

Abstract
Background and Purpose
ω3-polyunsaturated fatty acids (ω3-PUFAs) are known to exert anti-inflammatory effects in various disease models although their direct targets are only poorly characterized.
Experimental approach
Here we report on two new cPLA2 inhibitors, the ω3-derivatives AVX001 and AVX002, and their effects on inflammatory PGE2 production in cultures of renal mesangial cells.
Key Results
AVX001 and AVX002 dose-dependently inhibited the group IVA cytosolic phospholipase A2 (cPLA2) in an in vitro activity assay with similar IC50 values for AVX001 and AVX002, whereas the known cPLA2 inhibitor AACOCF3 was less potent and docosahexaenoic acid (DHA) was inactive. In renal mesangial cells, AVX001 and AVX002 suppressed IL-1β-induced PGE2 synthesis. Mechanistically, this effect occurred by a down-regulation of IL-1β-induced group IIA-sPLA2 protein expression, mRNA expression and promoter activity. A similar but less potent effect was seen with AACOCF3 and no effect was seen with DHA. As gene expression of sPLA2 is known to be regulated by the transcription factor NF-κB, we further investigated NF-κB activation. Both compounds prevented NF-κB activation by blocking degradation of the inhibitor of κB.
Conclusions and implications
These data show for the first time that the novel cPLA2 inhibitors AVX001 and AVX002 exert an anti-inflammatory effect in cultures of renal mesangial cells and reduce the pro-inflammatory mediator PGE2 through an inhibitory effect on NF-κB activation. Therefore, these compounds may represent promising novel drugs for the treatment of inflammatory disorders.
Keywords: AVX001, AVX002, cPLA2, prostaglandins, kidney, mesangial cell, inflammation
Introduction
Mesangial cells are specialized smooth muscle-like cells located in the renal glomerulus and are not only involved in the regulation of the glomerular filtration rate and in the preservation of the structural integrity of the glomerulus, but also play a central role in most pathological processes of the renal glomerulus (Kashgarian and Sterzel, 1992; Pfeilschifter, 1994; Gómez-Guerrero et al., 2005). Upon activation by a variety of pro-inflammatory cytokines, mesangial cells respond with three prominent reactions which are all hallmarks of many forms of glomerulonephritis: (i) increased proliferation; (ii) increased mediator production, including cytokines, chemokines, NO, superoxide radicals and PGs; and (iii) increased extracellular matrix production (Kashgarian and Sterzel, 1992; Pfeilschifter, 1994). The detailed mechanisms underlying these cellular responses are still not completely understood.
One important event in the inflammatory reaction and the rate-limiting step in the generation of PGs is the activation of a PLA2, which hydrolyzes the sn-2 ester bond of substrate phospholipids and thereby generates arachidonic acid and lysophospholipids (Schaloske and Dennis, 2006). Arachidonic acid is then further converted by either COXs or lipoxygenases and downstream enzymes to the eicosanoids including PGs, TXs and LTs.
So far, 15 groups and many subgroups of PLA2s have been identified which include five distinct types of enzymes, that is, secreted PLA2s (sPLA2), the cytosolic PLA2s (cPLA2), the Ca2+-independent PLA2s (iPLA2), the PAF acetylhydrolases (AHs) and the lysosomal PLA2s (Schaloske and Dennis, 2006).
As cPLA2 preferentially hydrolyzes arachidonic acid-containing phospholipids at the sn-2 position, this enzyme is thought to be one key enzyme in inflammatory eicosanoid formation.
In renal mesangial cells, four PLA2 subtypes are expressed either constitutively or inducibly. These include the cPLA2, the iPLA2, and the group IIA and V sPLA2 (Schalkwijk et al., 1992; Gronich et al., 1994; Akiba et al., 1998; van der Helm et al., 2000). In previous studies on mesangial cells, it was shown that the drastic increase of cytokine-triggered PGE2 formation involved both IIA-sPLA2 and cPLA2 activation (Pfeilschifter et al., 1993). By using the specific sPLA2 inhibitor CGP43187 or by using a neutralizing antibody against IIA-sPLA2, about 80% of the cytokine-triggered PGE2 formation was depleted, suggesting that the remaining small amount of approx. 20% of PGE2 derives from cytokine-stimulated cPLA2 activity (Pfeilschifter et al., 1993). Indeed, the cPLA2 is not only acutely activated by IL-1 (Gronich et al., 1994), but also up-regulated upon prolonged IL-1 treatment by a transcriptional mechanism (Lin et al., 1992; Schalkwijk et al., 1993). Furthermore, a cross-communication exists between the different PLA2s. Thus, in mesangial cells, we previously showed that the IIA-sPLA2, acting from the outside of cells, is able to activate the cPLA2 intracellularly via a PKC and MAPK-dependent mechanism (Huwiler et al., 1997). In mouse P388D1 macrophages, it was shown that cPLA2 also contributes to V-sPLA2 activation (Balsinde et al., 1998). All these data suggest that the different PLA2s regulate each other and are critically participating in pro-inflammatory PGE2 formation.
In this study, we have identified and characterized two novel direct cPLA2 inhibitors, the ω3-polyunsaturated fatty acid (PUFA) derivatives AVX001 and AVX002. We showed that in cultures of renal mesangial cells, AVX001 and AVX002 down-regulated cytokine-stimulated PGE2 formation through a mechanism that involved the blocking of cytokine-triggered and cPLA2-dependent NF-κB activation and subsequent gene transcription of sPLA2. These data suggest that AVX001 and AVX002 could serve as novel anti-inflammatory drugs.
Methods
AVX001 and AVX002
Both compounds were synthesized and characterized according to Holmeide and Skattebol (2000) and kindly provided by Dr Inger Reidun Aukrust and Dr Marcel Sandberg (Synthetica AS, Norway). The chemical structures are indicated in Figure 1. Both compounds were analysed by HPLC for purity (Figure 2). For this, all peaks in the chromatogram were taken for area integration using an integrated HPLC software. The purities were determined as 97% for AVX001 and 92.7% for AVX002. Both compounds were stored at −80°C at a 20 mM stock solution in DMSO under argon gas to minimize oxidation.
Figure 1.
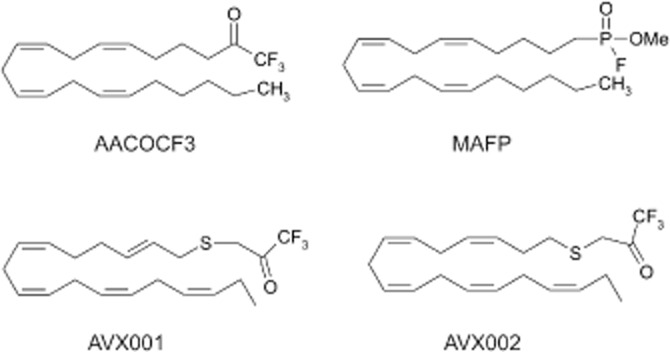
Chemical structure of various cPLA2 inhibitors.
Figure 2.

HPLC profiles of AVX001 and AVX002. The peak at 4.924 min (left panel) corresponds to AVX001. The peak at 4.599 min (right panel) corresponds to AVX002.
Cell culture
Rat renal mesangial cells were isolated, characterized and cultured as previously described (Pfeilschifter et al., 1984). For the experiments performed in this study, cells between passages 8–30 were used.
