

Abstract
BACKGROUND AND PURPOSE
Pharmacological activation of cannabinoid CB1 and CB2 receptors is a therapeutic strategy to treat chronic and inflammatory pain. It was recently reported that a mixture of natural triterpenes α- and β-amyrin bound selectively to CB1 receptors with a subnanomolar Ki value (133 pM). Orally administered α/β-amyrin inhibited inflammatory and persistent neuropathic pain in mice through both CB1 and CB2 receptors. Here, we investigated effects of amyrins on the major components of the endocannabinoid system.
EXPERIMENTAL APPROACH
We measured CB receptor binding interactions of α- and β-amyrin in validated binding assays using hCB1 and hCB2 transfected CHO-K1 cells. Effects on endocannabinoid transport in U937 cells and breakdown using homogenates of BV2 cells and pig brain, as well as purified enzymes, were also studied.
KEY RESULTS
There was no binding of either α- or β-amyrin to hCB receptors in our assays (Ki > 10 µM). The triterpene β-amyrin potently inhibited 2-arachidonoyl glycerol (2-AG) hydrolysis in pig brain homogenates, but not that of anandamide. Although β-amyrin only weakly inhibited purified human monoacylglycerol lipase (MAGL), it also inhibited α,β-hydrolases and more potently inhibited 2-AG breakdown than α-amyrin and the MAGL inhibitor pristimerin in BV2 cell and pig brain homogenates.
CONCLUSIONS AND IMPLICATIONS
We propose that β-amyrin exerts its analgesic and anti-inflammatory pharmacological effects via indirect cannabimimetic mechanisms by inhibiting the degradation of the endocannabinoid 2-AG without interacting directly with CB receptors. Triterpenoids appear to offer a very broad and largely unexplored scaffold for inhibitors of the enzymic degradation of 2-AG.
LINKED ARTICLES
This article is part of a themed section on Cannabinoids. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.167.issue-8
Keywords: amyrin, 2-AG, CB1 receptor, endocannabinoid system, hydrolases, monoacyl glycerol lipase, pristimerin, triterpene
Introduction
Cannabinoid CB1 receptors exert a range of central and peripheral pharmacological effects including analgesia (Pacher et al., 2006; Guindon and Hohmann, 2008; Bisogno and Di Marzo, 2010; receptor nomenclature follows Alexander et al., 2011). Activation of CB1 receptors in the CNS typically leads to pronounced psychomodulatory effects, including inhibition of locomotion, catalepsy, reduction of body temperature and antinociception (the tetrad effect) (Fride et al., 2006; Pertwee, 2009; Pertwee et al., 2010). While there is a wide range of synthetic CB1 receptor agonists, Δ9-tetrahydrocannabinol (Δ9-THC) from the plant Cannabis sativa L and closely related secondary metabolites are the only natural products known so far to potently activate the CB1 receptor (Gertsch et al., 2010). We were therefore intrigued by a recent study by Da Silva et al. (2011) reporting the ultrapotent and highly selective interaction (activation) of a 1:1 mixture of the triterpenes α/β-amyrin with the CB1 receptor without causing behavioural effects. The pentacyclic triterpene β-amyrin is a primary cyclization product derived from squalene (Rees et al., 1968) and thus a canonical and widely occurring triterpenoid precursor for a number of pentacyclic terpenes. Anti-inflammatory and antinociceptive properties of β-amyrin have been reported previously (Akihisa et al., 1996; Holanda Pinto et al., 2008; Melo et al., 2010; 2011), but a feasible mechanism of action is lacking. In the study by da Silva et al., oral α/β-amyrin chronically administered at 30 mg·kg−1 inhibited mechanical and thermal hyperalgesia and inflammation induced by complete Freund's adjuvant and by partial sciatic nerve ligation. Remarkably, in this study, the α/β-amyrin mixture exhibited a very potent binding to CB1 receptors with a Ki value of 133 pM, which is approximately 200–300-fold more potent than Δ9-THC. Moreover, in contrast to Δ9-THC and the endogenous ligand 2-arachidonoyol glycerol (2-AG), α/β-amyrin showed an unusual 15 000-fold binding selectivity to CB1 receptors over CB2 receptors (Da Silva et al., 2011). Notably, there are relatively few subnanomolar CB1 receptor ligands known, such as, for example CE-178253 and CP-945,598, which were synthesized at Pfizer (Griffith et al., 2009; Hadcock et al., 2010), JWH182 from Huffman Laboratories (Huffman et al., 2005), or AM4030a and others from the Makriyannis laboratory (Papahatjis et al., 1998; Thakur et al., 2002). Thus, the finding that a canonical plant triterpene exhibits such potent and selective binding interactions with CB1 over CB2 receptors is of great interest. The authors concluded that the CB1 receptor interaction (as detected in their binding assays) was the major mechanism of action. Although not shown in their study, they assumed that α/β-amyrin activates CB1 receptors. In their report, pretreatment with both CB1 or CB2 antagonists and gene knockdowns of both receptors significantly inhibited the antinociceptive effect of α/β-amyrin, indicating direct or indirect effects on CB receptors.
The study by da Silva et al. (2011) raises several basic questions that need to be addressed before classifying the amyrins as novel CB1 receptor-selective agonists. In order to understand the actual mechanism of action, α-amyrin (vicinal methyls in A ring) and β-amyrin (geminal methyls in A ring) should be studied separately as both compounds are commercially available in purities >95% from Sigma-Aldrich and other providers. The CB1 and CB2 receptor binding interactions were performed in rat brain and mouse spleen homogenates, respectively, using the specific radioligands [3H]-SR141716A (rimonabant) and [3H]-CP55,940. Because both tissues potentially show a high degree of non-specific binding and the radioligands may have additional targets (Breivogel, 2006), it is important to reproduce the ultrapotent CB1 receptor binding interaction in more defined recombinant systems, using additional controls. Because CB1 receptor agonists typically trigger a Gi/o protein-mediated signalling, it would be interesting to know whether α/β-amyrins are full or partial agonists. In light of the CB receptor non-specific pharmacology (the antinociceptive effects were inhibited by both CB1 and CB2 receptor antagonists), which is in conflict with a purely CB1 receptor-mediated mechanism, also, other targets related to endocannabinoid breakdown should be considered. In the present study, we have addressed these issues and now provide an alternative cannabimimetic mechanism of action of α/β-amyrin, which is able to explain, at least in part, the pharmacology of the amyrins. Our results show a potent inhibition of 2-AG hydrolysis by β-amyrin in brain tissue and point out a significant difference in the inhibitory potency between the two structural isomers.
Methods
Cell culture
Human leukemia monocytic lymphoma U937 cells were purchased from American Type Culture Collection (Manassas, VA, USA) and were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 g·mL−1 fungizone (amphotericin B), 100 units·mL−1 penicillin, 100 g·mL−1 streptomycin and 2 mM L-glutamine (all from Invitrogen, Carlsbad, CA, USA). The hCB1- or hCB2-expressing CHO-K1 cells (Raduner et al., 2006) were grown in the same medium as the U937 cells, but supplemented with 400 µg·mL−1 G418 (10131-027; Invitrogen). Cells were grown in a humidified incubator at 37°C and 5% CO2.
Membrane preparation
CHO-K1 cells stably transfected with hCB1 or hCB2 receptors were seeded in a T150 cm2 flask until reaching confluence and then collected by scraping. The cell suspension was resuspended in 1.5 mL of homogenization solution (50 mM Tris-HCl, 1 mM EDTA, 3 mM MgCl2, pH 7.5) and homogenized on ice using a Dounce homogenizer with 50 gentle strokes. The cell homogenate was then centrifuged at 800 ×g for 8 min at 4°C. The supernatant was collected (membrane fraction 1) and the pellet resuspended in 700 µL of homogenization solution, passed through a syringe 10 times, and centrifuged at 800 ×g for 8 min at 4°C. The supernatant was collected (membrane fraction 2) while the pellet underwent the same operations described earlier. The supernatant was collected (membrane fraction 3) and pooled with membrane fraction 2 (membrane fraction 4). Membrane fractions 1 and 4 were then centrifuged at 3500 ×g for 10 min at 4°C, the supernatant was discarded and both pellets resuspended in homogenization solution to a final volume of 600 µL (‘crude membrane fraction’). Protein concentration was determined using the Bradford method and aliquots stored at −80°C.
