

Abstract
Background and purpose
Pyrazole derivatives have recently been suggested as selective blockers of transient receptor potential cation (TRPC) channels but their ability to distinguish between the TRPC and Orai pore complexes is ill-defined. This study was designed to characterize a series of pyrazole derivatives in terms of TRPC/Orai selectivity and to delineate consequences of selective suppression of these pathways for mast cell activation.
Experimental approach
Pyrazoles were generated by microwave-assisted synthesis and tested for effects on Ca2+ entry by Fura-2 imaging and membrane currents by patch-clamp recording. Experiments were performed in HEK293 cells overexpressing TRPC3 and in RBL-2H3 mast cells, which express classical store-operated Ca2+ entry mediated by Orai channels. The consequences of inhibitory effects on Ca2+ signalling in RBL-2H3 cells were investigated at the level of both degranulation and nuclear factor of activated T-cells activation.
Key Results
Pyr3, a previously suggested selective inhibitor of TRPC3, inhibited Orai1- and TRPC3-mediated Ca2+ entry and currents as well as mast cell activation with similar potency. By contrast, Pyr6 exhibited a 37-fold higher potency to inhibit Orai1-mediated Ca2+ entry as compared with TRPC3-mediated Ca2+ entry and potently suppressed mast cell activation. The novel pyrazole Pyr10 displayed substantial selectivity for TRPC3-mediated responses (18-fold) and the selective block of TRPC3 channels by Pyr10 barely affected mast cell activation.
Conclusions and Implications
The pyrazole derivatives Pyr6 and Pyr10 are able to distinguish between TRPC and Orai-mediated Ca2+ entry and may serve as useful tools for the analysis of cellular functions of the underlying Ca2+ channels.
Keywords: pyrazole Ca2+ channel blockers, transient receptor potential, Orai, store-operated Ca2+ entry, receptor operated Ca2+ entry, NFAT signalling, mast cell degranulation
Introduction
Changes in cytosolic Ca2+ concentration control a broad range of cell- and tissue-specific processes reaching from B-cell activation, mast cell degranulation and cardiac pathologies, to cell proliferation and gene expression. Ca2+ entry via plasma membrane channels can be mediated by a diverse array of extra- and intracellular stimuli (Berridge et al., 2000). Characterization of the mechanisms that govern Ca2+ channel function has resulted in a commonly accepted distinction between ‘receptor operated Ca2+ entry’ (ROCE) pathways that take place in response to receptor agonist/ligand-induced phospholipase C-mediated phosphoinositol-4,5-bisphosphate hydrolysis formation (Hofmann et al., 1999; Lemonnier et al., 2008) and ‘store operated Ca2+ entry’ (SOCE), which is activated as a consequence of depletion of endoplasmic reticulum (ER) Ca2+ stores. Until the discovery of stromal interaction molecule 1 (STIM1) and Orai1 as key components of the latter process (Zhang et al., 2005; Prakriya et al., 2006), the family of canonical transient receptor potential channels (TRPCs) (Pedersen et al., 2005; Nilius et al., 2007; Abramowitz and Birnbaumer, 2009) has been considered the prime candidates for both Ca2+ entry pathways. Due to inherent overlap and crosstalk of the two mechanisms as well as the paucity of model systems that unequivocally lack one of these components, a clear-cut distinction between Orai1-meditated SOCE and TRPC-mediated ROCE appears difficult. Moreover, physical interactions between these two channel proteins in either a direct or indirect way has been proposed (Liao et al., 2007; Jardin et al., 2008; Yuan et al., 2009; Woodard et al., 2010; Cheng et al., 2011), and both Ca2+ influx pathways are tightly linked to downstream gene transcription via nuclear factor of activated T-cells (NFAT) (Sinkins et al., 2004; Kar et al., 2011).
Therefore, specific and potent pharmacological tools are highly desirable for further analysis of the contribution of either protein species or Ca2+ channel complexes to Ca2+ signalling and downstream cellular events. Inorganic blockers like Gd3+ or La3+ had been used extensively for this purpose as they are considered to interact specifically with TRPC channel pores (Trebak et al., 2002). Similarly, organic compounds such as SKF-96365 (Harteneck and Gollasch, 2011), 2-APB (DeHaven et al., 2008) and SK66 (Ng et al., 2008) have also been used due to their blocking effects on TRPC channels and store-operated Ca2+ conductance, with half-maximal concentrations in the low to intermediate micromolar range. All these pharmacological tools share common drawbacks in terms of lacking specificity for blocking a certain Ca2+ entry channel. The blockers not only lack selectivity for subtypes of TRPC channels (Harteneck and Gollasch, 2011), but exert, in addition, complex effects on Ca2+ entry and currents mediated by the STIM1/Orai1 SOCE pathway (DeHaven et al., 2008).
3,5-Bis(trifluoromethyl)pyrazole derivatives (BTPs), specifcally BTP2/Pyr2, have been proposed as potent small molecule inhibitors of both SOCE and TRPC-mediated ROCE (Zitt et al., 2004; He et al., 2005). Historically, this tool evolved from a class of immunosuppressants analogous to cyclosporine A or so called ‘-limus drugs’ such as sirolimus and tacrolimus (FK506) (Djuric et al., 2000; Chen et al., 2002; Ishikawa et al., 2003). Although primarily affecting NFAT activation and as a consequence cytokine production in immune cells, experiments showed that BTPs are capable of blocking store depletion-activated Ca2+ entry into a wide variety of cells at nanomolar to low micromolar concentrations with appreciable selectivity over voltage-gated Ca2+ entry. For a general review on BTPs as SOCE blockers, see Sweeney et al. (2009).
Recently, Pyr3, a pyrazole derivate, has been proposed as a highly subtype-specific inhibitor of TRPC3 activity (Kiyonaka et al., 2009). Specificity over other TRPC family members and other TRP subtypes in transfected HEK293 cells has been clearly demonstrated and the tricholoroarylic amide bond-linked side group was identified as essential for this property. Moreover, electron drawing side groups in C3 position of the pyrazole ring of BTP and Pyrs were proposed as key structural determinants of the inhibitory effect (Law et al., 2011). Studies demonstrating BTP2/Pyr2 as an inhibitor of receptor-operated TRPC functions, thus lacking selectivity for SOCE (He et al., 2005) and, in turn, Pyr3 as a potent inhibitor of SOCE (Salmon and Ahluwalia, 2010; Kim et al., 2011) have raised doubts about the suitability of these compounds for pharmacological differentiation of these Ca2+ entry mechanisms and support the hypothesis of a substantial overlap of these pathways and/or contribution of TRPC3 to SOCE. Applying a recently published synthesis strategy to generate pyrazole derivatives (Obermayer et al., 2011), we characterized the selectivity of four pyrazole compounds, including a new structure, designated as Pyr10, in cell systems that express high levels of well-characterized receptor-operated or store-operated Ca2+ channels. We employed HEK293 cells overexpressing TRPC3 as a ROCE model and native RBL-2H3 mast cells as an established Orai-mediated SOCE model (Di Capite and Parekh, 2009).