In vitro cPLA2 activity assay
Recombinant human cPLA2 enzyme was used for an in vitro activity assay as described by Wijkander and Sundler (#b1001), with some modifications according to Lucas and Dennis (2005). Enzyme with inhibitor (in DMSO, final concentration 1%) or solvent alone was pre-incubated in assay buffer (1 mM EDTA, 80 mM KCl and 10 mM HEPES (pH 7.4) containing 1.56 mM CaCl2, and 2.36 mM dithiothreitol for 80 s at 37°C and 10 min at 25°C. Lipid vesicles were prepared by drying 4.2 nmol of L-α-1-palmitoyl-2-arachidonyl-[arachidonyl-1-14C]-phosphatidylcholine under a stream of nitrogen. Dried lipids were resuspended in 2 mL assay buffer and sonicated twice for 7 min (setting: output 3.5 and 50% duty cycles) in a Branson Sonifier 250 (Branson Ultrasonic Corporation, Danbury, CT, USA). Sonicated lipid vesicles were added to the reaction to a final concentration of 0.2 μM. The reaction mixture was incubated for 1 h at 37°C and stopped by addition of 1.7 mL chloroform/methanol/37% KCl/ 0.45 M BHT/ 0.33 M AA (2:1:0.01:0.015:0.005, by vol). After phase separation, the lower phase was transferred to a glass tube, dried under nitrogen, and resuspended in chloroform/methanol (9:1, v v−1), and applied to a silicagel TLC. Free [1-14C]-arachidonic acid and L-α-1-palmitoyl-2-arachidonyl-[arachidonyl-1-14C]-phosphatidylcholine (Perkin Elmer, Waltham, MA, USA) were separated and analysed as described by Anthonsen et al. (2001).
PGE2 determination
Confluent mesangial cells in 24-well plates were pretreated for 90 min in the presence or absence of the inhibitors before stimulation for 24 h in a volume of 0.5 mL with IL-1β (1 nM) to induce PGE2 formation. Thereafter, equal volumes of supernatants were subjected to a PGE2-ELISA according to the manufacturer's instructions. Data were calculated as pg of PGE2 per 1.3 × 105 cells which was the cell number per well.
Cell stimulation and Western blot analysis
Confluent mesangial cells were stimulated as indicated in the figure legends in DMEM supplemented with 0.1 mg·mL−1 of fatty acid-free BSA and 10 mM HEPES. After stimulation, the supernatant was taken for detection of secreted IIA-sPLA2. The cell monolayers were homogenized in lysis buffer and processed exactly as previously described (Xin et al., 2004). Cell lysates were taken for protein determination. Lysates containing 60 μg of protein, were separated by SDS-PAGE, transferred to nitrocellulose membranes and subjected for Western blot analysis using antibodies as indicated in the figure legends. Bands were detected by enhanced chemiluminescence method as recommended by the manufacturer.
Detection of secreted IIA-sPLA2
Equal volumes of supernatants derived from the same number of cells were taken for protein precipitation using 7% (w v−1) of trichloroacetic acid. Precipitated proteins were redissolved in SDS-Laemmli buffer without dithiothreitol and subjected to SDS-PAGE (15% acrylamide gel), transferred to nitrocellulose membranes and immunostained by using a monoclonal antibody against rat IIA-sPLA2 at a dilution of 1:60 in 0.01% milk powder containing PBS as previously described (Petry et al., 2004). GAPDH was stained and densitometrically evaluated in the corresponding cell lysates and used for normalization by calculating the ratio between secreted sPLA2 and cytosolic GAPDH for each sample.
Quantitative PCR analysis
Total RNA (2 μg) was used for reverse transcriptase (RT)-PCR (first-strand cDNA synthesis kit, MBI Fermentas, St.Leon-Rot, Germany) using random hexamer primers for amplification. mRNA levels were determined by quantitative real-time PCR. The following primers were used: rat sPLA2-IIA: forward: GCC AAA TCT CCT GCT CTA CAA ACC, reverse: ACT GGG CGT CTT CCC TTT GC; 18S RNA (forward: CGA TTC CGT GGG TGG TGG TG, reverse: CAT GCC AGA GTC TCG TTC GTT ATC); iQ™ SYBR® Green Supermix was from Bio-Rad (Hercules, CA, USA). The cycling conditions were as follows: initial activation step (95°C for 3 min), followed by 40 cycles of denaturation (95°C for 15 s) and annealing (58°C for 1 min). PCR products were detected by monitoring the increase in fluorescence with iCycler, iQ™5 Multicolor Real-Time PCR Detection System, Bio-Rad. The Bio-Rad iQ5 Standard Edition Optical System Software Version 2.0 was used to analyse real-time and end point fluorescence.
Cell transfection and luciferase reporter gene assay
A 2.67 kb fragment of the rat sPLA2-IIA promoter was cloned according to a previous report (Scholz-Pedretti et al., 2002) and fused to a luciferase reporter gene. Cells were cultured in 12-well plates and transfected with 0.3 μg of the promoter-containing plasmid plus 0.03 μg of a plasmid containing the renilla luciferase gene by using Effectene Reagent (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Thereafter, the transfection medium was removed and cells were stimulated as indicated. Values for the relative gene promoter activities were calculated from the ratio of firefly/renilla luciferase activities.
Statistical analysis
Statistical analysis was performed using one-way ANOVA followed by a Bonferroni's post hoc test for multiple comparisons (GraphPad InStat version 3.00 for Windows NT, GraphPad Software, San Diego, CA, USA).
Chemicals
IL-1β was from Cell Concept GmbH (Umkirch, Deutschland); hyperfilm MP and horseradish-coupled secondary antibodies were from GE Healthcare Systems (Freiburg, Germany); AACOCF3, docosahexaenoic acid (DHA) and the PPARγ antagonist G3335 were from Merck Biosciences (Schwalbach, Germany); the monoclonal antibody against rat IIA-sPLA2 was generated and characterized as previously described (Aarsman et al., 1989); the IκB-α antibody was from Cell Signalling (Frankfurt am Main, Germany); the PGE2 ELISA was from Assay Designs, BIOTREND Chemikalien GmbH (Köln, Germany); all cell culture nutrients were from Invitrogen/Life Technologies (Karlsruhe, Germany).
Results
AVX001 and AVX002 inhibit cPLA2 activity in vitro and reduce cytokine-stimulated PGE2 formation in rat renal mesangial cells
Previously, it was shown that the ω6-PUFA derivatives arachidonyl-trifluoromethyl ketone (AACOCF3, ATK; Figure 1) (Street et al., 1993) and methyl-arachidonyl fluorophosphonate (MAFP; Figure 1) (Lio et al., 1996) are direct inhibitors of the group IVA cPLA2 in vitro. In the present study, we tested the ability of AVX001 and AVX002 to inhibit cPLA2 activity. In these compounds the methylene group β to the carbonyl group of the ketone in AACOCF3 was replaced by a sulphur atom. Holmeide and Skattebol, (2000) speculated that this would make the carbonyl carbon more electrophilic and consequently the molecule a more potent inhibitor of cPLA2. In vitro activity assays revealed that AVX001 blocked cPLA2 activity in a concentration-dependent manner (Figure 3). Multiple experiments were taken for the determination of the IC50 value, which was calculated as 120 ± 58 nM (n = 28). The structurally similar compound AVX002 also blocked cPLA2 activity in vitro (Figure 3). The IC50 value was analysed from several experiments to be 126 ± 37 nM (n = 14). By comparison, AACOCF3 was a less potent inhibitor; it showed 30% inhibition at 0.3 μM and 72% inhibition at 1 μM. In contrast, the ω3-fatty acid DHA had no direct inhibitory effect on cPLA2 (Figure 3). These data suggest that not only ω6-PUFA derivatives, but also certain ω3-PUFA derivatives, such as AVX001 and AVX002, but not DHA, are effective direct cPLA2 inhibitors.