Radioligand binding assays on hCB1 and hCB2 receptors
Receptor-binding experiments were performed with membrane preparations as previously reported (Raduner et al., 2006; Gertsch et al., 2008). Briefly, 18 µg of crude membrane expressing hCB1 or hCB2 receptors (prepared as described earlier) were resuspended in 500 µL of binding buffer (50 mM Tris-HCl, 2.5 mM EDTA, 5 mM MgCl2, 0.5 mg·mL−1 fatty acid-free BSA, pH 7.4) in silanized glass tubes and co-incubated with different concentrations of α-amyrin, β-amyrin or WIN55,512-2 and 0.5 nM of [3H]-CP-55 940 (168 Ci·mmol−1) for 2 h at 30°C with shaking. Non-specific binding of the radioligand was determined in the presence of 10 µM of WIN55,512-2. Non-specific binding was around 5%. After the incubation time, membrane suspensions were rapidly filtered through a 0.05% polyethyleneimine presoaked 96-well microplate bonded with GF/B glass fibre filters (UniFilter-96 GF/B, PerkinElmer Life Sciences) under vacuum and washed 12 times with 167 µL of ice-cold washing buffer (50 mM Tris-HCl, 2.5 mM EGTA, 5 mM MgCl2, 0.5% fatty acid free BSA, pH 7.4). Filters were added to 40 µL of MicroScint20 scintillation liquid and radioactivity measured with the 1450 MicroBeta Trilux top counter (PerkinElmer Life Sciences). Data were collected from three independent experiments performed in triplicate and the non-specific binding was subtracted. Results were expressed as [3H]-CP-55 940 bound as % of binding in vehicle-treated samples.
Fatty acid amide hydrolase (FAAH) activity assay
The experiments were performed as previously described (Chicca et al., 2009). Briefly, FAAH activity, which is responsible for most of the anandamide (AEA) breakdown, was assessed using a cellular homogenate from fresh pig brains obtained from the slaughterhouse. Different concentrations of α-amyrin, β-amyrin or 1 µM PMSF (as the positive control) in 2.5 µL DMSO were preincubated at 37°C for 20 min with 195 µL of diluted pig brain homogenate in 10 mM Tris–HCl and 1 mM EDTA, pH 7.6 (corresponding to 200 µg of total protein) and 1% w/v fatty acid-free BSA (Sigma, St. Louis, MO, USA). Successively, 2.5 µL of non-radioactive AEA and [ethanolamine-1-3H]-AEA (40–60 Ci·mmol−1) mixture was added to the homogenate (final concentration in the assay was 2 µM) and incubated for 10 min at 37°C with shaking. After the incubation time, 400 µL of a methanol : chloroform mixture 1:1 (v/v) was added at each sample, vigorously vortexed and centrifuged at 10 000 × g for 10 min at 4°C to separate aqueous and organic phases. The radioactivity associated with the hydrolysis product [3H]-ethanolamine was measured after addition of 3 mL of Ultima Gold scintillation liquid (PerkinElmer Life Sciences) to the aqueous phase, using a Tri-Carb 2100 TR scintillation counter (PerkinElmer Life Sciences). Data were collected from three independent experiments performed in triplicate and results were expressed as FAAH activity, relative to that in vehicle-treated samples (= 100%).
Assay of [3H]-2-oleoyl glycerol (2-OG) hydrolysis in pig brain homogenate
The experiments were performed as recently described (Björklund et al., 2010) with some minor changes. In brief, 195 µL (corresponding to 200 µg of total protein) of diluted pig brain homogenate in 10 mM Tris–HCl, 1 mM EDTA, plus 0.1% w/v fatty acid-free BSA pH 7.4 was preincubated for 30 min at 37°C with different concentrations (2.5 µL) of α-amyrin, β-amyrin, JZL184 or the mixture of α,β-hydrolases (ABHD) -6 and −12 inhibitors (10 µM WWL70 and 20 µM THL, respectively) plus 1 µM JZL184 as the positive control for the maximal inhibition of 2-AG hydrolysis. After the preincubation, 2.5 µL of nonradioactive (2-OG) and [1,2,3-3H]-2-OG (60 Ci·mmol−1) mixture was added to the homogenate (final concentration in the assay was 10 µM) and incubated for 10 min at 37°C with shaking. Successively, 400 µL of a methanol : chloroform mixture 1:1 (v/v) was added and, after vigorous vortexing, aqueous and organic phases were separated by centrifugation at 10 000 × g for 10 min at 4°C. The radioactivity associated with [3H]-glycerol formation was measured after addition of 3 mL of Ultima Gold scintillation liquid (PerkinElmer Life Sciences) to the aqueous phase, using a Tri-Carb 2100 TR scintillation counter (PerkinElmer Life Sciences). Data were collected from three independent experiments performed in triplicate and results were expressed as [3H]-glycerol formation, relative to that in vehicle-treated samples (=100%).
[3H]-2-OG hydrolysis in homogenates of BV2 cells
The method was adapted from Marrs et al. (2010). In brief, BV2 cell homogenates (100 µg of total protein) were added to silanized plastic tubes containing 100 µL of buffer (10 mM Tris-HCl, 1 mM EDTA (pH 7.4) supplemented with 0.1% fatty acid-free BSA. The homogenate was preincubated for 30 min at 37°C with shaking in presence of 1 µM URB597 (to block FAAH activity) and different concentrations of amyrins, JZL184, WWL70, pristimerin or vehicle. Subsequently 2.5 nM of [3H]-2-OG was added and the tubes incubated for 15 min at 37°C with shaking. The reaction was stopped by adding 300 µL of a methanol : chloroform mixture 1:1 (v/v) and vortexing. To separate organic and aqueous phases, the tubes were centrifuged for 10 min at 10 000 × g at 4°C. The upper aqueous phase was transferred to scintillation tubes andmixed with 3 mL of Ultima Gold scintillation liquid (PerkinElmer Life Sciences). Radioactivity was measured using a Beckman LS6500 scintillation counter. Data were collected from three independent experiments each performed in triplicate and results were expressed as [3H]-glycerol formation, relative to that in vehicle-treated samples (=100%).
Monoacylglycerol lipase (MAGL) activity assay
Purified recombinant human MAGL (hMAGL) was purchased from Cayman Chemicals. MAGL esterase activity was assessed as previously reported (King et al., 2007; 2009a; Björklund et al., 2010). The reaction consisted of 196 µL assay buffer (10 mM Tris-HCl 1 mM EDTA pH 7.4, plus 0.1% fatty acid-free BSA) containing 25 ng of purified hMAGL and 2 µL of JZL184, α-amyrin, β-amyrin, pristimerin or vehicle and 2.5 nM [3H]-2-OG (2 µL). To assess the time-dependency of MAGL inhibition, the reaction was incubated for periods between 1 and 60 min. To assess the reversibility of MAGL inhibition, we performed an assay reported previously (King et al., 2009b). The reaction mixture was incubated for 15 min at 37°C and then stopped by adding 400 µL of chloroform : methanol mixture 1:1 (v/v). Tubes were vortexed and centrifuged for 10 min at 10 000 × g at 4°C. Finally, the upper aqueous phase was transferred in scintillation tubes and mixed with 3 mL of Ultima Gold scintillation liquid (PerkinElmer Life Sciences). Radioactivity was measured using a Beckman LS6500 scintillation counter. Data were collected from two independent experiments performed in triplicates and the results were expressed as % [3H]-glycerol formation, relative to that in vehicle-treated samples (=100%).
[3H]-2-AG uptake into U937 cells
The uptake of [1,2,3-3H]-2-arachidonyl glycerol, (20–40 Ci·mmol−1) in intact cells was performed using U937 cells. Briefly, 106 cells were re-suspended in 500 µL of serum-free medium in silanized plastic tubes and preincubated with different concentrations of α-amyrin, β-amyrin or JZL184 for 30 min at 37°C. Then, a mixture of 2-AG/[3H]-2-AG to a final concentration of 1 µM, was added to the cells and incubated for 5 min at 37°C. The uptake process was stopped by placing the tubes on ice and rapidly centrifuging them at 800 × g for 5 min at 4°C. The supernatant was collected and transferred into 1 mL of a methanol : chloroform mixture 1:1 (v/v), while the pellet was re-suspended in ice-cold PBS plus 1% of fatty acid-free BSA and centrifuged at 800 × g for 5 min at 4°C (washing step). The washing solution was discarded while the cell pellet was re-suspended in 250 µL of ice-cold PBS and transferred into 500 µL of a methanol : chloroform mixture 1:1 (v/v), vortexed vigorously, sonicated on ice-cold water bath for 5 min and finally centrifuged at 10 000 × g for 10 min at 4°C. The aqueous phase was pooled with the aqueous phase extracted from the supernatant and transferred in a scintillation tube while the lipophilic phase was transferred in a separated tube. The radioactivity associated with the intracellular [3H]-2-AG or the hydrolysis product [3H]-glycerol was measured by adding 3 mL of Ultima Gold scintillation liquid (PerkinElmer Life Sciences) using a Tri-Carb 2100 TR scintillation counter (PerkinElmer Life Sciences). Data were collected from three independent experiments each performed in triplicates. Results were expressed as [3H]-glycerol formation (or [3H]-2-AG intracellular accumulation), relative to that in vehicle-treated samples (=100%).