Our results demonstrate the ability of two pyrazole derivatives to discriminate between the classical Orai-mediated, highly Ca2+ selective signalling pathway and the phospholipase C-dependent Ca2+ entry-mediated by TRPC channels, specifically by TRPC3. We present Pyr6 and Pyr10 as valuable tools to dissect these signalling pathways.
Methods
DNA, cell culture and transfection
RBL-2H3 and HEK293 cells were cultivated in DMEM medium (Invitrogen, LifeTechnologies, Vienna, Austria) supplemented with 10% FBS. HEK293 cells seeded out in adequate density were transiently transfected by lipofection using FuGENE® (Roche, Vienna, Austria) according to manufacturer's protocol with an n-terminally YFP-tagged TRPC3 cDNA clone to be used as ROCE model or with a CFP-tagged STIM1 and YFP-tagged Orai1 clone to reconstitute the Ca2+ release-activated current (CRAC) pore in HEK293 cells (Muik et al., 2008). For NFAT translocation experiments, RBL-2H3 cells were electroporated with a GFP-tagged NFAT construct (at 300 V and 275 μF) with 20 μg of DNA 12–18 h before and seeded out on glass coverslips. For characterization of Pyr6 and Pyr10 selectivity for TRPC channels, n-terminally GFP-tagged murine TRPC6, n-terminally YFP-tagged TRPC5 (Schindl et al., 2008) and c-terminally YFP-tagged TRPC4beta (Graziani et al., 2010) were used. Detailed procedures and TRPC3 and NFAT constructs have been described previously (Poteser et al., 2011).
All measurements were performed at room temperature.
Reagents and synthesis of pyrazole compounds
If not mentioned otherwise, chemicals were purchased from Sigma Aldrich (Vienna, Austria). For synthesis of the pyrazole compounds, a recently published, optimized three-step microwave procedure was used (Glasnov et al., 2009; Obermayer et al., 2011). In a standard cyclocondensation reaction between 4-nitrophenylhydrazine hydrochloride and corresponding enone or 1,3-dicarbonyl compound under acidic conditions, the desired 1-(4-nitrophenyl)-1H-pyrazoles could be obtained within a short reaction time using microwave heating (160°C, 5 min). The latter products are further reduced into the corresponding anilines in a catalytic transfer hydrogenation procedure with cyclohexene as hydrogen donor over Pd/C (160°C, 2–5 min reaction time). To generate the final Pyr-compounds, two straightforward synthetic routes were available – reaction with an acid in the presence of PCl3 to conveniently obtain amide-bond derivatives Pyr2, Pyr3 and Pyr6 (150°C, 5 min microwave heating), or reaction with corresponding sulfonyl chlorides in the presence of pyridine as a base (100°C, 5 min microwave heating) to obtain Pyr10. Molecular properties of the compounds were calculated using Molinspiration Property Calculation Service (http://www.molinspiration.com).
Electrophysiology
Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus, Hugo Sachs, March-Hugstetten, Germany; resistance 3–5 MΩ). Currents were recorded at room temperature using a List EPC7 patch-clamp amplifier (HEKA Instruments, Lambrecht, Germany). Signals were low-pass filtered at 3 and 10 kHz and digitized with 5 kHz. For HEK293 cells, voltage-clamp protocols (voltage ramps from −130 to +80 mV, holding potential 0 mV) were controlled by pClamp software (Axon Instruments, Molecular Devices, Biberach, Germany). Extracellular solution (ECS) contained (in mM) 140 NaCl, 2 CaCl2, 2 MgCl2, 10 glucose, pH adjusted to 7.4 with NaOH. The pipette solution (ICS) contained (in mM) 120 caesium methanesulphonate, 20 CsCl, 15 HEPES, 5 MgCl2, 3 EGTA, pH adjusted to 7.3 with CsOH. To activate the TRPC3 channel, current cells were challenged with 100 μM carbachol or 100 μM 1-oleoyl-2-acetyl-sn-glycerol (OAG). For RBL-2H3 cells CRAC measurement, standard protocols and buffers were modified from Derler et al. (2009). In brief voltage ramps from −90 to +90 mV over 1 s (holding potential +30 mV) were applied and controlled by pClamp software. ECS contained (in mM) 130 NaCl, 5 CsCl, 1 MgCl2, 10 HEPES, 10 glucose, 20 CaCl2 at pH 7.4. ICS was comprised of 3.5 MgCl2, 145 caesium methanesulphonate, 8 NaCl, 10 HEPES, 20 EGTA at pH 7.2. Experiments in HEK-293 cells expressing STIM1 and Orai1 to reconstitute the CRAC pore were done as in Muik et al. (2008). ECS contained (in mM) 145 NaCl, 5 CsCl, 1 MgCl2, 10 HEPES, 10 Glucose, 10 CaCl2 at pH 7.4. ICS was comprised of 3.5 MgCl2, 145 caesium methanesulphonate, 8 NaCl, 10 HEPES, 20 EGTA at pH 7.2. For measuring the sodium currents in divalent-free conditions, protocols and buffers from Bergsmann et al. (2011) were used. If not mentioned otherwise for the experiments, cells were pre-incubated for 3 min and measured in the presence of either 3 μM Pyr2, Pyr3, Pyr6 or Pyr10.