Figure 3.
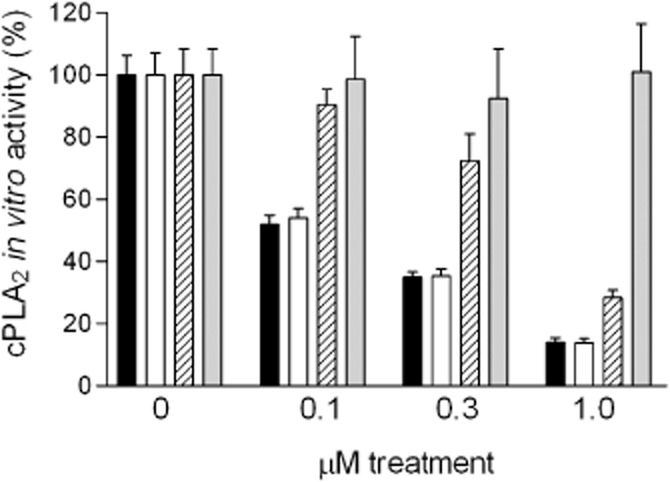
Direct inhibitory effect of AVX001, AVX002, AACOCF3 and DHA on cPLA2 activity in vitro. Recombinant cPLA2 enzyme was incubated with either solvent (DMSO, final concentration 1%, 0) or the indicated concentrations of AVX001 (solid columns) or AVX002 (open columns), AACOCF3 (hatched columns), and DHA (grey columns), and taken for an in vitro activity assay as described in the Methods section. cPLA2 enzyme activity is given as % of control value (activity in the absence of inhibitors). The results shown are representative of at least three independent experiments, and data represent mean of duplicate determinations.
To further test the effectiveness of AVX001 and AVX002 in a cell culture system, rat renal mesangial cells were used, as they represent a good model system to investigate molecular inflammatory mechanisms (Pfeilschifter, 1994; Gómez-Guerrero et al., 2005).
Treatment of mesangial cells with IL-1β led to a high production of PGE2 confirming many previous reports of our group (Pfeilschifter et al., 1989; 1990; 1993). The average amount of PGE2 in different experiments varied from 0–55 pg per 1.3 × 105 cells in unstimulated cells, to 2359–3751 pg per 1.3 × 105 cells in IL-1β-stimulated cells. Due to this variability, which seemed to depend on the cell passages, the maximal IL-1β-stimulated values were always set to 100%. In the presence of AVX001, PGE2 formation was concentration-dependently reduced with a 90% inhibition at 10 μM (Figure 4). A similar inhibitory effect was also seen for AVX002 (Figure 4). AACOCF3 also reduced PGE2 levels but was less potent, and DHA even at 20 μM had no significant effect (Figure 4).
Figure 4.
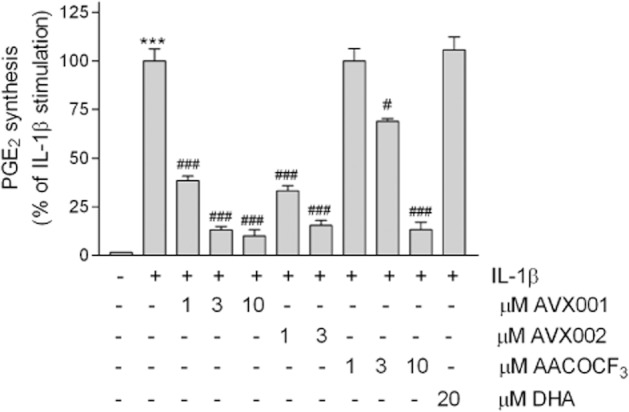
Effect of AVX001 and AVX002 on IL-1β-stimulated PGE2 formation in mesangial cells. Quiescent cells were stimulated for 24 h with either DMEM (-), and IL-1β (1 nM) in the absence (-) or presence of the indicated concentrations (in μM) of AVX001, AVX002, AACOCF3 and DHA. The inhibitors were all added as a pretreatment for 90 min before stimulation. Supernatants were collected and taken for PGE2 quantification by using an ELISA. Data are expressed as % of maximal IL-1β-stimulated PGE2 and are means ± SD (n = 3).***P < 0.001 considered statistically significant when compared with the control values; #P < 0.05, ###P < 0.001 when compared with the IL-1β-stimulated values.
As we previously showed that the cytokine-induced PGE2 formation in mesangial cells involves both sPLA2 and cPLA2 activation (Pfeilschifter et al., 1993), we next investigated the effect of AVX001 on sPLA2 protein and mRNA expressions. As seen in Figure 5, AVX001 and AVX002 both concentration-dependently down-regulated the sPLA2 protein expression paralleling the reduced PGE2 formation. To verify that supernatants derived from equal cell amounts, corresponding cell monolayers were lysed and stained for GAPDH protein expression (Figure 5, inset, lower panels) and used for normalization of secreted proteins. A similar reducing effect on sPLA2 protein expression was also seen for AACOCF3, whereas DHA had no significant effect (Figure 5A). IIA-sPLA2 mRNA expression was also reduced by AVX001 and AVX002 (Figure 5B). Again, AACOCF3 was less potent and DHA was ineffective at reducing IIA-sPLA2 mRNA expression (Figure 5B). Notably, higher concentrations of AVX001 and AVX002 were needed to down-regulate the sPLA2 protein and mRNA expressions compared with the reduction in PGE2.
Figure 5.