Quantification of AEA and 2-AG levels by GC-MS
The quantification of the endogenous AEA and 2-AG level was performed according to a published method (Marazzi et al., 2011). Briefly, 2.5 mL of diluted pig brain homogenate in 10 mM Tris-HCl and 1 mM EDTA, pH 7.6 (corresponding to 1.8 mg of protein) was incubated with α-amyrin, β-amyrin, PMSF or JZL184 for 30 min at 37°C with shaking. After incubation, lipids were extracted by Folch extraction (Folch et al., 1957), transferring the brain homogenate to 15 mL of ice-cold chloroform/methanol 2:1 containing internal standards (the final mixture was 4:2:1 /CHCl3 : CH3OH : H2O, v/v/v). The suspension was vortexed vigorously, sonicated for 5 min on ice-cold water bath and centrifuged for 10 min at 500 ×g. The organic phase was transferred to a glass vial, dried under N2, reconstituted in 1 mL ethanol, diluted with 9 mL of water and finally extracted by solid-phase extraction with C-18 Sep-Pak cartridge (Waters AG, Zug, Switzerland). The internal standards used for the quantification were: N-arachidonoylethanolamine-d4 1 ng·µL−1 (25 ng per sample), 2-arachidonoylglycerol-d5 8 ng·µL−1 (200 ng per sample). Derivatization of the hydroxyl groups was performed with the silylating agent dimethylisopropylsilyl imidazole at room temperature for 1 h as previously reported (Obata et al., 2003). The samples were analysed by GC-electron ionization (EI)-MS using an Agilent 6890 N GC equipped with a 30 m HP-5MS column and a 5975 C EI-MS with triple-axis detector. Helium was used as carrier gas at a constant flow rate of 1.5 mL·min−1 with splitless injection at an inlet temperature of 250°C. Optimal separation of the analytes was achieved with the following oven programme: initial temperature of 150°C for 1 min, followed by an increase to 280°C at 8°C·min−1 with a final time of 20 min. Because of acyl group migration of 2-AG during lipid extraction and purification, a significant amount of 1-AG was formed. Therefore, the peak areas of 1-AG and 2-AG were combined for all quantifications.
Data analysis
Results are expressed as mean values ± SEM for each examined group. Statistical significance of differences between groups was determined by the Student's t-test (paired t-test) with GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA, USA). Outliers in a series of identical experiments were determined by Grubb's test (ESD method) with α set to 0.05. Statistical differences between treated and vehicle control groups were determined by Student's t-test for dependent samples. Differences between the analysed samples were considered as significant if P≤ 0.05.
Materials
[Ethanolamine-1-3H]-anandamide (3H-AEA) (40–60 Ci·mmol−1) and [1,2,3-3H]-2-arachidonyl glycerol (20–40 Ci·mmol−1) were obtained from American Radiolabeled Chemicals Inc. (Saint Louis, MO, USA), and the radioligand [3H]-CP-55 940 (168 Ci·mmol−1) was obtained from PerkinElmer Life Sciences (Waltham, MA, USA). WIN55,212-2, AEA, 2-arachidonoyl glycerol (2-AG), JZL184, WWL70 and tetrahydrolipstatin (THL) were obtained from Cayman Chemicals Europe (Tallin, Estonia). Pristimerin and α-amyrin were purchased from Fluka Sigma-Aldrich (Buchs, Switzerland) while the structural isomer β-amyrin (∼95%) was kindly provided by Professor Dr. Giovanni Appendino. The natural products α-amyrin and β-amyrin were also purchased from Extrasynthese (Lyon, France) with a purity of ≥98.0%. Purity in stock solutions was checked by GC-MS. In the experiments, all batches were used to rule out differences between different sources.
Results
First, the two structural isomers of amyrin (Figure 1) were tested in radioligand binding assays for both cannabinoid receptors using membrane preparations obtained from CHO-K1 cells stably transfected with hCB1 or hCB2 receptors, respectively. β-Amyrin induced only a slight partial displacement of the radioligand from CB1 receptors at 1 µM (approximately 25%), while α-amyrin inhibited radioligand binding at concentrations ≥10 µM (Figure 2A). As shown in Figure 2B, both α- and β-amyrin did not strongly displace [3H]-CP55940 from CB2 receptors at concentrations below 10 µM. Thus, in our hands, α- and β-amyrin are not or are very weak CB1 receptor ligands. Because the study by da Silva et al. (2011) showed a very consistent CB receptor-dependent antinociceptive effect in vivo, we tested whether amyrins interact with other major components of the endocannabinoid system. We used two different methods to address potential effects on AEA and 2-AG hydrolysis: the classical radioactivity-based assays using [3H]-AEA and [3H]-2-AG and absolute GC-MS quantification of the endogenous endocannabinoid levels in brain homogenate. Both α- and β-amyrin were assessed for FAAH activity in the concentration range of 0.1 nM–10 µM and the results obtained by both methods showed no inhibition of AEA hydrolysis (Figure 3). PMSF was used as positive control showing a complete reduction of [3H]-ethanolamine formation in radioactivity experiments, as well as an accumulation of the endogenous AEA in the pig brain homogenate (Figure 3). Next, we evaluated the effects of α- and β-amyrin on 2-AG hydrolysis in the same homogenate. As shown in Figure 4, β-amyrin exhibited a concentration-dependent reduction of [3H]-glycerol formation starting at 10 nM and reaching the maximal effect of 35–40% inhibition at 10 µM. Likewise, α-amyrin led to a slight (about 5–8%), but significant 2-AG hydrolysis inhibition at 10 nM, but reaching only 20% of the maximal inhibition effect at 10 µM (Figure 4A). In the same system, JZL184 was used as a positive control for the inhibition of MAGL, the main enzyme involved in 2-AG hydrolysis in the brain (Labar et al., 2010; Lichtman et al., 2010). The concentration–inhibition curves of JZL184 and β-amyrin showed a similar trend without major differences in maximal inhibition at 10 µM. A further positive control included 1 µM of JZL184 in combination with 10 µM of WWL70 and 20 µM of THL [inhibitors of ABHD-6 and −12, respectively, two enzymes recently identified to hydrolyse 2-AG in the brain (Blankman et al., 2007; Marrs et al., 2010)]. This combination reduced [3H]-glycerol formation to 20% of vehicle control, confirming that MAGL, unlike FAAH for AEA, is not the only enzyme involved in 2-AG hydrolysis in the brain. Thus, β-amyrin inhibited 2-AG hydrolysis in a concentration-dependent manner, similar to that of JZL184, although not reaching the same maximal inhibition in pig brain homogenates.
Figure 1.
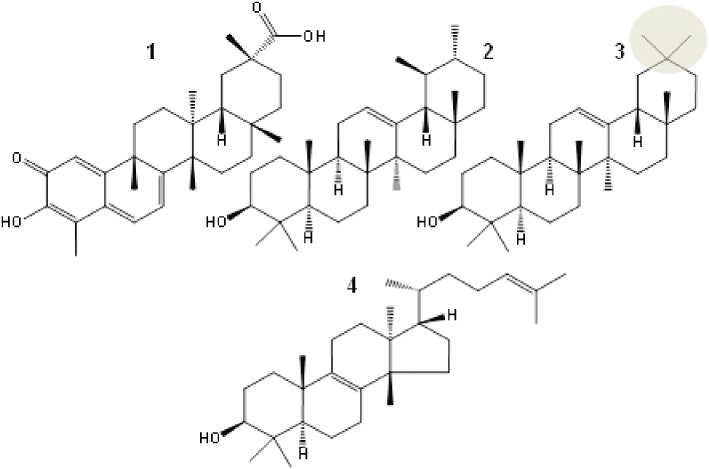
Chemical structures of the triterpenes pristimerin (1), α-amyrin (2), β-amyrin (3) and euphol (4), all of which inhibit 2-AG hydrolysis. β-Amyrin (3) with geminal methyl groups in ring A (grey circle) is the most effective inhibitor of 2-AG degradation in cellular extracts.