Measurement of intercellular Ca2+
Cells were loaded with 2 μM Fura-2-AM (Molecular Probes, LifeTechnologies, Vienna, Austria) for 45 min in Optimem® medium (Invitrogen) and washed. Cells were continuously perfused with Ca2+ free ECS buffer for any cell type as above and either challenged by depletion of intracellular Ca2+ stores with 1 μM thapsigargin (RBL-2H3) for 5 min, or by acute application of 100 μM carbachol (HEK293). Pyrazole compounds were supplied in corresponding concentration in the buffer at least 5 min before the start of the measurement. Agonists as well as inhibitors remained present continuously. For Ca2+ re-addition, 2 mM extracellular CaCl2 was added. Excitation light was supplied via a Polychrome II polychromator (TILL Photonics, Gräfeling, Germany) and emission was detected by a Sensicam CCDcamera (PCO Computer Optics, Kelheim, Germany). Ca2+-sensitive fluorescence ratios (340 nm/380 nm excitation; 510 nm emission) were recorded and analysed by using Axon Imaging Workbench (Axon Instruments).
NFAT translocation
For NFAT imaging experiments, coverslips with transfected RBL-2H3 cells were transferred into nominally Ca2+-free RBL-2H3 buffer (described previously), and incubated with thapsigargin (1 μM) for 5 min to deplete the internal Ca2+ stores. NFAT translocation was triggered by adding 2 mM extracellular CaCl2. Pyrazole compounds were present in all buffers at 10 μM. Basal NFAT localization was assessed from cells before store depletion.
GFP-NFAT translocation was monitored (488 nm laser excitation) with standard fluorescence microscopy (Zeiss Axiovert equipped with Coolsnap HQ, Zeiss, Oberkochen, Germany). Nuclear/cytosol fluorescence intensity ratios of cells were calculated with ImageJ software (Bethesda, MA, USA; http://imagej.nih.gov/ij/, 1997–2012). Nuclei were delineated in phase contrast images and visualized by DAPI staining. DAPI (AppliChem, Darmstadt, Germany) was used according to the manufacturer's recommendations on para-formaldehyde fixed cells.
Degranulation assay
Degranulation of RBL-2H3 cells was measured by determining the level of secreted β-hexosaminidase similar to Law et al. (2011). Cells were seeded out in 24-well plates and grown to confluency. All subsequent incubations were done at 37°C. After being washed with 2 mM CaCl2 containing RBL-2H3 buffer (described previously), cells were incubated with either 3 or 10 μM pyrazole compounds for 15 min. Ionomycin was added to a final concentration of 0.4 μM per well and the incubation continued for a further 30 min. Aliquots (30 μL) of each well's supernatant were transferred to 96-well plates containing 50 μL of 1.3 mg·mL−1 p-nitrophenyl-n-acetyl-β-d-glucosaminide in 100 mM Na-citrate buffer (pH 4.5) as substrate. Aliquots of untreated and triton incubated (final concentration 2% triton, used in 1:10 dilution for substrate assay) cells were used as basal and maximal granula content references. Control experiments were performed to test the potential interference of compounds with ionomycin function by elevating the ionomycin concentration 10-fold, which results in Ca2+ entry mediated mainly by direct Ca2+ transport via ionomycin pores in the plasma membrane. After 45 min, the enzymatic reaction was stopped with 50 μL 0.4 M glycine buffer (at pH 10.7). Absorbance was measured at 405 nm in a plate reader.
Drug target nomenclature, curve fitting, data evaluation and statistics
Nomenclature for drug targets conforms to BJP's Guide to Receptors and Channels (Alexander et al. 2011). Dose-response curves were fitted according a standard 4-parameter logistic equitation using Sigma Plot 12®. Data are presented as mean values ± SEM and were tested for statistical significance using one-way ANOVA in Sigma Plot® (Systat Software, Erkrath, Germany). *Indicates P < 0.05, **P < 0.01 and ***P < 0.001.
Results
Nomenclature and structure of pyrazole compounds
Pyrazole compounds were designated according to the previous literature and for novel structures following order of synthesis in our laboratory. Correct chemical nomenclature is Pyr2/BTP2 – N-(4-(3,5-bis(trifluoromethyl)-1H-pyrazole-1-yl)phenyl)-4-methyl-1,2,3-thiadiazole-5-carboxamide; Pyr3 – ethyl 1-(4-(2,3,3-trichloroacrylamido)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate; Pyr6 – N-(4-(3,5-bis(trifluoromethyl)-1H-pyrazole-1-yl)phenyl)-3-fluoroisonicotinamide; Pyr10 – N-(4-(3,5-bis(trifluoromethyl)-1H-pyrazole-1-yl)phenyl)-4-methylbenzenesulfonamide (Obermayer et al., 2011). As illustrated in Figure 1, Pyr2, Pyr6 and Pyr10 share the common backbone of BTPs with a trifluoromethyl-group on position C3 and C5 of the pyrazole ring, whereas Pyr3 lacks this at position C3 and is substituted with a carboxylate group on position C4 of the pyrazole ring. Estimating the octanol-water-coefficient (logP) of the four compounds by determining the hydro-/lipophilic behaviour revealed two groups: Pyr2, Pyr3 and Pyr6 (values 3.87, 3.89 and 3.84) versus Pyr10 with a higher value of 5.14. Calculation of the total molecular polar surface area as a second indicator of membrane permeability yielded 59.81 Å (Pyr6), 63.99 Å (Pyr10), 72.71 Å (Pyr2) and 73.23 Å (Pyr3), and did not suggest substantial differences in membrane permeability among these compounds.
Figure 1.
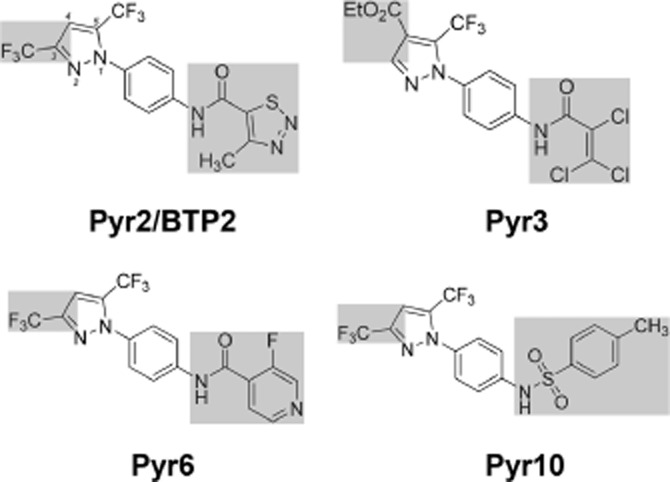
Chemical structures of the pyrazole compounds tested. Molecular structures suggested of importance for Ca2+ channel blocking activity are highlighted.
