST) EST Profile Viewer. Available at: http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.75110.
- Navarro G, Carriba P, Gandia J, Ciruela F, Casado V, Cortes A, et al. Detections of heteromers formed by cannabinoid CB1, dopamine D2, and adenosine A2A G-protein- coupled receptors by combining bimolecular fluorescence complementation and bioluminescence energy transfer. ScientificWorldJournal. 2008;11:1088–1097. doi: 10.1100/tsw.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro HA, Howard JL, Pollard GT, Carroll FI. Positive allosteric modulation of the human cannabinoid (CB) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nortarnicola M, Messa C, Orlando A, Bifulco M, Laezza C, Gazzerro P, et al. Estrogenic induction of cannabinoid CB1 receptor in human colon cancer cell lines. Scand J Gastroenterol. 2008;43:66–72. doi: 10.1080/00365520701559011. [DOI] [PubMed] [Google Scholar]
- Pacheco DF, Klein A, Perez AC, Pacheco CM, de Francischi JN, Reis GM, et al. Central antinociception induced by mu-opioid receptor agonist morphine, but not delta- or kappa-, is mediated by cannabinoid CB1 receptor. Br J Pharmacol. 2009;158:225–231. doi: 10.1111/j.1476-5381.2009.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo FA, Angelini M, Cottone E, Virgili M, Franzoni MF, Mosconi G, et al. Involvement of endocannabinoid CB1 receptor in the modulation of stress responses related to xenoestrogen exposure. Ann N Y Acad Sci. 2009;1163:504–507. doi: 10.1111/j.1749-6632.2009.04455.x. [DOI] [PubMed] [Google Scholar]
- Paria BC, Das SK, Dey SK. The preimplantation mouse embryo is a target for cannabinoind ligand-receptor signalling. Proc Natl Acad Sci U S A. 1995;92:9460–9464. doi: 10.1073/pnas.92.21.9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos MR, Sagredo O, Fernandez-Ruiz J. The endocannabinoid system in Huntington's disease. Curr Pharm Des. 2008;14:2317–2325. doi: 10.2174/138161208785740108. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto JG, Hornby KR, Jones DG, Murphy KM. Developmental changes in GABAergic mechanisms in human visual cortex across the lifespan. Front Cell Neurosci. 2010;4:1–12. doi: 10.3389/fncel.2010.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proto MC, Gazzerro P, Di Croce L, Santoro A, Malfitano AM, Pisanti S, et al. Interaction of endocannabinoid system and steroid hormones in the control of colon cancer cell growth. J Cell Physiol. 2011;27:250–258. doi: 10.1002/jcp.22727. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Haskó G, Huffman JW, Mackie K, Pacher P. CB2 cannabinoid receptor agonists attenuate TNF-alpha-induced human vascular smooth muscle cell proliferation and migration. Br J Pharmacol. 2008;153:347–357. doi: 10.1038/sj.bjp.0707569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Calandra B, Shire D, Bouaboula M, Oustric D, Barth F, et al. Characterization of two cloned human CB1 receptor isoforms. J Pharmacol Exp Ther. 1996;278:871–878. [PubMed] [Google Scholar]
- Romero J, Garcia-Palomero E, Berrendero F, Garcia-Gil J, Hernandez ML, Ramos JA, et al. Atypical location of cannabinoid receptors in white matter areas during rat brain development. Synapse. 1997;26:317–323. doi: 10.1002/(SICI)1098-2396(199707)26:3<317::AID-SYN12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Rozenfeld R, Bushlin I, Gomes I, Tzavaras N, Gupta A, Neves S, et al. Receptor heteromerization expands the repertoire of cannabinoid signaling in rodent neurons. PLoS ONE. 2012;7:e29239. doi: 10.1371/journal.pone.0029239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryberg E, Vu HK, Larsson N, Groblewski T, Hjorth S, Elebring T, et al. Identification and characterisation of a novel splice variant of the human CB1receptor. FEBS Lett. 2005;579:259–264. doi: 10.1016/j.febslet.2004.11.085. [DOI] [PubMed] [Google Scholar]
- Sarzani R, Bordicchia M, Marcucci P, Bedetta S, Santini S, Giovagnoli A, et al. Altered pattern of cannabinoid 1 receptor expression in adipose tissue of dysmetabolic and overweight patients. Metabolism. 2009;58:361–367. doi: 10.1016/j.metabol.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Shire D, Calandra B, Rinaldi-Carmona M, Oustric D, Pessègue B, Bonnin-Cabanne O, et al. Molecular cloning, expression and function of the murine CB2 peripheral cannabinoid receptor. Biochim Biophys Acta. 1996;1307:132–136. doi: 10.1016/0167-4781(96)00047-4. [DOI] [PubMed] [Google Scholar]