Figure 2.
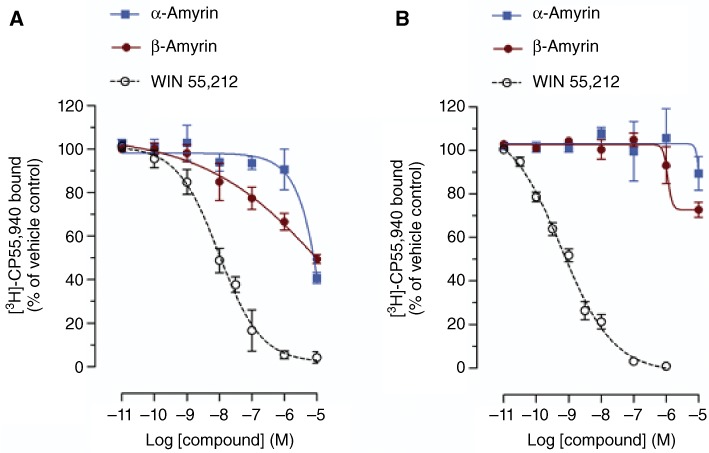
Radioligand binding assay using CHO-K1 cells stably transfected with (A) hCB1 and (B) hCB2 receptors and 0.5 nM [3H]-CP55,940. The potent non-specific CB receptor ligand WIN55,212-2 was used as a positive control. Although β-amyrin displaced ∼30% at 1 µM of [3H]-CP55,940 at the CB1 receptor, no IC50 was reached at concentrations <10 µM. A racemic α,β-amyrin 1:1 mixture was assessed at 10 µM and did not exhibit significantly higher [3H]-CP55,940 displacement than the isomers tested separately (data not shown). Data show means ± SEM, n= 12 from four independent experiments.
Figure 3.
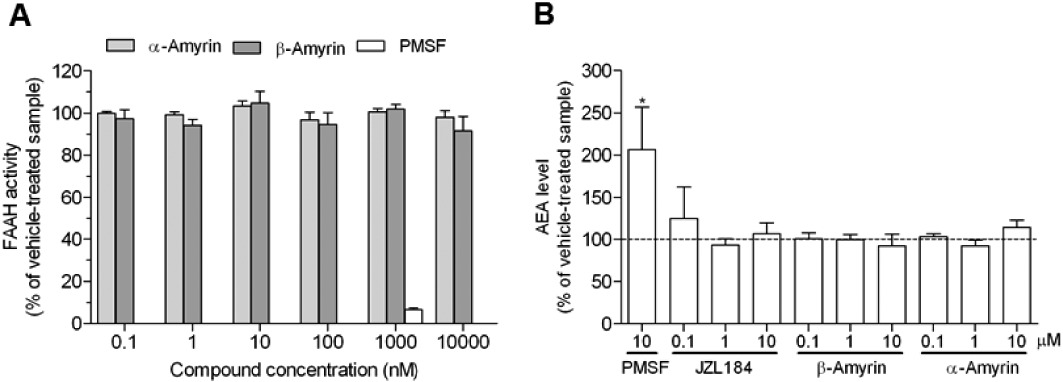
Neither α- nor β-amyrin show inhibition of AEA hydrolysis. (A) FAAH activity was measured by incubating 200 µg of pig brain homogenate with 2 µM of the AEA/[3H]-AEA mixture for 10 min at 37°C after 20 min preincubation with amyrins, PMSF or vehicle. (B) Absolute quantification of the AEA level by GC-MS in 1.8 mg pig brain homogenate after 30 min incubation at 37°C with amyrins, PMSF or vehicle. Data show means ± SEM of nine measurements from three independent experiments. *P < 0.05, significantly different from vehicle-treated samples.
Figure 4.
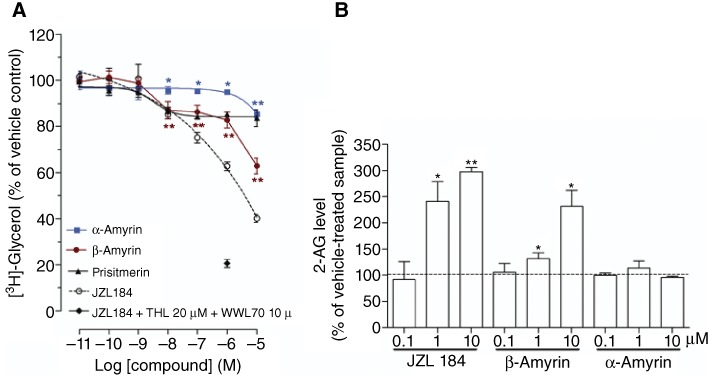
Inhibition of 2-AG hydrolysis by amyrins. (A) Dose-dependent inhibition of [3H]-glycerol formation in pig brain homogenate after 30 min pretreatment with amyrins, JZL184, JZL184 1 µM + 10 µM WWL70 + 20 µM THL or vehicle, followed by 10 min incubation at 37°C with 10 µM of the 2-AG/[3H]-2-AG mix. (B) Absolute quantification of the 2-AG level by GC-MS in 1.8 mg pig brain homogenate after 30 min incubation with amyrins, JZL184 or vehicle at 37°C. Data show means ± SEM, n= 9 from three independent experiments. *P < 0.05, **P < 0.01, significantly different from vehicle-treated samples (=100%).
To further investigate the inhibition of 2-AG hydrolysis by the amyrins in a more physiological setting, we incubated pig brain homogenates with amyrins, JZL184, or vehicle at 37°C for 30 min without adding any 2-AG and then quantified the endocannabinoid levels by GC-MS. As shown in Figure 4B, β-amyrin led to a significant inhibition of endogenous 2-AG hydrolysis, as shown by the increased 2-AG levels (140 and 240% at 1 and 10 µM, respectively). Similarly, JZL184 treatment produced an increase of 2-AG levels of 240 and 300% at 1 and 10 µM, respectively. Unlike the experiments with radio-labelled substrate, in this more complex homogenate, neither β-amyrin nor JZL184 produced a significant inhibition of 2-AG hydrolysis at 100 nM (Figure 4B). This may be due to the higher detection limit for endogenous 2-AG compared with radioactive assays. In keeping with this hypothesis, α-amyrin did not show any significant accumulation of endogenous 2-AG up to 10 µM, while a slight, but significant reduction of [3H]-glycerol formation was observed in the radioactive experiments (Figure 4B).
The results obtained showed significant inhibition of 2-AG hydrolysis by β-amyrin in vitro. Therefore, we further investigated whether this molecule could interact with 2-AG trafficking and degradation in cells. We assessed the intracellular accumulation of [3H]-2AG as well as the amount of [3H]-glycerol formed in U937 cells. As expected for a 2-AG hydrolysis inhibitor, β-amyrin treatment resulted in a significant concentration-dependent increase of intracellular 2-AG compared with vehicle control in the nanomolar and sub-nanomolar concentrations (Figure 5A). Noteworthy, α-amyrin also induced a similar concentration-dependent 2-AG accumulation curve, but with lower efficacy than β-amyrin, in agreement with its weaker inhibition of 2-AG hydrolysis. The MAGL inhibitor JZL184, which was tested in the same conditions, led to a curve identical to that of β-amyrin in the nanomolar concentrations, further supporting MAGL inhibition by the triterpene. Interestingly, unlike JZL184, both α- and β-amyrin at higher concentrations (1 and 10 µM) partially or completely lost the ability to increase intracellular accumulation of 2-AG, but without altering the inhibition of [3H]-glycerol formation (Figure 5A and B). In fact, the two structural isomers inhibited [3H]-glycerol formation in a concentration-dependent manner up to 10 µM without losing efficacy. In agreement with our 2-AG hydrolysis experiments, β-amyrin was more potent than α-amyrin (Figure 5B). As an additional positive control, we tested the triterpene pristimerin, which is a reversible inhibitor of MAGL (King et al., 2009a). The effect of the non-selective inhibitor of hydrolases, methoxy arachidonyl fluorophosphonate (MAFP), was also measured in the [3H]-2-AG uptake assay. 10 µM of MAFP led to the same intracellular [3H]-2-AG accumulation as the maximal effect obtained with JZL184 (approximately 200%) (Figure 5A). In contrast, MAFP treatment led to a stronger inhibition than JZL184 of the [3H]-glycerol production (75 and 55%, respectively) (Figure 5B). The latter difference can be explained by the broader spectrum of hydrolase inhibition by MAFP at 10 µM, which in vitro was shown to fully inhibit MAGL and FAAH and, at least partially, also ABHD-6 and ABHD-12 (Hoover et al., 2008). In the same [3H]-2-AG uptake assay, we also assessed the effect of the triterpene MAGL inhibitor pristimerin. As shown in Figure 5, 10 µM pristimerin produced a 25% inhibition of [3H]-glycerol formation and a 125% accumulation of intracellular [3H]-2-AG, thus being less potent than β-amyrin in this assay.