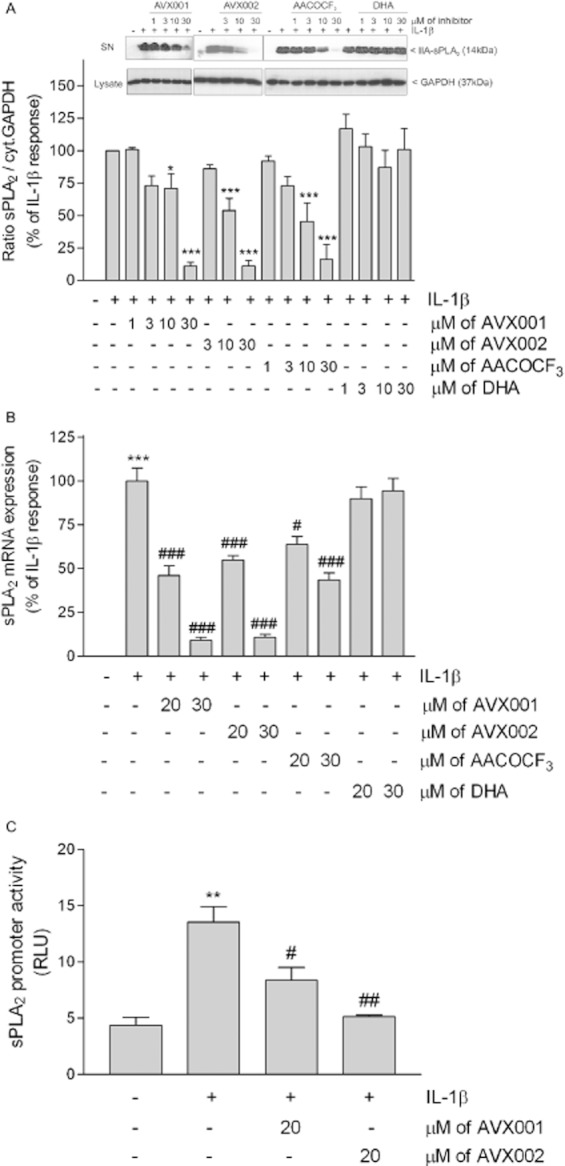
Effect of AVX001 and AVX002 on IL-1β-stimulated sPLA2 protein (A), mRNA expression (B) and promoter activity (C) in mesangial cells. Quiescent cells were stimulated with either DMEM (-), or IL-1β (1 nM) in the absence (-) or presence of the indicated concentrations of AVX001, AVX002, AACOCF3 and DHA (all inhibitors were added as a pretreatment for 90 min before stimulation). (A) Supernatants were taken for protein precipitation, separated by SDS-PAGE, transfered to membranes and subjected to Western blot analysis using a monoclonal antibody against rat sPLA2. Corresponding cell lysates were stained for GAPDH and used for normalization. Bands were densitometrically evaluated and the ratio between secreted sPLA2 and cytosolic GAPDH was calculated. Results are expressed as % of IL-1β stimulation. Data are means ± SD (n = 3–5). The inset shows representative Western blots of sPLA2 (upper panels) from supernatants (SN) and GAPDH (lower panels) from cell lysates. (B) Cells were taken for RNA extraction and subjected to quantitative PCR analysis of rat IIA-sPLA2 and 18S RNA. ΔΔCt values were calculated and results are expressed as % of maximal IL-1β-stimulated response and are means ± SD (n = 3–5). (C) Cells were transfected with the sPLA2 promoter construct plus a plasmid coding for Renilla luciferase. After transfection, cells were stimulated for 24 h with vehicle (-), IL-1β (1 nM), or IL-1β in the absence or presence of AVX001 or AVX002 (both at 20 μM). Both inhibitors were added as a pretreatment 90 min before stimulation. sPLA2 promoter activity was calculated and results are expressed as relative luciferase units (RLU) and are means ± SD (n = 3). **P < 0.01, ***P < 0.001 considered statistically significant when compared with the control values; #P < 0.05, ##P < 0.01, ###P < 0.001 when compared with the IL-1β-stimulated values.
To see whether this effect on sPLA2 protein and mRNA expression was due to a reducing effect on sPLA2 promoter activation and subsequent gene transcription, luciferase reporter gene assays were performed. A 2.26 kb fragment of the rat IIA-sPLA2 promoter was cloned according to Scholz-Pedretti et al. (2002) and ligated into a luciferase-containing vector (pGL3) and used to transfect mesangial cells. AVX001 and AVX002 at 20 μM significantly reduced IL-1β-stimulated promoter activity (Figure 5C), suggesting that cPLA2-regulated transcription factors are essential for sPLA2 gene transcription.
AVX inhibitors block cytokine-stimulated NF-κB activity in mesangial cells
As it is well known that the transcription factor NF-κB is crucially involved in sPLA2 gene transcription under inflammatory conditions (Walker et al., 1995), we further studied whether the AVX inhibitors had an effect on NF-κB activation. NF-κB activation was measured indirectly by Western blot analysis of the inhibitor of κB (IκB) protein expression. Short-term stimulation of cells for 15 min with IL-1β revealed a marked degradation of IκB protein (Figure 6), which is a key event in NF-κB activation that releases NF-κB from its sequestration in the cytoplasm and allows its nuclear translocation. This down-regulating effect of IL-1β on IκB was reverted by the IκB inhibitor Bay11-7085, but also by AVX001 and AVX002 (Figure 6).
Figure 6.
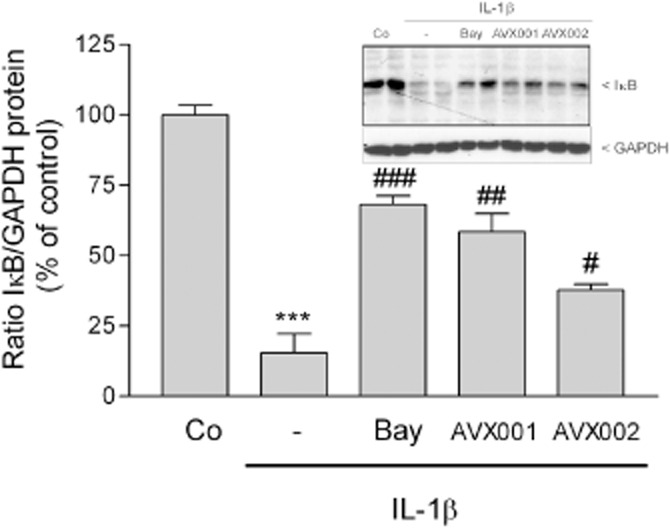
Effect of AVX inhibitors on IL-1β-stimulated NF-κB activation in mesangial cells. Quiescent cells were stimulated for 15 min with either vehicle (Co), or IL-1β (1 nM) in the absence (-) or presence of Bay11-7085 (Bay, 10 μM), AVX001 or AVX002 (both at 30 μM), pretreated for 90 min. Thereafter, cell lysates were separated by SDS-PAGE, transferred to nitrocellulose and subjected to a Western blot analysis using a polyclonal antibody against IκBα (inset, upper panel) and GAPDH (inset, lower panel). Bands corresponding to IκB and GAPDH were densitometrically evaluated and the ratio between IκB and GAPDH was calculated for each sample. Results are depicted as % of control stimulations and are means ± SD (n = 3). ***P < 0.001 considered statistically significant when compared with the control values; #P < 0.05, ##P < 0.01, ###P < 0.001 when compared with the IL-1β-stimulated values. The inset shows a representative experiment in duplicates.
AVX001 and AVX002 do not act through PPARγ activation
As AVX001 and AVX002 are structurally derived from ω3-fatty acids, and it has been reported previously that ω3-PUFAs are ligands and activators of PPAR (Stahl et al., 2010), which has been attributed an anti-inflammatory effect (Straus and Glass, 2007), we further tested whether the reducing effect of AVX compounds on PGE2 is mediated through PPARγ activation. Indeed, we found that in the presence of the PPARγ antagonist G3335, the AVX001 and AVX002 effects were partially reverted (Figure 7). However, the IL-1β effect was also increased to the same extent in the presence of G3335 (Figure 7), supporting the conclusion that there is a general stimulating effect of the PPARγ antagonist on the PGE2 pathway rather than a specific antagonistic effect on the AVX action.
Figure 7.
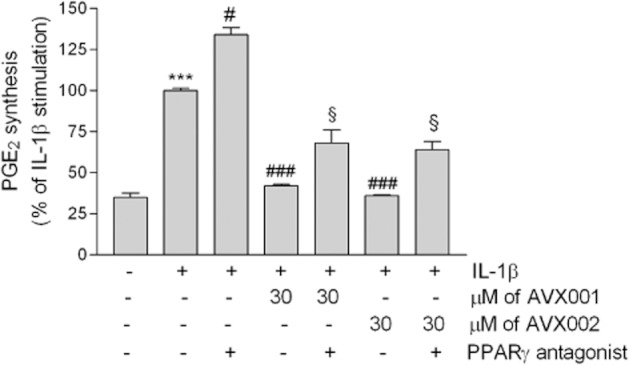
Effect of the PPARγ antagonist G3335 on AVX001 and AVX002 reduced PGE2 formation in mesangial cells. Quiescent cells were stimulated for 24 h with either DMEM (-), and IL-1β (1 nM) in the absence (-) or presence of 30 μM AVX001 or AVX002, in the absence or presence of 10 μM of G3335. The inhibitors were all added as a pretreatment for 90 min before stimulation. Supernatants were collected and taken for PGE2 quantification by using an ELISA. Data are expressed as % of maximal IL-1β-stimulated PGE2 and are means ± SD (n = 3). ***P < 0.001 considered statistically significant when compared with the control values; #P < 0.05, ###P < 0.001 when compared with the IL-1β-stimulated values; §P < 0.05 when compared with the corresponding AVX001 or AVX002 values.