Potency and selectivity of pyrazole compounds in ROCE and SOCE model systems
The group of Mori reported a high selectivity of Pyr3 for TRPC3 channels as compared to channels formed by other TRPC species (Kiyonaka et al., 2009). The potency and selectivity of this compound for classical SOCE, which was repeatedly suggested to overlap with TRPC signalling, has so far not been delineated. By contrast, another pyrazole, Pyr2 (BTP2), is commonly accepted as a pharmacological tool as well as an inhibitor of the classical SOCE pathways mediated by Orai1 (Zitt et al., 2004). Here we set out to compare the TRPC/Orai1 selectivity of Pyr2 and Pyr3 along with Pyr6, and a newly synthesized structure designated as Pyr10 (Figure 1). The inhibitory potential of these four compounds on TRPC3-ROCE and SOCE was tested by measuring Ca2+ entry into stimulated cells using Fura-2 and classical Ca2+ re-addition protocols. As a ROCE model, HEK293 cells overexpressing YFP-tagged TRPC3, as a typical lipid/second messenger-controlled TRPC, were used. TRPC3 channels were activated by stimulating endogenous muscarinic receptors with 100 μM carbachol (Mundell and Benovic, 2000; Thyagarajan et al., 2001). As a SOCE model native RBL-2H3 cells were employed, which display the prototypical STIM1/Orai1-mediated SOCE based on the classical CRAC conductance (Hoth and Penner, 1992; Calloway et al., 2009). In this system, SOCE was activated by passively depleting the intracellular stores with thapsigargin before Ca2+ re-addition.
Table 1 and Figure 2 show the calculated IC50 values obtained from fitted dose-response curves. Notably, Pyr3 lacked selectivity for TRPC3-ROCE in this test and displayed a similar potency for SOCE inhibition in native RBL-2H3 cells like Pyr2, which, in line with earlier reports, was found to be sevenfold [0.85 orders of magnitude (OM)] more potent in the SOCE than in the TRPC3-ROCE model (Sweeney et al., 2009). Direct inhibition of the STIM1/Orai1-mediated CRAC currents was confirmed by reconstitution of the CRAC pore in HEK293 cells. Inhibition of CRAC elicited by passive store depletion using EGTA (10 mM) in the pipette solution, was observed upon acute administration in a rapid and dose-dependent manner. This effect was not readily reversed upon washout and was also evident in a divalent-free condition (Supporting Information Figure S1) representing monovalent permeation through CRAC channels (Hoth and Penner, 1993).
Table 1.
IC50 of Ca2+ influx inhibition by pyrazoles in carbachol-stimulated YFP-TRPC3-transfected HEK293 cells for ROCE or thapsigargin-depleted native RBL-2H3 cells for SOCE
| Pyrazole | IC50 (TRPC3-ROCE) (μM) | IC50 (SOCE) (μM) |
|---|---|---|
| Pyr2 | 4.21 | 0.59 |
| Pyr3 | 0.54 | 0.54 |
| Pyr6 | 18.46 | 0.49 |
| Pyr10 | 0.72 | 13.08 |
Values were calculated by fitting the data shown in Figure 2 with a 4-parameter logistic function Data points were derived from 3–5 individual experiments and a total of 29–80 cells.
Figure 2.

Concentration-dependence of Ca2+ entry inhibition by pyrazoles in TRPC3 overexpressing HEK293 cells and in native RBL-2H3 mast cells representing model systems of TRPC3-ROCE and SOCE. Fura-2 Ca2+ imaging experiments for YFP-TRPC3 transfected cells (ROCE model, blue symbols and line) or native RBL-2H3 (SOCE model, red symbols and line) and fitted dose-response curves. Inhibition is presented as percentage of the peak Ca2+ entry level measured in the absence of pyrazole compounds. Mean values ± SEM are given. Each value was derived from 3–5 individual experiments and a total of 29–80 cells. HEK293 YFP-TRPC3 cells were challenged with carbachol (100 μM) to stimulate ROCE. Native RBL-2H3 cells were incubated with thapsigargin (1 μM) before the experiment to elucidate SOCE by depleting intracellular Ca2+ stores.
The most striking selectivity was obtained with Pyr6 and Pyr10. In line with earlier reports suggesting Pyr6 is a selective SOCE inhibitor (Yonetoku et al., 2008; Sweeney et al., 2009), Pyr6 displayed 37-fold (1.58 OM) higher potency for RBL SOCE than for TRPC3 ROCE, with an IC50 comparable to that of Pyr2 and Pyr3. Interestingly, the sulphonamide-substituted compound Pyr10, by contrast, showed substantial selectivity for TRPC3-ROCE, being 18-fold (1.25 OM) more potent as compared with the SOCE model.
Pyr6 and Pyr10 – tools to distinguish between TRPC3 ROCE and STIM1/Orai1 SOCE
As Pyr6 and Pyr10 affected cellular Ca2+ handling in a divergent manner, we investigated their effects on TRPC and CRAC channels more directly by electrophysiology. In line with the Fura-2 imaging results, pre-incubation of cells with Pyr2 and Pyr3 at 3 μM completely eliminated the Ca2+ entry-mediating conductances in both cell types (Figure 3 and Table 2). In contrast, Pyr6 and Pyr10 interfered with these conductances in a selective manner. While completely inhibiting CRAC currents, Pyr6 at 3 μM diminished TRPC3 currents to only 52%. Pyr10 (3 μM) eliminated TRPC3 currents but suppressed SOCE in RBL-2H3 cells to only 60% of control. These results suggest Pyr6 and Pyr10 are potential tools for selectively affecting TRPC3 or Orai1 channels and demonstrate that Pyr3 clearly lacks selectivity. Inhibition of TRPC3 by Pyr10 was also observed when the channel was activated directly by the lipid mediator OAG (Supporting Information Fig. S2), demonstrating that this effect is not dependent on intracellular Ca2+ stores.
Figure 3.

Differences in the selectivity of Pyr6 and Pyr10 for blocking TRPC3- and STIM1/Orai1-mediated membrane currents. (A) Top panel: time course of currents measured at −90 mv (n ≥ 7 experiments for each condition) after incubation with pyrazoles for 5 min and stimulation of HEK-293 cells transiently expressing TRPC3 with carbachol (100 μM). Lower panel: representative I-V relationship of carbachol-stimulated currents in cells pretreated with pyrazole compound versus control. (B) Top panel: time course of CRAC currents in native RBL-2H3 cells after incubation with pyrazoles and store depletion with EGTA in the patch pipette (n ≥ 6). Lower panel: representative I-V relationship of EGTA-induced currents in store-depleted RBL-2H3 cells, pretreated with pyrazole compound, versus control. Mean values ± SEM are given. In all experiments, pyrazoles were administrated at 3 μM concentrations. Symbols/colours: filled black square/black trace – untreated control; open orange square/orange trace – Pyr2; filled green triangle/green trace – Pyr3; filled red circle/red trace – Pyr6; filled blue circles/blue trace – Pyr10.