Figure 5.
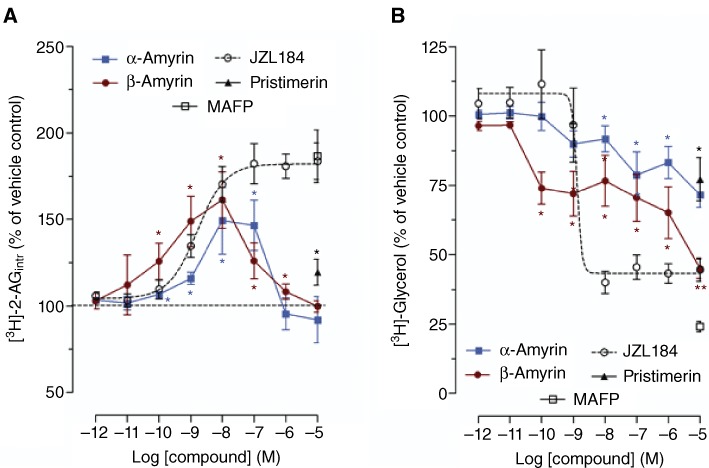
The effect of amyrins on 2-AG trafficking and breakdown in intact U937 cells. (A) [3H]-2-AG intracellular levels and (B) [3H]-glycerol formation in U937 cells pretreated with amyrins, JZL184 or vehicle and then incubated with 1 µM 2-AG/[3H]2-AG mix for 5 min. The effect of pristimerin and MAFP is shown only at a concentration of 10 µM. The radioactive counts in the vehicle controls were approximately 9000 and 300 dpm for [3H]-glycerol formation and intracellular [3H]-2-AG, respectively. Data show means ± SEM, n= 9 from three independent experiments. *P < 0.05, **P < 0.01, significantly different from vehicle-treated samples (=100%).
The unexpected reduction of [3H]-2-AG accumulation by the amyrins at higher concentrations (Figure 5A) could be the result of ‘off-target’ effects such as, for example an interaction with a putative 2-AG membrane transporter. In order to address this hypothesis, we measured the effect of α- and β-amyrin on [3H]-2-AG uptake and degradation in U937 cells in presence of MAGL inhibition by 10 µM JZL184, which does not affect 2-AG uptake (Fowler and Ghafouri, 2008). The data indicate that at low concentrations (0.1–1 nM), β-amyrin still led to a slight but significant increase of intracellular [3H]-2-AG (10–20%) over the 10 µM JZL184-treated cells, while at higher concentrations (0.1–10 µM) a significant decrease of the intracellular [3H]-2-AG (25–40%) as compared with JZL184-treated cells is shown (Figure 6A). Similarly, U937 cells treated with α-amyrin in the presence of 10 µM JZL184 did not accumulate intracellular [3H]-2-AG at low concentrations, whereas at higher concentrations, a reduction of [3H]-2-AG uptake (10–25%) was observed (Figure 6B). As shown in Supporting Information Figure S1, inhibition of [3H]-glycerol formation via [3H]-2-AG hydrolysis was independent of JZL184 treatment. This suggests that the reduction of [3H]-glycerol formation is dependent on inhibition of both 2-AG hydrolysis and cellular uptake. Intriguingly, at 0.1 nM β-amyrin, [3H]-glycerol formation was significantly inhibited, compared with that in cells treated with JZL184 (10 µM). This clearly suggested an additional 2-AG hydrolysis inhibition independent of MAGL. On the basis of this potential non-MAGL-mediated inhibition of 2-AG hydrolysis, we measured the MAGL and non-MAGL components by using purified hMAGL and mouse microglial BV2 cells, which lack MAGL, but still retain 2-AG hydrolysis via ABHDs [mainly ABHD-6 (Marrs et al., 2010)]. As shown in Figure 7A, β-amyrin inhibited purified hMAGL more potently than α-amyrin (see IC50 values in Table 1). Pristimerin and JZL184 produced a total inhibition of MAGL (Figure 7A), with IC50 values (Table 1), in concordance with published values (King et al., 2009a; Long et al., 2009). As shown in Figure 7B, in BV2 cell homogenates, α- and β-amyrin induced a partial but significant inhibition (10–20%) of [3H]-2-AG hydrolysis at concentrations ≥100 nM indicating additional non-MAGL targets. Importantly, JZL184 did not inhibit [3H]-2-AG hydrolysis in this system below 10 µM, which is in agreement with its selectivity towards MAGL (Long et al., 2009). The triterpene pristimerin only produced a very weak partial inhibition (<10%) at 10 µM, reflecting its MAGL selectivity (Figure 7B). The assay was validated using the ABHD-6 inhibitor WWL70, which showed an almost complete inhibition of [3H]-2-AG hydrolysis (75–80%) at 10 µM (Figure 7B).
Figure 6.
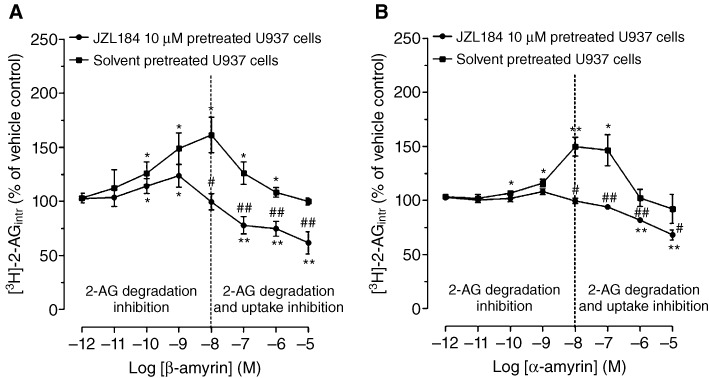
The effect of amyrins on 2-AG uptake in intact U937 cells pretreated with 10 µM JZL184. Intracellular [3H]-2-AG levels in U937 cells pretreated with (A) β-amyrin plus JZL184 or β-amyrin plus vehicle, (B) α-amyrin plus JZL184 or α-amyrin plus vehicle incubated with 1 µM 2-AG/[3H]-2-AG for 5 min. Data show mean values ± SEM, n= 12 from four independent experiments. *P < 0.05, **P < 0.01, significantly different from vehicle control (=100%). #P < 0.05, ##P < 0.01, JZL184 plus amyrin-treated cells significantly different from amyrin-treated cells (=100%).
Figure 7.
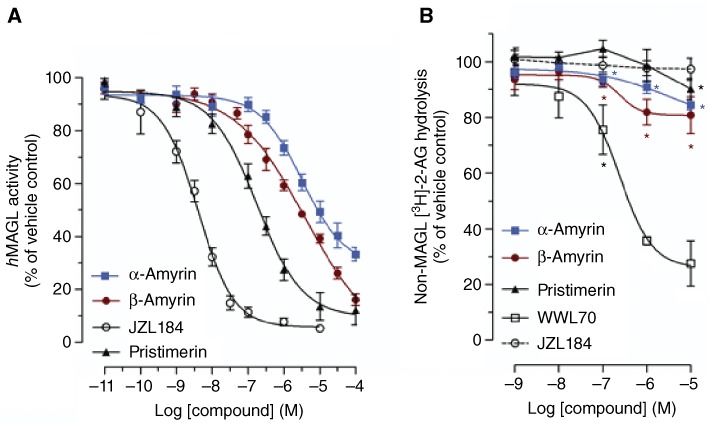
MAGL and non-MAGL components in the inhibition of 2-AG hydrolysis by α- and β-amyrin. (A) Concentration-dependent inhibition of purified hMAGL activity by α-amyrin, β-amyrin, pristimerin and JZL184. (B) Concentration-dependent inhibition of [3H]-2-AG hydrolysis in BV2 cell homogenate in presence of 1 µM URB597 and different concentrations of α-amyrin, β-amyrin, pristimerin, WWL70 and JZL184. Data show mean values ± SEM, n= 12 from four independent experiments. *P < 0.05, **P < 0.01 significantly different from vehicle control (=100%).