Discussion
In the past few years, ω3-PUFAs have attracted a lot of interest due to accumulating evidence for their beneficial effects in various inflammatory diseases. These include rheumatoid arthritis, asthma, colitis, atherosclerosis, neurodegenerative diseases and even certain forms of cancer (Calder, 2005; Calon and Cole, 2007; Edwards and O'Flaherty, 2008).
Various possibilities for the mechanism of these anti-inflammatory effects were forwarded. For example, an increased incorporation of ω3-PUFA species into phospholipids may occur at the expense of arachidonic acid. Consequently, they replace arachidonic acid as a substrate for COXs and lipoxygenases resulting in reduced formation of PGE2 (Bagga et al., 2003), TXA2 (Krämer et al., 1996), LTB4 and LTE4 (Whelan et al., 1991). Instead, an increased generation of other less active PG and LT subspecies occurs (Terano et al., 1986; Krämer et al., 1996; Li et al., 1997; Bagga et al., 2003). Furthermore, it was proposed that ω3-PUFA may change the membrane lipid composition and thereby directly affect the functions of immune cells including the phagocytotic activity of macrophages, T cell signalling and proliferation, and antigen presentation activity of dendritic cells (Costa Rosa et al., 1996; Sanderson et al., 1997; Calder 2008).
Various previous studies have shown that ω3-PUFAs are able to down-regulate the gene transcription of pro-inflammatory and growth-promoting genes by interfering with transcription factors. In this regard, Liu et al., (2001) showed that in a mouse epidermal cell system, ω3-PUFAs, but not the ω6-arachidonic acid, effectively inhibited growth factor-triggered activator protein-1 transactivation. Moreover, ω3-PUFAs down-regulated cytokine-triggered NF-κB activation and subsequent gene transcription in endothelial cells (McGuinness et al., 2008) as well as in macrophages (Lo et al., 1999). However, the direct PUFA target was not addressed in any of these studies.
Intriguingly, it was recently shown that ω3-PUFAs can act as ligands of PPARs and thereby can modulate a multitude of pro- and anti-inflammatory genes (Forman et al., 1997; Price et al., 2000; Stahl et al., 2010). In fact, the appreciated lipid-lowering effect of fish oil supplementation (McKenney and Sica, 2007) is mainly explained by the action of ω3-PUFAs or metabolites as PPAR agonists (Forman et al., 1997; Xu et al., 1999). ω3-PUFAs can also act as ligands for the orphan receptor GPR120 and it was shown that this action contributes to its anti-inflammatory and insulin-sensitizing effects in macrophages (Oh et al., 2010).
Our data now clearly show that certain ω3-PUFA derivatives, such as AVX001 and AVX002, can also directly inhibit cPLA2 in vitro as well as in intact cells. As a consequence, they are able to down-regulate cytokine-stimulated PGE2 formation in mesangial cells and thereby reduce an inflammatory reaction. Mechanistically, we further showed that cPLA2 is involved in the cross-regulation of sPLA2-IIA expression via NF-κB activation. This is suggested by our findings that the direct cPLA2 inhibitors AVX001 and AVX002 not only down-regulated cytokine-induced sPLA2 promoter activities (Figure 5C), mRNA expression (Figure 5B), protein expression (Figure 5A) and subsequent activity (Figure 4), but also, as a more upstream event, blocked IκB degradation and subsequent NF-κB activation (Figure 6). Notably, higher concentrations of AVX001 and AVX002 were needed to down-regulate the sPLA2 protein and mRNA expressions compared with the reduction of PGE2. Their more potent effect on PGE2 formation than on sPLA2 expression may be derived from the dual action of AVX compounds, that is, the direct inhibition of cPLA2 (this enzyme accounts for approx. 20% of the cytokine-induced PGE2 formation according to Pfeilschifter et al., 1993), and the partial down-regulation of sPLA2 expression (this enzyme accounts for the remaining 80% of cytokine-induced PGE2 formation according to Pfeilschifter et al., 1993).
It is also worth noting that we previously reported that in the human keratinocyte cell line HaCaT, three previously known cPLA2 inhibitors, AACOCF3, MAFP and the trifluoromethyl ketone analogue of EPA (EPACOCF3), all inhibited TNFα-induced NF-κB activation and ICAM-1 expression (Thommesen et al., 1998). The reduced NF-κB activity could be reversed by the addition of an excess of exogenous arachidonic acid (Anthonsen et al., 2001), which further corroborates the involvement of cPLA2 in NF-κB activation. Additional evidence that cPLA2 is positively regulating NF-κB activation was also presented by Camandola et al., (1996). These authors showed that in promonocytic U937 cells, arachidonic acid stimulated NF-κB activation by a mechanism involving the metabolization of arachidonic acid to PGs and LTs.
However, when considering the fact that ω3-PUFAs can act as PPAR agonists, and the fact that the IIA-sPLA2 gene contains functional PPAR binding elements in its promoter sequence (Scholz-Pedretti et al., 2002), and in view of the recent report that parts of the anti-inflammatory effect of PPARs may be mediated by direct negative interference with transcription factors such as NF-κB, AP-1 and C/EBP (Genolet et al., 2004), it is possible that the observed reducing effect of the AVX compounds on PGE2 formation reported in this study also involves additional mechanisms besides the direct inhibition of cPLA2 such as PPAR activation. We therefore used a PPARγ antagonist to see whether the AVX001- and AVX002-mediated effects on PGE2 could be reversed. Indeed, the down-regulated PGE2 formation was increased in the presence of a PPARγ antagonist (Figure 7). However, as the IL-1β-triggered PGE2 formation was also increased by the PPARγ antagonist, we concluded that there is more a general PGE2 stimulating effect induced by PPARγ antagonism than a specific effect on AVX action.
PLA2 activation is the rate-limiting step in the generation of eicosanoids. It is also the initial step in the generation of PAF, which is a further potent inflammatory mediator. Therefore, the pharmacological inhibition of PLA2 is considered an attractive target to block inflammatory processes and should, at least theoretically, be a more effective approach than blocking one of the downstream enzymes like the COXs. Unfortunately, the development of PLA2 inhibitors has been hampered by the fact that too many subtypes of PLA2 exist, that are involved in a redundant manner in the inflammatory reaction (Murakami and Kudo, 2004; Cummings, 2007; Lambeau and Gelb, 2008).
Very few PLA2 inhibitors have been developed which also exerted in vivo efficacy in various inflammatory animal models (reviewed in Huwiler and Pfeilschifter, 2009). These include an inhibitor of IIA-sPLA2, LY311727 (Schevitz et al., 1995), which proved effective in a rat model of inflammatory pain (Svensson et al., 2005). The synthesis of novel IIA-sPLA2 inhibitors that also exert in vivo activity and reduce carrageenan-induced oedema formation in rats with a similar potency to indomethacin, at a dose of 10 mg·kg−1, has recently been described (Boukli et al., 2008).