Table 2.
Peak TRPC3-ROCE and SOCE currents in the absence and presence of 3 μM pyrazoles
| Pyrazole | ITRPC3-ROCE (pA/pF) | Statistical significance | ISOCE (pA/pF) | Statistical significance |
|---|---|---|---|---|
| Pyr2 | −1.54 ± 0.53 | * | −1.13 ± 0.16 | *** |
| Pyr3 | −2.27 ± 0.96 | * | −1.45 ± 0.19 | *** |
| Pyr6 | −9.50 ± 2.66 | n.a. | −1.31 ± 0.15 | *** |
| Pyr10 | −1.01 ± 0.57 | * | −4.49 ± 0.75 | n.a. |
| no pyrazole | −18.50 ± 3.47 | * | −7.50 ± 0.64 | *** |
Currents were measured at maximal carbachol activation of ROCE in YFP-TRPC3-transfected HEK-293 cells or at −85 mV 300 s after perforating the cell for fully developed SOCE in native RBL-2H3 cells. Statistical significance was calculated compared with Pyr6-incubated cells for TRPC3-ROCE and compared with Pyr10 for SOCE. Values are mean values of net-currents (measured current minus basal current) ± SEM. (*P < 0.05, ***P < 0.001). n.a., not available.
Physiological consequences of Pyr-mediated inhibition of SOCE in RBL-2H3 cells
Due to the key role for Ca2+, which acts as an important second messenger for transduction of plasma membrane signals to cellular functions including gene transcription, the effects of the four compounds on NFAT translocation and mast cell degranulation were examined. As shown in Figure 4A,B, SOCE inhibition by 10 μM Pyr2, Pyr3 or Pyr6 clearly inhibited NFAT translocation, whereas the selective TRPC3-ROCE inhibitor Pyr10 failed to suppress NFAT activation significantly.
Figure 4.

Pyrazol effects on NFAT activation and degranulation in RBL-2H3 cells. (A) Mean values ± SEM of NFAT nuclear to cytosolic ratio (n ≥ 18 cells for each condition) Values were determined after depletion of intracellular Ca2+ stores with thapsigargin and re-addition of extracellular Ca2+ for control (thapsigargin only). Pyrazole-treated (10 μM) cells as indicated are compared with basal condition (dashed line). (B) Representative images of NFAT localization (GFP), localization of the nuclei (DAPI staining), an overlay of both and DIC microscopy images for basal, control and pyrazole-treated cells after paraformaldehyde fixation. (C) Mean values ± SEM of degranulation (n = 3 experiments from different passages) shown for control (ionomycin 0.4 μM only), for the effect of Pyr3 on cells stimulated by a 10-fold higher concentration of ionomycin (control for lack of interference of the pyrazole with ionomycin pore formation) and pyrazole-treated cells (3 μM – dark grey; 10 μM – light grey) stimulated with 0.4 μM ionomycin to initiate SOCE. Basal degranulation is indicated by a dashed line. Asterisks indicate statistical significant differences and refer to levels measured at 10 μM concentration of pyrazoles. (**P < 0.01, ***P < 0.001).
The degranulation of RBL-2H3 initiated by ionomycin was inhibited by all the pyrazole compounds, with Pyr10 being the weakest inhibitor. The potent SOCE inhibitors Pyr2, Pyr3 and Pyr6 substantially reduced degranulation at 3 μM and suppressed responses down to basal levels at 10 μM, while Pyr10 (10 μM) prevented degranulation to only 65% of control. Therefore, these results clearly demonstrate that Orai1-mediated SOCE is a master regulator of degranulation and Ca2+-dependent transcriptional control in RBL-2H3 cells but the TRPC permeation pathways do not significantly contribute to this process in these cells.
Discussion and conclusion
Pharmacological dissection of SOCE and ROCE pathways
In view of the currently incomplete understanding of the molecular structures involved in agonist/receptor-operated control of Ca2+ entry into many native tissues, it is highly desirable to identify potent inhibitors for specific Ca2+ channel pore structures that are controlled via receptor-phospholipase C-dependent mechanisms. A principal problem is the typical simultaneous activation of pathways activated by Ca2+ store depletion and by second messenger mechanisms such as generation or depletion of lipid mediators. These mechanisms not only overlap upon stimulation of PLC but may also both involve TRPC proteins as essential components (Putney, 2004). Nonetheless, pharmacological dissection of these mechanisms appears possible, based on different pore complexes of receptor/second messenger-operated and store-operated channels. Prototypical molecules mediating these Ca2+ entry mechanism are on the one hand TRPC3, which, upon overexpression, forms diacylglycerol-regulated non-selective cation channels and on the other hand Orai1, which forms a highly Ca2+ selective channel activated by interaction of the ER Ca2+ sensor STIM1 in response to a reduction in ER Ca2+ levels. Here we tested several potential blockers of TRPC and Orai channels for selectivity, including a recently synthesized, novel pyrazole compound termed Pyr10. We report the ability of two pyrazole compounds, Pyr6 and Pyr10, to discriminate between receptor-operated TRPC3 and native STIM1/Orai1 channels. At low micromolar concentrations, the novel structure Pyr10 completely eliminated TRPC3 currents as well as Ca2+ entry while exerting modest effects on Orai-mediated responses. Interestingly, CRAC currents displayed a somewhat higher sensitivity to Pyr10, as expected from Ca2+ entry measurements. Pyr10 (3 μM) inhibited CRAC currents by about 50%, while Ca2+ entry was reduced by only 30%. This difference could be based on the different experimental conditions, which to some extent affect channel regulation and may modulate drug sensitivity. Pyr6 displayed inverse selectivity, resulting in weak inhibition of TRPC3 currents at concentrations that eliminated Orai-mediated currents. Interestingly, the recently proposed TRPC3-selective blocker Pyr3 (Kiyonaka et al., 2009), as well as Pyr2, a previously suggested inhibitor of store-operated Ca2+ entry and inhibitor of the classical CRAC current in immune cells, was barely able to distinguish between TRPC3 and Orai1 (Zitt et al., 2004; He et al., 2005; Kiyonaka et al., 2009). Our current findings confirm the principle activity of pyrazole derivatives as inhibitors of CRAC currents and, thus, of Orai channels. For Pyr3, we found that this compound rapidly suppresses both native CRAC currents as well as heterologously reconstituted CRAC currents upon acute administration (Supporting Information Fig. S1A,B). The rapid inhibitory effect of Pyr3 observed upon extracellular administration may be interpreted as an interaction of the pyrazole with an extracellular target site at the Orai/CRAC channel complex. A similar conclusion was reached in a study characterizing the effects of Pyr2 on CRAC channels in T lymphocytes (Zitt et al., 2004). The observation that pyrazoles are able to inhibit CRAC channels after complete activation in response to store depletion, argues against suppression of the channel's activation process. Nonetheless, interference of the pyrazoles with inactivation or certain regulatory processes cannot be excluded at present. Consistent with inhibition of Orai channel activity, Pyr2, Pyr3 or Pyr6 substantially inhibited typical Orai downstream signalling events in RBL mast cells (NFAT activation and degranulation) activated by passive store depletion. Unequivocally, Pyr2 and Pyr3 also inhibit TRPC-mediated Ca2+ entry (He et al., 2005; Kiyonaka et al., 2009), and investigations performed in human neutrophils (Salmon and Ahluwalia, 2010), pancreatic and salivary gland acinar cells (Kim et al., 2011) demonstrated inhibition of SOCE by Pyr3, which was interpreted as a contribution of TRPC proteins to the SOCE phenomena (Salmon and Ahluwalia, 2011). However, insufficient selectivity of Pyr3 in terms of discrimination between TRPC and Orai channel pores as demonstrated here, weakens this conclusion.