Table 1.
IC50 values of amyrins, pristimerin and JZL184 calculated from the hMAGL inhibition curves
| Compound | IC50 (nM) values ± SEM |
|---|---|
| α-Amyrin | 9300 ± 1200 |
| β-Amyrin | 2800 ± 500 |
| Pristimerin | 204 ± 16.2 |
| JZL184 | 3.9 ± 1.0 |
Our data showed that β-amyrin exerts a significant mixed action on enzymes hydrolysing 2-AG, targeting MAGL and ABHDs. Becauase MAGL is the main enzyme involved in 2-AG hydrolysis (Blankman et al., 2007) we next assessed the mechanism of action of β-amyrin towards MAGL. As shown in Figure 8A, 3 µM of β-amyrin produced a rapid (1 min) and continuous inhibition of MAGL activity. In Figure 8B, we show the interaction of β-amyrin with purified hMAGL using a rapid dilution assay (King et al., 2009b). Under these conditions, the incubation of MAGL with the covalent inhibitor MAFP resulted in the formation of an enzyme-inhibitor complex that was resistant to dilution of the assay mixture. By contrast, rapid dilution of the MAGL-β-amyrin mixture resulted in an almost full recovery of the enzymatic activity, suggesting a reversible inhibition. In Figure 8C, further kinetic studies demonstrated that β-amyrin decreased the maximal catalytic velocity (Vmax in pmol·min·µg−1) of MAGL from 43.9 ± 8.6 (vehicle) to 24.3 ± 4.6 (3 µM β-amyrin) without influencing its Michaelis–Menten constant (Km: 1.31 ± 0.98 µM for the vehicle and 1.19 ± 0.67 µM for 3 µM β-amyrin). Together, these data show that β-amyrin inhibited MAGL activity in a rapid, reversible and non-competitive manner, similar to that previously shown for the structurally related, but more potent triterpene pristimerin (King et al., 2009a).
Figure 8.
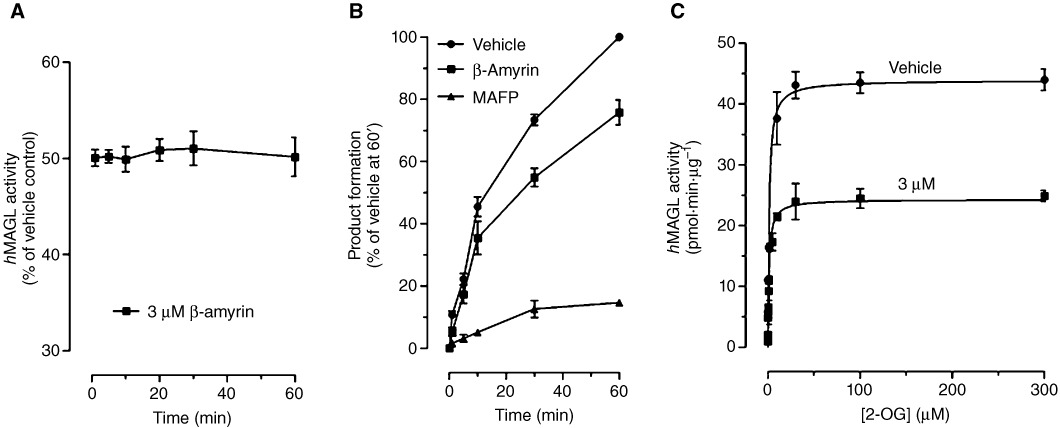
Mechanism of action of β-amyrin-mediated MAGL inhibition. (A) Time course of MAGL inhibition by 3 µM β-amyrin. (B) Rapid dilution assays of MAGL in the presence of vehicle, β-amyrin or MAFP. Results are expressed as percentage of product generated after 60 min incubation with vehicle. (C) Michaelis–Menten analysis of the MAGL reaction (measured as pmol·min−1·µg protein−1) in the presence of vehicle or 3 µM of β-amyrin. Data show mean values ± SEM, n= 9 from three independent experiments.
Discussion and conclusions
Although we cannot fully explain the discrepancy between the ultrapotent CB1 receptor interaction of α/β-amyrin reported (Da Silva et al., 2011) and the lack of CB1 receptor interaction in this study, it is likely to be related to methodology. Da Silva et al. used membranes prepared from rat brain (containing CB1 receptors) or rat spleen (containing CB2 receptors). The view that these tissues express either CB1 or CB2 receptors, respectively, is too simple as brain tissues are known to also contain CB2 receptors (Onaivi, 2011) and spleen contains CB1 receptors (Noe et al., 2001). Moreover, in their study, the radioligands [3H]-SR141716A (rimonabant) and [3H]-CP55,940 were used, which both potentially interact with other targets in these tissues; for example SR141716A may directly interact with GABAA and GPR55 receptors (Anavi-Goffer et al., 2011; Baur et al., 2011). Notably, the binding curves published for α/β-amyrin were not sigmoidal, but were rather linear and only the specific binding was indicated whereas the actual binding (including % non-specific binding) was not shown (Da Silva et al., 2011). In addition, we could also speculate that the CB1 receptor binding of the inverse agonist [3H]-SR141617A might be affected by amyrins via some unidentified interactions at different receptor sites. Thus, potential methodological issues cannot be excluded. Although we did not use rat, but human cannabinoid receptors, it is unlikely that the discrepancy was only due to species differences. We used an established and validated CB receptor binding assay (Gertsch et al., 2008) and our positive control WIN55,212-2 showed a Ki value in the concentration range reported previously (McPartland et al., 2007). Using our experimental system, we were unable to detect significant CB receptor binding in independent experiments.
In a profiling of α-amyrin and β-amyrin on other targets within the endocannabinoid system (endocannabinoid transport across membranes and hydrolytic degradation), we found significant inhibitory effects on 2-AG hydrolysis by amyrins. The geminal methyls (β-amyrin) in the A ring were more effective than the vicinal methyl groups (α-amyrin). The finding that β-amyrin inhibits 2-AG hydrolysis in different biological preparations may also explain the CB1 receptor binding of β-amyrin reported by da Silva et al. The brain homogenate used for CB1 receptor binding may lead to a significant rise of background 2-AG levels (Sugiura et al., 2001), which is the most abundant endocannabinoid in the brain, and could thus interfere with the relative [3H]-SR141716A binding. In fact, rat brain membranes retain some 2-AG hydrolytic enzymes such as ABHDs, which are integral cell membrane proteins (Blankman et al., 2007; Ahn et al., 2008), and MAGL, which is found associated with the inner cell membrane leaflet (Labar et al., 2010). Indeed, da Silva et al. reports that rats were killed by decapitation before preparing the brain membranes (Da Silva et al., 2011). This procedure has been shown to produce a strong accumulation of 2-AG in the rat brain to up to 1.54 nmol g−1 of tissue 30 s after decapitation (Sugiura et al., 2001), thus leading to even a higher 2-AG content in the membrane preparations used for the CB1 receptor binding assay. That the inhibition of 2-AG breakdown in rat brain homogenates was able to interfere with [3H]-SR141716A binding cannot be excluded at this point. In the paper by da Silva et al., pre-incubation of the rat brain membranes before adding the amyrins could have removed the residual 2-AG through the action of the retained hydrolytic activity.