Also, the cPLA2 inhibitors MAFP and AACOCF3 were shown to reduce thermal hyperalgesia induced by carrageenan- or formalin-induced flinching in rats (Lucas et al., 2005), suggesting that not only the IIA-sPLA2, but also the cPLA2, is involved in the molecular mechanisms of nociception. Furthermore, it was shown that AACOCF3 reduced chronic inflammatory responses in mice; AACOCF3 given i.p. inhibited phorbol ester-induced chronic ear oedema (Malaviya et al., 2006). Oral application of AACOCF3 also prevented the development of airway hyperresponsiveness in a mouse asthma model (Malaviya et al., 2006) and reduced acute lung injury induced by septic syndrome in mice (Nagase et al., 2003). AACOCF3 also has beneficial effects in a mouse model of experimental autoimmune encephalomyelitis, which is an inflammatory demyelinating disease of the CNS that results in CNS lesions (Kalyvas and David, 2004). However, due to the fact that AACOCF3 is not exclusively selective for the cPLA2 (Ackermann et al., 1995) and, additionally, a cell lytic activity has been reported (Risse et al., 2002), the various in vivo activities of AACOCF3 must be viewed with caution. Recently, second-generation cPLA2 inhibitors have become available such as 2-oxoamides, which exert potent cPLA2 inhibitory effects in vitro and have proved effective in first in vivo models of inflammation and pain (Kokotos et al., 2004). All these data strongly suggest that cPLA2 is a valid target to treat inflammatory diseases, and, therefore, the development of novel, more selective cPLA2 inhibitors, is of utmost importance.
AACOCF3 has the characteristic of being a slow binding inhibitor of cPLA2 (Street et al., 1993). The trifluoromethyl ketone group turned out to be especially important for inhibition since substitution of this group by either CONH2, CHO, COCH3 or CH(OH)CF3 led to a loss of inhibitory potency (Street et al., 1993). Holmeide and Skattebol (2000) speculated that the replacement of the methylene group β to the carbonyl group of the ketone with a sulphur atom would make the carbonyl carbon more electrophilic, and consequently, the molecule a more potent inhibitor of cPLA2. This assumption is indeed confirmed by our study that showed a more potent inhibitory effect of the sulphur-containing AVX001 and AVX002 when compared to the AACOCF3, which originally was reported to inhibit cPLA2 by 78% at 1.6 mol% of AACOCF3 in the cPLA2 assay system (Riendeau et al., 1994). A further point that needs attention is the different potencies of AVX001 and AVX002 in a cell-free in vitro system compared with their potencies in a cellular system. Riendeau et al. (1994) already highlighted the issue that in a cellular system, higher concentrations of AACOCF3 were needed when compared with the in vitro system. This is due to the fact that AACOCF3 is a slow binding inhibitor of cPLA2, meaning that it takes many minutes to exert its full inhibitory potency (Street et al., 1993). As arachidonic acid release in cells is a rapid process taking place within minutes, relatively high concentrations are required to inhibit cPLA2. A second point concerns its stability. Riendeau et al. (1994) showed the conversion of the ketone of AACOCF3 to its non-inhibitory alcohol in cells, which may become particularly relevant in long-term stimulation settings and could reduce the inhibitory potential. Furthermore, PUFAs are known to be easily oxidized, which can additionally reduce their inhibitory effect. Similar explanations may also be true for the AVX001 and AVX002, although the metabolism of these compounds in vivo is still unknown.
In summary, in this study we have described two novel cPLA2 inhibitors, the ω3-PUFA derivatives AVX001 and AVX002, which are effective at reducing inflammatory PGE2 synthesis in mesangial cells. These two compounds are certainly very promising candidates to be tested in in vivo disease models, such as in models of chronic inflammatory kidney disease. Also, further studies are needed to characterize the pharmacokinetic properties of these novel cPLA2 inhibitors.
Acknowledgments
We thank Dr Inger Reidun Aukrust and Dr Marcel Sandberg (Synthetica AS, Oslo, Norway) for synthesis of AVX001 and AVX002. We also thank Mari Saether for excellent technical assistance. This work was supported by the Swiss National Science Foundation, the German Research Foundation (GRK757/2, FOG 784, HU842/5-1), the Norwegian Research Council and Avexxin AS.
Glossary
- AACOCF3, ATK
arachidonyl-trifluoromethyl ketone
- AVX001
1-octadeca-2,6,9,12,15-pentaenylsulfanyl-propan-2-one
- AVX002
1-octadeca-3,6,9,12,15-pentaenylsulfanyl-propan-2-one
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- IκB
inhibitor of κB
- MAFP
methyl-arachidonyl fluorophosphonate
- PAF-AH
PAF acetylhydrolase
- PUFA
polyunsaturated fatty acid
- RT-PCR
reverse transcriptase-PCR
Statement of conflict of interest
BJ is a stockholder of Avexxin AS.
References
- Aarsman AJ, de Jong JG, Arnoldussen E, Neys FW, van Wassenaar PD, Van den Bosch H. Immunoaffinity purification, partial sequence, and subcellular localization of rat liver phospholipase A2. J Biol Chem. 1989;264:10008–10014. [PubMed] [Google Scholar]
- Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- Akiba S, Hayama M, Sato T. Inhibition of Ca2+-independent phospholipase A2 by bromoenol lactone attenuates prostaglandin generation induced by interleukin-1 beta and dibutyryl cAMP in rat mesangial cells. FEBS Lett. 1998;437:225–228. doi: 10.1016/s0014-5793(98)01236-8. [DOI] [PubMed] [Google Scholar]
- Anthonsen MW, Solhaug A, Johansen B. Functional coupling between secretory and cytosolic phospholipase A2 modulates tumor necrosis factor-alpha- and interleukin-1beta-induced NF-kappa B activation. J Biol Chem. 2001;276:30527–30536. doi: 10.1074/jbc.M008481200. [DOI] [PubMed] [Google Scholar]
- Bagga D, Wang L, Farias-Eisner R, Glaspy JA, Reddy ST. Differential effects of prostaglandin derived from omega-6 and omega-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc Natl Acad Sci USA. 2003;100:1751–1756. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsinde J, Balboa MA, Dennis EA. Functional coupling between secretory phospholipase A2 and cyclooxygenase-2 and its regulation by cytosolic group IV phospholipase A2. Proc Natl Acad Sci USA. 1998;95:7951–7956. doi: 10.1073/pnas.95.14.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukli L, Touaibia M, Meddad-Belhabich N, Djimdé A, Park CH, Kim JJ, et al. Design of new potent and selective secretory phospholipase A2 inhibitors. Part 5: synthesis and biological activity of 1-alkyl-4-[4,5-dihydro-1,2,4-[4H]-oxadiazol-5-one-3-ylmethylbenz-4'-yl(oyl)] piperazines. Bioorg Med Chem. 2008;16:1242–1253. doi: 10.1016/j.bmc.2007.10.077. [DOI] [PubMed] [Google Scholar]
- Calder PC. Polyunsaturated fatty acids and inflammation. Biochem Soc Trans. 2005;33:423–427. doi: 10.1042/BST0330423. [DOI] [PubMed] [Google Scholar]