The observed selectivity of Pyr6 and Pyr10 suggests that these compounds may be useful to identify and analyse TRPC- and Orai-mediated conductances in native tissues. Our results obtained in RBL-2H3 mast cells and STIM1/Orai1-expressing HEK293 cells were in line with the concept that store-operated Ca2+ entry in these two cell systems occurs via the same channels, which are characterized by sensitivity to Pyr6 being clearly higher than to Pyr10. By contrast, TRPC3 homomeric pore structures are highly sensitive to Pyr10 but weakly sensitive to Pyr6. Importantly, a test for inhibitory effects of Pyr6 and Pyr10 on homomeric channels of other TRPC isoforms such as TRPC4, 5 and 6, revealed a low potency (IC50 > 10 μM) at these channels, indicating that Pyr10 exhibits a significant TRPC subtype selectivity (Supporting Information Fig. S3).
We suggest a pyrazole sensitivity of Pyr6 > Pyr10 as a characteristic of Orai1-mediated Ca2+ entry. As RBL-2H3 cells express TRPC genes including TRPC3 (Ma et al., 2008), our results may be taken as an indication that TRPC3 does not contribute to store-operated Ca2+ entry in mast cells. Nonetheless, we cannot exclude contribution of a TRPC3 containing channel complex in local Ca2+ signalling events that are not detectable as global cellular Ca2+ changes but could be pivotal for certain downstream signalling processes as recently reported for cardiac TRPC3s (Poteser et al., 2011). Our finding of NFAT translocation being highly sensitivity to Pyr6 but not to Pyr10 is in line with the concept that in RBL-2H3 cells, NFAT is mainly controlled via the CRAC/Orai Ca2+ entry pathway, as recently suggested by Kar et al. (2011). It is of note that pyrazole structures have initially been recognized as effective inhibitors of NFAT signalling via an ill-defined mechanism downstream of Ca2+ signalling (Djuric et al., 2000; Trevillyan et al., 2001). Here we report that the inhibitory effects of Pyr6 and Pyr10 correlate well with their efficacy as CRAC/Orai1 inhibitors. The observation that Pyr6 was more potent than Pyr10 as a suppressant of mast cell degranulation corroborates the view that Orai channels represent the main source of Ca2+ for exocytosis in RBL-2H3 cells (Ma et al., 2008). Consistent with previous reports, Pyr6 was found to be highly effective as an inhibitor of immune cell transcriptional activation and cytokine production (Ishikawa et al., 2003; Birsan et al., 2004; Shirakawa et al., 2010; Law et al., 2011), underscoring the potential value of this chemical structure for the development of potent immune modulators (Chen et al., 2002; Zitt et al., 2004).
Structural basis of selective modulation of Ca2+ signalling by pyrazoles
The structural basis of Ca2+ entry block by pyrazoles has recently been analysed. The C3 position in the pyrazole ring was recognized as a critical determinant of the inhibitory effects on mast cell degranulation, most likely corresponding to inhibition of SOCE. Block of SOCE apparently requires substitution with bulky, electron-drawing groups in C3. However, a similar substitution in C5, as present in Pyr3, by itself was found to be insufficient to provide SOCE blocking activity. (Law et al., 2011) Our finding of a high SOCE inhibiting potency of Pyr3 indicates that the introduction of an ethyl-carboxylate in C4 of Pyr3 restores inhibitory potency, although appropriate C3 substitution is lacking. Interestingly, substitution in C4 was suggested of importance for the Pyr3 potency regarding inhibition of TRPC family members (Kiyonaka et al., 2009). Moreover, this later study also proposed the amid-bond linked side-group as pivotal for TRPC subtype selectivity, with the tricholoraryl-substitution in Pyr3 as the TRPC3 selectivity encompassing structural element. This amid-bond linked side group is, according to our results, also a potential structural determinant for the CRAC/SOCE inhibitory action, as this structural feature is lacking in the weak SOCE inhibitor Pyr10. Notably, Pyr10 contains a sulphonamide-linked side-group at the BTP backbone. One might speculate about substantial changes within the molecule generated by different interactions between the backbone and the side-group resulting in structures that discriminate between the two channel types. As electronegativity and polarity of the amine-linked side groups in Pyr3 and Pyr10 are similar, it is tempting to speculate that these structures are essential for inhibition of TRPC3-ROCE with high potency. Although a direct interaction of pyrazoles with TRPC3, presumably involving association to an extracellular binding domain, appears likely and has been demonstrated for Pyr3 (Kiyonaka et al., 2009), the exact molecular basis of pyrazole action at the TRPC channel remains to be elucidated.