Pristimerin, another structurally related pentacyclic triterpenoid is a potent reversible inhibitor of MAGL activity (King et al., 2009a), which is in agreement with our finding that amyrin inhibits 2-AG breakdown. In our hands, pristimerin was less potent in inhibiting 2-AG breakdown than β-amyrin in brain tissue, despite the fact that in purified enzyme assays, pristimerin was an approximately 10-fold more potent inhibitor of hMAGL (Table 1 and Figure 7A). This could be explained by the fact that the amyrins, but not pristimerin, also inhibit ABHDs, as shown in the experiments using BV2 cell homogenates where MAGL is not expressed (Marrs et al., 2010). Thus, relatively weak and partial simultaneous inhibition of MAGL and ABHDs appears to be synergistic, leading to significant pharmacological inhibition of 2-AG metabolism. That triterpenoids may be a yet unexploited source for MAGL inhibitors is also shown by euphol, which is a different plant tetracyclic terpene that has been reported to inhibit MAGL, albeit less potently than pristimerin (King et al., 2009a). Interestingly, Dutra et al. from the same laboratory in which α/β-amyrin was found to be an ultrapotent CB1 receptor ligand recently reported that the triterpene euphol is an ultrapotent CB2 receptor-selective agonist with a subnanomolar Ki value (Dutra et al., 2011). In this study again rat spleen was used and no functional data were reported. As observed with α/β-amyrin, the antinociceptive effects (neuropathic pain model) of euphol could be inhibited by both CB1 and CB2 receptor antagonists, pointing to an indirect cannabimimetic effect via the previously described inhibition of MAGL by euphol (King et al., 2009a). In our ongoing CB receptor screening activities using natural product libraries (Gertsch et al., 2006; 2010; Leonti et al., 2010), we have so far not found any triterpenoid with significant CB receptor affinity towards the classical CP55,940 binding site (i.e. targeting the endocannabinoid binding pocket). However, a sulphur containing steroid with a triterpenoid-like steroid scaffold from a marine sponge was recently reported to selectively bind to the CB2 receptor with a Ki value at low micromolar concentrations (Chianese et al., 2011). Nevertheless, CB receptor ligands from steroids or triterpenes are generally lacking. Overall, plant triterpenoids, many of which are found in medicinal plants and known to exert significant anti-inflammatory and antinociceptive effects (Ríos, 2010), appear to offer a very broad and largely unexplored scaffold for potent inhibitors of 2-AG metabolism by non-selectively targeting MAGL and ABHDs. Future studies will address this hypothesis.
A recent study reported the pharmacokinetic profile of amyrin in rats (Ching et al., 2011). In this study, the absolute oral bioavailability of β-amyrin was in the range of 0.8–3.8%, depending on purity and admixtures. Although only partly bioavailable, β-amyrin showed a very long terminal elimination half-life (t1/2λz= 610 ± 179 min) and extremely slow clearance (Cl = 2.04 ± 0.24 mL·min·kg−1) (Ching et al., 2011). The data by da Silva et al. suggest that ∼0.3 mg kg−1 of bioavailable α/β-amyrin from a total of 30 mg·kg−1 orally administered dose is enough to exert significant in vivo effects when administered chronically, indicating either tissue accumulation (depot effect) or high potency. Of note, the specific MAGL inhibitor JZL184 is only poorly orally bioavailable (unpublished data) and triterpenoid MAGL/ABHDs inhibitors like β-amyrin may thus provide novel tool compounds. Because β-amyrin is a widespread constituent of many anti-inflammatory medicinal plants and vegetable foods, MAGL inhibition could contribute to the network pharmacology exerted by anti-inflammatory botanical drugs (Gertsch, 2011). Given that Nature produces a wide variety of distinct tetracyclic and pentacyclic terpenes, their potential effects on MAGL need to be addressed systematically in future studies.
Acknowledgments
This work was supported by the Swiss National Science Foundation NCCR TransCure and the by the Novartis Research Foundation grant 10B48. The study was within the framework of the COST action Chemical Biology with Natural Products. We would like to thank Giovanni Appendino, Università degli Studi del Piemonte Orientale, for kindly providing β-amyrin. Some AEA transport assays that are not shown were performed by Simon Nicolussi in our laboratory.
Glossary
- Δ9-THC
Δ9-tetrahydrocannabinol
- CP55940
-(–)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- ECS
endocannabinoid system
- FAAH
fatty acid amide hydrolase
- JZL184
4-nitrophenyl-4-[bis(1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate
- MAFP
methyl arachidonylfluorophosphonate
- MAGL
monoacylglycerol lipase
- SR141716A
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
- THL
tetrahydrolipstatin
- Tmax
time to maximal concentration in plasma (pharmacokinetic parameter)
- WIN55
212-2, (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo-[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphtaleneylmethanone
Conflict of interest
The authors state no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 The effect of amyrins on glycerol formation in U937 cells pretreated with 10 μM JZL184. [3H]-glycerol levels in U937 cells pretreated with (A) β-amyrin plus JZL184 or β-amyrin + vehicle (B) α-amyrin + JZL184 or α-amyrin + vehicle and then incubated with 1 μM 2-AG/[3H]2-AG mix for 5 min. Data show means ± SEM of 12 measurements from 4 independent experiments. *P < 0.05, **P < 0.01 amyrins treated cells versus vehicle-treated cells; #P < 0.05, amyrins plus JZL184-treated cells versus amyrins plus vehicle-treated cells.
References
- Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akihisa T, Yasukawa K, Oinuma H, Kasahara Y, Yamanouchi S, Takido M, et al. Triterpene alcohols from the flowers of compositae and their anti-inflammatory effects. Phytochemistry. 1996;43:1255–1260. doi: 10.1016/s0031-9422(96)00343-3. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anavi-Goffer S, Baillie G, Irving AJ, Gertsch J, Greig IR, Pertwee RG, et al. Modulation of L-α-lysophosphatidylinositol/GPR55 MAP kinase signalling by cannabinoids. J Biol Chem. 2011;287:91–104. doi: 10.1074/jbc.M111.296020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur R, Gertsch J, Sigel E. The cannabinoid receptor CB1 antagonists rimonabant (SR141716) and AM251 directly potentiate GABA(A) receptors. Br J Pharmacol. 2011;165:2479–2484. doi: 10.1111/j.1476-5381.2011.01405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Di Marzo V. Cannabinoid receptors and endocannabinoids: role in neuroinflammatory and neurodegenerative disorders. CNS Neurol Disord Drug Targets. 2010;5:564–573. doi: 10.2174/187152710793361568. [DOI] [PubMed] [Google Scholar]
- Björklund E, Noren E, Nilsson J, Fowler CJ. Inhibition of monoacylglycerol lipase by troglitazone, N-arachidonoyldopamine and the irreversible inhibitor JZL184: comparison of two different assays. Br J Pharmacol. 2010;161:1512–1526. doi: 10.1111/j.1476-5381.2010.00974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel C. Cannabinoid receptor binding to membrane homogenates and cannabinoid-stimulated [35S]GTP-S binding to membrane homogenates or intact cultured cells. Marijuana and cannabinoid research. Methods Mol Med. 2006;123:149–162. doi: 10.1385/1-59259-999-0:149. [DOI] [PubMed] [Google Scholar]
- Chianese G, Fattorusso E, Taglialatela-Scafati O, Bavestrello G, Calcinai B, Dien HA, et al. Desulfohaplosamate, a new phosphate-containing steroid from Dasychalina sp., is a selective cannabinoid CB2 receptor ligand. Steroids. 2011;76:998–1002. doi: 10.1016/j.steroids.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Chicca A, Raduner S, Pellati F, Strompen T, Altmann KH, Schoop R, et al. Synergistic immunomopharmacological effects of N-alkylamides in Echinacea purpurea herbal extracts. Int Immunopharmacol. 2009;9:850–858. doi: 10.1016/j.intimp.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Ching J, Lin HS, Tan CH, Koh HL. Quantification of α- and β-amyrin in rat plasma by gas chromatography-mass spectrometry: application to preclinical pharmacokinetic study. J Mass Spectrom. 2011;46:457–464. doi: 10.1002/jms.1912. [DOI] [PubMed] [Google Scholar]