- Silverdale MA, McGuire S, McInnes A, Crossman AR, Brotchie JM. Striatal cannabinoid CB1 receptor mRNA expression is decreased in the reserpine-treated rat model of Parkinson's disease. Exp Neurol. 2001;169:400–406. doi: 10.1006/exnr.2001.7649. [DOI] [PubMed] [Google Scholar]
- Smith TH, Sim-Selley LJ, Selley DE. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? Br J Pharmacol. 2010;160:454–466. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner H, Bonner TI, Zimmer AM, Kitai ST, Zimmer A. Altered gene expression in striatal projection neurons in CB1 cannabinoid receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5786–5790. doi: 10.1073/pnas.96.10.5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Wagner-Miller J, Hutchens J, Mackie K. Differential signaling in human cannabinoid CB1 receptors and their splice variants in autaptic hippocampal neurons. Br J Pharmacol. 2012;165:2660–2671. doi: 10.1111/j.1476-5381.2011.01744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash B, Thiruchelvan K, Ahuja M, Suppiramaniam V, Dhanasekaran M. Methampethamine-induced neurotoxicity: the road to Parkinson's disease. Pharmacol Rep. 2009;61:966–977. doi: 10.1016/s1734-1140(09)70158-6. [DOI] [PubMed] [Google Scholar]
- Uriguen L, Garcia-Fuster MJ, Callado LF, Morentin B, La Harpe R, Cassado V, et al. Immunodensity and mRNA expression of A2Aadenosine, D2 dopamine, and CB1 cannabinoid receptors in post-mortem frontal cortex of subjects with schizophrenia: effect of antipsychotic treatment. Psychopharmacology (Berl) 2009;206:313–324. doi: 10.1007/s00213-009-1608-2. [DOI] [PubMed] [Google Scholar]
- Vinod KY, Arango V, Xie S, Kassir SA, Mann JJ, Cooper TB, et al. Elevated levels of endocannabinoids and CB1 receptor-mediated G-protein signalling in the prefrontal cortex of alcoholic suicide victims. Biol Psychiatry. 2005;57:480–486. doi: 10.1016/j.biopsych.2004.11.033. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Laine J, Simon A, Roland A, Leterrier C, Lenkei Z. The type 1 cannabinoid receptor is highly expressed in embryonic cortical projection neurons and negatively regulates neurite growth in vitro. Eur J Neurosci. 2008;28:1705–1718. doi: 10.1111/j.1460-9568.2008.06484.x. [DOI] [PubMed] [Google Scholar]
- Wang D, Wang H, Ning W, Backlund MG, Dey SK, DuBois RN. Loss of cannabinoid receptor 1 accelerates intestinal tumor growth. Cancer Res. 2008;68:6468–6476. doi: 10.1158/0008-5472.CAN-08-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlake TM, Howlett AC, Bonner TI, Matsuda LA, Herkenham M. Cannabinoid receptor binding and messenger RNA expression in human brain: an in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer's brains. Neuroscience. 1994;63:637–652. doi: 10.1016/0306-4522(94)90511-8. [DOI] [PubMed] [Google Scholar]
- Xiao JC, Jewell JP, Lin LS, Hagmann WK, Fong TM, Shen CP. Similar in vitro pharmacology of human cannabinoid CB1 receptor variants expressed in CHO cells. Brain Res. 2008;1238:36–43. doi: 10.1016/j.brainres.2008.08.027. [DOI] [PubMed] [Google Scholar]
- Zeng BY, Dass B, Owen A, Rose S, Cannizzaro C, Tel BC, et al. Chronic L-DOPA treatment increases striatal cannabinoid CB1 receptor mRNA expression in 6-hydroxydopamine-lesioned rats. Neurosci Lett. 1999;276:71–74. doi: 10.1016/s0304-3940(99)00762-4. [DOI] [PubMed] [Google Scholar]
- Zhang F, Hong S, Stone V, Smith PJ. Expression of cannabinoid CB1 receptors in models of diabetic neuropathy. J Pharmacol Exp Ther. 2007;323:508–515. doi: 10.1124/jpet.107.128272. [DOI] [PubMed] [Google Scholar]
- Zhang P-W, Ishiguro H, Ohtsuki T, Hess J, Carillo F, Walther D, et al. Human cannabinoid receptor 1: 5' exons, candidate regulatory regions, polymorphisms, haplotypes and association with polysubstance abuse. Mol Psychiatry. 2004;9:916–931. doi: 10.1038/sj.mp.4001560. [DOI] [PubMed] [Google Scholar]
- Zhuang S, Kittler J, Grigorenko EV, Kirby MT, Sim LJ, Hampson RE. Effects of long- term exposure to delta9-THC on expression of cannabinoid receptor (CB1) mRNA in different rat brain regions. Brain Res Mol Brain Res. 1998;62:141–149. doi: 10.1016/s0169-328x(98)00232-0. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased motality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurolo E, Iyer AM, Spliet WG, Van Rijen PC, Troost D, Gorter JA, et al. CB1 and CB2 cannabinoid receptor expression during development and in epileptogenic development pathologies. Neuroscience. 2010;170:28–41. doi: 10.1016/j.neuroscience.2010.07.004. [DOI] [PubMed] [Google Scholar]