- Calder PC. Polyunsaturated fatty acids, inflammatory processes and inflammatory bowel diseases. Mol Nutr Food Res. 2008;52:885–897. doi: 10.1002/mnfr.200700289. [DOI] [PubMed] [Google Scholar]
- Calon F, Cole G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: evidence from animal studies. Prostaglandins Leukot Essent Fatty Acids. 2007;77:287–293. doi: 10.1016/j.plefa.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Camandola S, Leonarduzzi G, Musso T, Varesio L, Carini R, Scavazza A, et al. Nuclear factor κB is activated by arachidonic acid but not by eicosapentaenoic acid. Biochem Biophys Res Commun. 1996;229:643–647. doi: 10.1006/bbrc.1996.1857. [DOI] [PubMed] [Google Scholar]
- Costa Rosa LF, Safi DA, Guimarães AR. The effect of N-3 PUFA rich diet upon macrophage and lymphocyte metabolism and function. Biochem Mol Biol Int. 1996;40:833–842. doi: 10.1080/15216549600201443. [DOI] [PubMed] [Google Scholar]
- Cummings BS. Phospholipase A2 as targets for anti-cancer drugs. Biochem Pharmacol. 2007;74:949–959. doi: 10.1016/j.bcp.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Edwards IJ, O'Flaherty JT. Omega-3 fatty acids and PPARgamma in cancer. PPAR Res. 2008;2008:358052. doi: 10.1155/2008/358052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors α and δ. Proc Natl Acad Sci USA. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genolet R, Wahli W, Michalik L. PPARs as drug targets to modulate inflammatory responses? Curr Drug Targets Inflamm Allergy. 2004;3:361–375. doi: 10.2174/1568010042634578. [DOI] [PubMed] [Google Scholar]
- Gómez-Guerrero C, Hernández-Vargas P, López-Franco O, Ortiz-Muñoz G, Egido J. Mesangial cells and glomerular inflammation: from the pathogenesis to novel therapeutic approaches. Curr Drug Targets Inflamm Allergy. 2005;4:341–351. doi: 10.2174/1568010054022169. [DOI] [PubMed] [Google Scholar]
- Gronich J, Konieczkowski M, Gelb MH, Nemenoff RA, Sedor JR. Interleukin 1 alpha causes rapid activation of cytosolic phospholipase A2 by phosphorylation in rat mesangial cells. J Clin Invest. 1994;93:1224–1233. doi: 10.1172/JCI117076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Helm HA, Aarsman AJ, Janssen MJ, Neys FW, van den Bosch H. Regulation of the expression of group IIA and group V secretory phospholipases A2 in rat mesangial cells. Biochim Biophys Acta. 2000;1484:215–224. doi: 10.1016/s1388-1981(00)00021-4. [DOI] [PubMed] [Google Scholar]
- Holmeide AK, Skattebol L. Syntheses of some polyunsaturated trifluoromethyl ketones as potential phospholipase A2 inhibitors. J Chem Soc Perkin Trans. 2000;1:2271–2276. [Google Scholar]
- Huwiler A, Pfeilschifter J. Lipids as targets for novel anti-inflammatory therapies. Pharmacol Rev. 2009;124:96–112. doi: 10.1016/j.pharmthera.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Staudt G, Kramer RM, Pfeilschifter J. Cross-talk between secretory phospholipase A2 and cytosolic phospholipase A2 in rat renal mesangial cells. Biochim Biophys Acta. 1997;1348:257–272. doi: 10.1016/s0005-2760(97)00073-8. [DOI] [PubMed] [Google Scholar]
- Kalyvas A, David S. Cytosolic phospholipase A2 plays a key role in the pathogenesis of multiple sclerosis-like disease. Neuron. 2004;41:323–335. doi: 10.1016/s0896-6273(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Kashgarian M, Sterzel RB. The pathobiology of the mesangium. Kidney Int. 1992;41:524–529. doi: 10.1038/ki.1992.74. [DOI] [PubMed] [Google Scholar]
- Kokotos G, Six DA, Loukas V, Smith T, Constantinou-Kokotou V, Hadjipavlou-Litina D, et al. Inhibition of group IVA cytosolic phospholipase A2 by novel 2-oxoamides in vitro, in cells, and in vivo. J Med Chem. 2004;47:3615–3628. doi: 10.1021/jm030485c. [DOI] [PubMed] [Google Scholar]
- Krämer HJ, Stevens J, Grimminger F, Seeger W. Fish oil fatty acids and human platelets: dose-dependent decrease in dienoic and increase in trienoic thromboxane generation. Biochem Pharmacol. 1996;52:1211–1217. doi: 10.1016/0006-2952(96)00473-x. [DOI] [PubMed] [Google Scholar]
- Lambeau G, Gelb MH. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem. 2008;77:495–520. doi: 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- Li Y, Kang JX, Leaf A. Differential effects of various eicosanoids on the production or prevention of arrhythmias in cultured neonatal rat cardiac myocytes. Prostaglandins. 1997;54:511–530. doi: 10.1016/s0090-6980(97)00122-6. [DOI] [PubMed] [Google Scholar]
- Lin LL, Lin AY, DeWitt DL. Interleukin-1 alpha induces the accumulation of cytosolic phospholipase A2 and the release of prostaglandin E2 in human fibroblasts. J Biol Chem. 1992;267:23451–23454. [PubMed] [Google Scholar]
- Lio YC, Reynolds LJ, Balsinde J, Dennis EA. Irreversible inhibition of Ca2+-independent phospholipase A2 by methyl arachidonyl fluorophosphonate. Biochim Biophys Acta. 1996;1302:55–60. doi: 10.1016/0005-2760(96)00002-1. [DOI] [PubMed] [Google Scholar]
- Liu G, Bibus DM, Bode AM, Ma WY, Holman RT, Dong Z. Omega 3 but not omega 6 fatty acids inhibit AP-1 activity and cell transformation in JB6 cells. Proc Natl Acad Sci U S A. 2001;98:7510–7515. doi: 10.1073/pnas.131195198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CJ, Chiu KC, Fu M, Lo R, Helton S. Fish oil decreases macrophage tumor necrosis factor gene transcription by altering the NFκB activity. J Surg Res. 1999;82:216–221. doi: 10.1006/jsre.1998.5524. [DOI] [PubMed] [Google Scholar]
- Lucas KK, Dennis EA. Distinguishing phospholipase A2 types in biological samples by employing group-specific assays in the presence of inhibitors. Prostaglandins Other Lipid Mediat. 2005;77:235–248. doi: 10.1016/j.prostaglandins.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Lucas KK, Svensson CI, Hua XY, Yaksh TL, Dennis EA. Spinal phospholipase A2 in inflammatory hyperalgesia: role of group IVA cPLA2. Br J Pharmacol. 2005;144:940–952. doi: 10.1038/sj.bjp.0706116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Ansell J, Hall L, Fahmy M, Argentieri RL, Olini GC, Jr, et al. Targeting cytosolic phospholipase A2 by arachidonyl trifluoromethyl ketone prevents chronic inflammation in mice. Eur J Pharmacol. 2006;539:195–204. doi: 10.1016/j.ejphar.2006.03.018. [DOI] [PubMed] [Google Scholar]
- McGuinness J, Byrne J, Condron C, McCarthy J, Bouchier-Hayes D, Redmond JM. Pretreatment with omega-3 fatty acid infusion to prevent leukocyte-endothelial injury responses seen in cardiac surgery. J Thorac Cardiovasc Surg. 2008;136:135–141. doi: 10.1016/j.jtcvs.2007.11.010. [DOI] [PubMed] [Google Scholar]
- McKenney JM, Sica D. Role of prescription omega-3 fatty acids in the treatment of hypertriglyceridemia. Pharmacotherapy. 2007;27:715–728. doi: 10.1592/phco.27.5.715. [DOI] [PubMed] [Google Scholar]
- Murakami M, Kudo I. Secretory phospholipase A2. Biol Pharm Bull. 2004;27:1158–1164. doi: 10.1248/bpb.27.1158. [DOI] [PubMed] [Google Scholar]