In this study, we obtained evidence for a possible value of pyrazoles as selective modulators of cellular Ca2+ handling, widening the view on both their therapeutic potential as immunosuppressants as well as their utility for experimental dissection of Ca2+ signalling pathways. In aggregate, we introduce a novel pharmacological approach to distinguish between second messenger-gated TRPC-mediated and store-operated, Orai-mediated Ca2+ entry using selective pyrazole compounds including Pyr10 as a novel TRPC3-selective inhibitor. The identified ability of certain pyrazole structures to discriminate between a receptor- and store-operated Ca2+ signalling pathway is expected to pave the way towards both better experimental analysis and understanding of Ca2+ entry mechanisms in native tissues and for the development of novel therapeutic strategies.
Acknowledgments
This work was funded by FWF (Austrian research fund) projects P21925-B19 (to K G), P22565-B18 (to C R) and the DK+ Metabolic and Cardiovascular Disease grant W1226-B18. We like to thank Renate Schmidt and Ines Neubacher for their technical assistance, Sonia Stürmer for support with preliminary experiments, as well as RH Kehlenbach and M Zhu for kindly providing the GFP-NFAT and GFP-TRPC6 construct.
Glossary
- BTP
3,5-bis(trifluoromethyl)pyrazole derivative
- CRAC
Ca2+ release activated current
- DAG
diacylglycerol
- ER
endoplasmatic reticulum
- NFAT
nuclear factor of activated T-cells
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- OM
orders of magnitude
- ROCE
receptor operated Ca2+ entry
- SOCE
store-operated Ca2+ entry
- STIM1
stromal interaction molecule 1
- TRPC channel
transient receptor potential canonical channel
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Pyr3 acutely blocks a reconstituted CRAC pore in a dose-dependent, non-reversible manner as well as affecting sodium influx through it. (A) Time course of measured currents (n = 4 cells) of acute Pyr3 inhibition of CRAC in a HEK293 cell system expressing STIM1 and Orai1 to reconstitute the CRAC pore. (B) Time course of acute Pyr3 inhibition (3μM) in native RBL-2H3 cells (n = 6 cells). (C) Time course of currents (n = 8 cells) with acute Pyr3 inhibition of a reconstituted CRAC pore. Pyrazole compound was applied for only a limited time to examine reversibility of inhibitory effect. (D) Time course of Pyr3-effect (10 μM) on divalent-free sodium current through CRAC-pore after activation of current in Ca2+ containing buffer (n = 4 cells). All values are mean values ± SEM.
Figure S2 Pyr10 potently blocks DAG-mediated TRPC3 membrane currents. Mean values of OAG-induced (100 μM) TRPC3 currents in HEK293 cells in the absence and presence of 3 μM Pyr10. Mean values ± SEM of (n = 6).
Figure S3 Selectivity of Pyr10 and Pyr6 on TRPCs. Average inhibition of peak calcium entry level measured by Fura 2 Ca2+ imaging at 10 μM Pyr6 or Pyr10 in HEK293 cells overexpressing TRPC4beta-YFP, YFP-TRPC5 or GFP-TRPC6. HEK293 cells were challenged with carbachol (100 μM) to stimulate calcium entry and pre-incubated for 5 min with corresponding pyrazole compounds. Values are presented as percentage of non-inhibited carbachol-stimulated cells from the same experiments and were derived from a total of 27–55 cells from three individual experiments.
References
- Abramowitz J, Birnbaumer L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009;23:297–328. doi: 10.1096/fj.08-119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsmann J, Derler I, Muik M, Frischauf I, Fahrner M, Pollheimer P, et al. Molecular determinants within N terminus of Orai3 protein that control channel activation and gating. J Biol Chem. 2011;286:31565–31575. doi: 10.1074/jbc.M111.227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Birsan T, Dambrin C, Marsh KC, Jacobsen W, Djuric SW, Mollison KW, et al. Preliminary in vivo pharmacokinetic and pharmacodynamic evaluation of a novel calcineurin-independent inhibitor of NFAT. Transpl Int. 2004;17:145–150. doi: 10.1007/s00147-003-0676-1. [DOI] [PubMed] [Google Scholar]
- Calloway N, Vig M, Kinet JP, Holowka D, Baird B. Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol Biol Cell. 2009;20:389–399. doi: 10.1091/mbc.E07-11-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Smith ML, Chiou GX, Ballaron S, Sheets MP, Gubbins E, et al. TH1 and TH2 cytokine inhibition by 3,5-bis(trifluoromethyl)pyrazoles, a novel class of immunomodulators. Cell Immunol. 2002;220:134–142. doi: 10.1016/s0008-8749(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca(2)+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca(2)+ signals required for specific cell functions. PLoS Biol. 2011;9:e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW., Jr Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem. 2008;283:19265–19273. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Fahrner M, Carugo O, Muik M, Bergsmann J, Schindl R, et al. Increased hydrophobicity at the N terminus/membrane interface impairs gating of the severe combined immunodeficiency-related ORAI1 mutant. J Biol Chem. 2009;284:15903–15915. doi: 10.1074/jbc.M808312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Capite J, Parekh AB. CRAC channels and Ca2+ signaling in mast cells. Immunol Rev. 2009;231:45–58. doi: 10.1111/j.1600-065X.2009.00808.x. [DOI] [PubMed] [Google Scholar]
- Djuric SW, BaMaung NY, Basha A, Liu H, Luly JR, Madar DJ, et al. 3,5-Bis(trifluoromethyl)pyrazoles: a novel class of NFAT transcription factor regulator. J Med Chem. 2000;43:2975–2981. doi: 10.1021/jm990615a. [DOI] [PubMed] [Google Scholar]
- Glasnov TN, Groschner K, Kappe CO. High-speed microwave-assisted synthesis of the trifluoromethylpyrazol-derived canonical transient receptor potential (TRPC) channel inhibitor Pyr3. ChemMedChem. 2009;4:1816–1818. doi: 10.1002/cmdc.200900304. [DOI] [PubMed] [Google Scholar]