- Da Silva KA, Paszcuk AF, Passos GF, Silva ES, Bento AF, Meotti FC, et al. Activation of cannabinoid receptors by the pentacyclic triterpene α,β-amyrin inhibits inflammatory and neuropathic persistent pain in mice. Pain. 2011;152:1872–1287. doi: 10.1016/j.pain.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Dutra RR, Silva KB, Bento AF, Paszcuk AF, Marcon R, Meiotti FC, et al. Euphol, a novel cannabinoid agonist, prevents inflammatory and neuropathic persistent pain in rodents. Planta Med. 2011;77:1254. [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Fowler CJ, Ghafouri N. Does the hydrolysis of 2-arachidonoylglycerol regulate its cellular uptake? Pharmacol Res. 2008;58:72–76. doi: 10.1016/j.phrs.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Fride E, Perchuk A, Hall FS, Uhl GR, Onaivi ES. Behavioural methods in cannabinoid research. Methods Mol Med. 2006;123:269–290. doi: 10.1385/1-59259-999-0:269. [DOI] [PubMed] [Google Scholar]
- Gertsch J. Botanical drugs, synergy, and network pharmacology: forth and back to intelligent mixtures. Planta Med. 2011;77:1086–1098. doi: 10.1055/s-0030-1270904. [DOI] [PubMed] [Google Scholar]
- Gertsch J, Raduner S, Altmann KH. New natural noncannabinoid ligands for cannabinoid type-2 (CB2) receptors. J Recept Signal Transduct Res. 2006;26:709–730. doi: 10.1080/10799890600942674. [DOI] [PubMed] [Google Scholar]
- Gertsch J, Leonti M, Raduner S, Racz I, Chen JZ, Xie XQ, et al. A. Beta-caryophyllene is a dietary cannabinoid. Proc Natl Acad Sci USA. 2008;105:9099–90104. doi: 10.1073/pnas.0803601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsch J, Pertwee RG, Di Marzo V. Phytocannabinoids beyond the Cannabis plant – do they exist? Br J Pharmacol. 2010;160:523–529. doi: 10.1111/j.1476-5381.2010.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith DA, Hadcock JR, Black SC, Iredale PA, Carpino PA, DaSilva-Jardine P, et al. Discovery of 1-[9-(4-chlorophenyl)-8-(2-chlorophenyl)-9H-purin-6-yl]-4-ethylaminopiperidine-4-carboxylic acid amide hydrochloride (CP-945,598), a novel, potent, and selective cannabinoid type 1 receptor antagonist. J Med Chem. 2009;52:234–237. doi: 10.1021/jm8012932. [DOI] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadcock JR, Carpino PA, Iredale PA, Dow RL, Gautreau D, Thiede L, et al. Quantitative in vitro and in vivo pharmacological profile of CE-178253, a potent and selective cannabinoid type 1 (CB1) receptor antagonist. BMC Pharmacol. 2010;10:9. doi: 10.1186/1471-2210-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holanda Pinto SA, Pinto LM, Guedes MA, Cunha GM, Chaves MH, Santos FA, et al. Antinoceptive effect of triterpenoid alpha,beta-amyrin in rats on orofacial pain induced by formalin and capsaicin. Phytomedicine. 2008;15:630–634. doi: 10.1016/j.phymed.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Hoover HS, Blankman JL, Niessen S, Cravatt BF. Selectivity of inhibitors of endocannabinoid biosynthesis evaluated by activity-based protein profiling. Bioorg Med Chem Lett. 2008;18:5838–5841. doi: 10.1016/j.bmcl.2008.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman JW, Zengin G, Wu M, Lu J, Hynd G, Bushell K, et al. Structure-activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid CB(1) and CB(2) receptors: steric and electronic effects of naphthoyl substituents. New highly selective CB(2) receptor agonists. Bioorg Med Chem. 2005;13:89–112. doi: 10.1016/j.bmc.2004.09.050. [DOI] [PubMed] [Google Scholar]
- King AR, Duranti A, Tontini A, Rivara S, Rosengarth A, Clapper JR, et al. URB602 inhibits monoacylglycerol lipase and selectively blocks 2-arachidonoylglycerol degradation in intact brain slices. Chem Biol. 2007;14:1357–1365. doi: 10.1016/j.chembiol.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AR, Dotsey EY, Lodola A, Jung KM, Ghomian A, Qiu Y, et al. Discovery of potent and reversible monoacylglycerol lipase inhibitors. Chem Biol. 2009a;16:1045–1052. doi: 10.1016/j.chembiol.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AR, Lodola A, Carmi C, Fu J, Mor M, Piomelli D. A critical cysteine residue in monoacylglycerol lipase is targeted by a new class of isothiazolinone-based enzyme inhibitors. Br J Pharmacol. 2009b;157:974–983. doi: 10.1111/j.1476-5381.2009.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labar G, Bauvois C, Borel F, Ferrer JL, Wouters J, Lambert DM. Crystal structure of the human monoacylglycerol lipase, a key factor in endocannabinoid signaling. Chembiochem. 2010;11:218–227. doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- Leonti M, Casu L, Raduner S, Cottiglia F, Floris C, Altmann KH, et al. Falcarinol is a covalent cannabinoid CB1 receptor antagonist and induces pro-allergic effects in skin. Biochem Pharmacol. 2010;79:1815–1826. doi: 10.1016/j.bcp.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Blankman JL, Cravatt BF. Endocannabinoid overload. Mol Pharmacol. 2010;78:993–995. doi: 10.1124/mol.110.069427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPartland JM, Glass M, Pertwee RG. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007;152:583–593. doi: 10.1038/sj.bjp.0707399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazzi J, Kleyer J, Paredes JM, Gertsch J. Endocannabinoid content in fetal bovine sera – unexpected effects on mononuclear cells and osteoclastogenesis. J Immunol Methods. 2011;373:219–228. doi: 10.1016/j.jim.2011.08.021. [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo CM, Carvalho KM, Neves JC, Morais TC, Rao VS, Santos FA, et al. Alpha,beta-amyrin, a natural triterpenoid ameliorates L-arginine-induced acute pancreatitis in rats. World J Gastroenterol. 2010;16:4272–4280. doi: 10.3748/wjg.v16.i34.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo CM, Morais TC, Tomé AR, Brito GA, Chaves MH, Rao VS, et al. Anti-inflammatory effect of α,β-amyrin, a triterpene from Protium heptaphyllum, on cerulein-induced acute pancreatitis in mice. Inflamm Res. 2011;60:673–681. doi: 10.1007/s00011-011-0321-x. [DOI] [PubMed] [Google Scholar]
- Noe SN, Newton C, Widen R, Friedman H, Klein TW. Modulation of CB1 mRNA upon activation of murine splenocytes. Adv Exp Med Bio. 2001;l493:215–221. doi: 10.1007/0-306-47611-8_25. [DOI] [PubMed] [Google Scholar]
- Obata T, Sakurai Y, Kase Y, Tanifuji Y, Horiguchi T. Simultaneous determination of endocannabinoids (arachidonylethanolamide and 2-arachidonylglycerol) and isoprostane (8-epiprostaglandin F2alpha) by gas chromatography-mass spectrometry-selected ion monitoring for medical samples. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;792:131–140. doi: 10.1016/s1570-0232(03)00311-8. [DOI] [PubMed] [Google Scholar]
- Onaivi ES. Commentary: functional Neuronal CB2 Cannabinoid Receptors in the CNS. Curr Neuropharmacol. 2011;9:205–208. doi: 10.2174/157015911795017416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papahatjis DP, Kourouli T, Abadji V, Goutopoulos A, Makriyannis A. Pharmacophoric requirements for cannabinoid side chains: multiple bond and C1′-substituted delta 8-tetrahydrocannabinols. J Med Chem. 1998;41:1195–1200. doi: 10.1021/jm970277i. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Emerging strategies for exploiting cannabinoid receptor agonists as medicines. Br J Pharmacol. 2009;156:397–411. doi: 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raduner S, Majewska A, Chen JZ, Xie XQ, Hamon J, Faller B, et al. Alkylamides from Echinacea are a new class of cannabinomimetics. Cannabinoid type 2 receptor-dependent and -independent immunomodulatory effects. J Biol Chem. 2006;281:14192–14206. doi: 10.1074/jbc.M601074200. [DOI] [PubMed] [Google Scholar]
- Rees HH, Britton G, Goodwin TW. The biosynthesis of beta-amyrin. Mechanism of squalene cyclization. Biochem J. 1968;106:659–665. doi: 10.1042/bj1060659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos JL. Effects of triterpenes on the immune system. J Ethnopharmacol. 2010;128:1–14. doi: 10.1016/j.jep.2009.12.045. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Yoshinaga N, Waku K. Rapid generation of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand, in rat brain after decapitation. Neurosci Lett. 2001;297:175–118. doi: 10.1016/s0304-3940(00)01691-8. [DOI] [PubMed] [Google Scholar]
- Thakur GA, Palmer SL, Harrington PE, Stergiades IA, Tius MA, Makriyannis A. Enantiomeric resolution of a novel chiral cannabinoid receptor ligand. J Biochem Biophys Methods. 2002;54:415–422. doi: 10.1016/s0165-022x(02)00144-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.