- Nagase T, Uozumi N, Aoki-Nagase T, Terwaki K, Ishii S, Tomita T, et al. A potent inhibitor of cytosolic phospholipase A2, arachidonyl trifluoromethyl ketone, attenuates LPS-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2003;284:L720–L726. doi: 10.1152/ajplung.00396.2002. [DOI] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142:687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petry C, Fritz G, Pfeilschifter J, Huwiler A. Inhibition of Rho modulates cytokine-induced prostaglandin E2 formation in renal mesangial cells. Biochim Biophys Acta. 2004;1636:108–118. doi: 10.1016/j.bbalip.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J. Mesangial cells orchestrate inflammation in the renal glomerulus. News Physiol Sci. 1994;9:271–276. [Google Scholar]
- Pfeilschifter J, Kurtz A, Bauer C. Activation of phospholipase C and prostaglandin synthesis by [arginine]vasopressin in cultures. Biochem J. 1984;223:855–859. doi: 10.1042/bj2230855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeilschifter J, Pignat W, Vosbeck K, Märki F. Interleukin 1 and tumor necrosis factor synergistically stimulate prostaglandin synthesis and phospholipase A2 release from rat renal mesangial cells. Biochem Biophys Res Commun. 1989;159:385–394. doi: 10.1016/0006-291x(89)90003-x. [DOI] [PubMed] [Google Scholar]
- Pfeilschifter J, Pignat W, Leighton J, Märki F, Vosbeck K, Alkan S. Transforming growth factor beta 2 differentially modulates interleukin-1 beta- and tumour-necrosis-factor-alpha-stimulated phospholipase A2 and prostaglandin E2 synthesis in rat renal mesangial cells. Biochem J. 1990;270:269–271. doi: 10.1042/bj2700269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeilschifter J, Schalkwijk C, Briner VA, van den Bosch H. Cytokine-stimulated secretion of group II phospholipase A2 by rat mesangial cells. Its contribution to arachidonic acid release and prostaglandin synthesis by cultured rat glomerular cells. J Clin Invest. 1993;92:2516–2523. doi: 10.1172/JCI116860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price PT, Nelson CM, Clarke SD. Omega-3 polyunsaturated fatty acid regulation of gene expression. Curr Opin Lipidol. 2000;11:3–7. doi: 10.1097/00041433-200002000-00002. [DOI] [PubMed] [Google Scholar]
- Riendeau D, Guay J, Weech PK, Laliberté F, Yergey J, Li C, et al. Arachidonyl trifluoromethyl ketone, a potent inhibitor of 85-kDa phospholipase A2, blocks production of arachidonate and 12-hydroxyeicosatetraenoic acid by calcium ionophore-challenged platelets. J Biol Chem. 1994;269:15619–15624. [PubMed] [Google Scholar]
- Risse D, Elfringhoff AS, Lehr M. Determination of the cell lytic properties of amphiphilic inhibitors of the cytosolic phospholipase A2 against human platelets by measuring the liberation of serotonin with high-performance liquid chromatography and fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;769:185–190. doi: 10.1016/s1570-0232(02)00013-2. [DOI] [PubMed] [Google Scholar]
- Sanderson P, MacPherson GG, Jenkins CH, Calder PC. Dietary fish oil diminishes the antigen presentation activity of rat dendritic cells. J Leukoc Biol. 1997;62:771–777. doi: 10.1002/jlb.62.6.771. [DOI] [PubMed] [Google Scholar]
- Schalkwijk C, Pfeilschifter J, Märki F, van den Bosch H. Interleukin-1 beta- and forskolin-induced synthesis and secretion of group II phospholipase A2 and prostaglandin E2 in rat mesangial cells is prevented by transforming growth factor-β2. J Biol Chem. 1992;267:8846–8851. [PubMed] [Google Scholar]
- Schalkwijk CG, Vervoordeldonk M, Pfeilschifter J, van den Bosch H. Interleukin-1 beta-induced cytosolic phospholipase A2 activity and protein synthesis is blocked by dexamethasone in rat mesangial cells. FEBS Lett. 1993;333:339–343. doi: 10.1016/0014-5793(93)80683-l. [DOI] [PubMed] [Google Scholar]
- Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Schevitz RW, Bach NJ, Carlson DG, Chirgadze NY, Clawson DK, Dillard RD, et al. Structure-based design of the first potent and selective inhibitor of human non-pancreatic secretory phospholipase A2. Nat Struct Biol. 1995;2:458–465. doi: 10.1038/nsb0695-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz-Pedretti K, Gans A, Beck KF, Pfeilschifter J, Kaszkin M. Potentiation of TNF-alpha-stimulated group IIA phospholipase A2 expression by peroxisome proliferator-activated receptor alpha activators in rat mesangial cells. J Am Soc Nephrol. 2002;13:611–620. doi: 10.1681/ASN.V133611. [DOI] [PubMed] [Google Scholar]
- Stahl A, Sapieha P, Connor KM, Sangiovanni JP, Chen J, Aderman CM, et al. Short communication: PPAR γ mediates a direct antiangiogenic effect of omega 3-PUFAs in proliferative retinopathy. Circ Res. 2010;107:495–500. doi: 10.1161/CIRCRESAHA.110.221317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Street IP, Lin HK, Laliberté F, Ghomashchi F, Wang Z, Perrier H, et al. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Lucas KK, Hua XY, Powell HC, Dennis EA, Yaksh TL. Spinal phospholipase A2 in inflammatory hyperalgesia: role of the small, secretory phospholipase A2. Neuroscience. 2005;133:543–553. doi: 10.1016/j.neuroscience.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Terano T, Salmon JA, Higgs GA, Moncada S. Eicosapentaenoic acid as a modulator of inflammation. Effect on prostaglandin and leukotriene synthesis. Biochem Pharmacol. 1986;35:779–785. doi: 10.1016/0006-2952(86)90246-7. [DOI] [PubMed] [Google Scholar]
- Thommesen L, Sjursen W, Gåsvik K, Hanssen W, Brekke OL, Skattebøl L, et al. Selective inhibitors of cytosolic or secretory phospholipase A2 block TNF-induced activation of transcription factor nuclear factor-κB and expression of ICAM-1. J Immunol. 1998;161:3421–3430. [PubMed] [Google Scholar]
- Walker G, Kunz D, Pignat W, van den Bosch H, Pfeilschifter J. Pyrrolidine dithiocarbamate differentially affects cytokine- and cAMP-induced expression of group II phospholipase A2 in rat renal mesangial cells. FEBS Lett. 1995;364:218–222. doi: 10.1016/0014-5793(95)00402-u. [DOI] [PubMed] [Google Scholar]
- Whelan J, Broughton KS, Lokesh B, Kinsella JE. In vivo formation of leukotriene E5 by murine peritoneal cells. Prostaglandins. 1991;41:29–42. doi: 10.1016/0090-6980(91)90102-l. [DOI] [PubMed] [Google Scholar]
- Wijkander J, Sundler R. An 100-kDa arachidonate-mobilizing phospholipase A2 in mouse spleen and the macrophage cell line J774. Purification, substrate interaction and phosphorylation by protein kinase C. Eur J Biochem. 1991;202:873–880. doi: 10.1111/j.1432-1033.1991.tb16445.x. [DOI] [PubMed] [Google Scholar]
- Xin C, Ren S, Kleuser B, Shabahang S, Eberhardt W, Radeke H, et al. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem. 2004;279:35255–35262. doi: 10.1074/jbc.M312091200. [DOI] [PubMed] [Google Scholar]
- Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]