- Graziani A, Poteser M, Heupel WM, Schleifer H, Krenn M, Drenckhahn D, et al. Cell-cell contact formation governs Ca2+ signaling by TRPC4 in the vascular endothelium: evidence for a regulatory TRPC4-beta-catenin interaction. J Biol Chem. 2010;285:4213–4223. doi: 10.1074/jbc.M109.060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harteneck C, Gollasch M. Pharmacological modulation of diacylglycerol-sensitive TRPC3/6/7 channels. Curr Pharm Biotechnol. 2011;12:35–41. doi: 10.2174/138920111793937943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He LP, Hewavitharana T, Soboloff J, Spassova MA, Gill DL. A functional link between store-operated and TRPC channels revealed by the 3,5-bis(trifluoromethyl)pyrazole derivative, BTP2. J Biol Chem. 2005;280:10997–11006. doi: 10.1074/jbc.M411797200. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa J, Ohga K, Yoshino T, Takezawa R, Ichikawa A, Kubota H, et al. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J Immunol. 2003;170:4441–4449. doi: 10.4049/jimmunol.170.9.4441. [DOI] [PubMed] [Google Scholar]
- Jardin I, Lopez JJ, Salido GM, Rosado JA. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J Biol Chem. 2008;283:25296–25304. doi: 10.1074/jbc.M802904200. [DOI] [PubMed] [Google Scholar]
- Kar P, Nelson C, Parekh AB. Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem. 2011;286:14795–14803. doi: 10.1074/jbc.M111.220582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Lee KP, Yang D, Shin DM, Abramowitz J, Kiyonaka S, et al. Genetic and pharmacologic inhibition of the Ca2+ influx channel TRPC3 protects secretory epithelia from Ca2+-dependent toxicity. Gastroenterology. 2011;140:2107–2115. doi: 10.1053/j.gastro.2011.02.052. 2115 e2101-2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci USA. 2009;106:5400–5405. doi: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Morales JL, Mottram LF, Iyer A, Peterson BR, August A. Structural requirements for the inhibition of calcium mobilization and mast cell activation by the pyrazole derivative BTP2. Int J Biochem Cell Biol. 2011;43:1228–1239. doi: 10.1016/j.biocel.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier L, Trebak M, Putney JW., Jr Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium. 2008;43:506–514. doi: 10.1016/j.ceca.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci USA. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Peng Z, Hiragun T, Iwaki S, Gilfillan AM, Beaven MA. Canonical transient receptor potential 5 channel in conjunction with Orai1 and STIM1 allows Sr2+ entry, optimal influx of Ca2+, and degranulation in a rat mast cell line. J Immunol. 2008;180:2233–2239. doi: 10.4049/jimmunol.180.4.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- Mundell SJ, Benovic JL. Selective regulation of endogenous G protein-coupled receptors by arrestins in HEK293 cells. J Biol Chem. 2000;275:12900–12908. doi: 10.1074/jbc.275.17.12900. [DOI] [PubMed] [Google Scholar]
- Ng SW, di Capite J, Singaravelu K, Parekh AB. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+ influx through Ca2+ release-activated Ca2+ channels. J Biol Chem. 2008;283:31348–31355. doi: 10.1074/jbc.M804942200. [DOI] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- Obermayer D, Glasnov TN, Kappe CO. Microwave-assisted and continuous flow multistep synthesis of 4-(pyrazol-1-yl)carboxanilides. J Org Chem. 2011;76:6657–6669. doi: 10.1021/jo2009824. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- Poteser M, Schleifer H, Lichtenegger M, Schernthaner M, Stockner T, Kappe CO, et al. PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc Natl Acad Sci USA. 2011;108:10556–10561. doi: 10.1073/pnas.1106183108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr The enigmatic TRPCs: multifunctional cation channels. Trends Cell Biol. 2004;14:282–286. doi: 10.1016/j.tcb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Salmon MD, Ahluwalia J. Discrimination between receptor- and store-operated Ca(2+) influx in human neutrophils. Cell Immunol. 2010;265:1–5. doi: 10.1016/j.cellimm.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Salmon MD, Ahluwalia J. Pharmacology of receptor operated calcium entry in human neutrophils. Int Immunopharmacol. 2011;11:145–148. doi: 10.1016/j.intimp.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Schindl R, Frischauf I, Kahr H, Fritsch R, Krenn M, Derndl A, et al. The first ankyrin-like repeat is the minimum indispensable key structure for functional assembly of homo- and heteromeric TRPC4/TRPC5 channels. Cell Calcium. 2008;43:260–269. doi: 10.1016/j.ceca.2007.05.015. [DOI] [PubMed] [Google Scholar]
- Shirakawa H, Sakimoto S, Nakao K, Sugishita A, Konno M, Iida S, et al. Transient receptor potential canonical 3 (TRPC3) mediates thrombin-induced astrocyte activation and upregulates its own expression in cortical astrocytes. J Neurosci. 2010;30:13116–13129. doi: 10.1523/JNEUROSCI.1890-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkins WG, Goel M, Estacion M, Schilling WP. Association of immunophilins with mammalian TRPC channels. J Biol Chem. 2004;279:34521–34529. doi: 10.1074/jbc.M401156200. [DOI] [PubMed] [Google Scholar]
- Sweeney ZK, Minatti A, Button DC, Patrick S. Small-molecule inhibitors of store-operated calcium entry. ChemMedChem. 2009;4:706–718. doi: 10.1002/cmdc.200800452. [DOI] [PubMed] [Google Scholar]
- Thyagarajan B, Poteser M, Romanin C, Kahr H, Zhu MX, Groschner K. Expression of Trp3 determines sensitivity of capacitative Ca2+ entry to nitric oxide and mitochondrial Ca2+ handling: evidence for a role of Trp3 as a subunit of capacitative Ca2+ entry channels. J Biol Chem. 2001;276:48149–48158. doi: 10.1074/jbc.M103977200. [DOI] [PubMed] [Google Scholar]
- Trebak M, Bird GS, McKay RR, Putney JW., Jr Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition. J Biol Chem. 2002;277:21617–21623. doi: 10.1074/jbc.M202549200. [DOI] [PubMed] [Google Scholar]
- Trevillyan JM, Chiou XG, Chen YW, Ballaron SJ, Sheets MP, Smith ML, et al. Potent inhibition of NFAT activation and T cell cytokine production by novel low molecular weight pyrazole compounds. J Biol Chem. 2001;276:48118–48126. doi: 10.1074/jbc.M107919200. [DOI] [PubMed] [Google Scholar]
- Woodard GE, Lopez JJ, Jardin I, Salido GM, Rosado JA. TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J Biol Chem. 2010;285:8045–8053. doi: 10.1074/jbc.M109.033605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonetoku Y, Kubota H, Miyazaki Y, Okamoto Y, Funatsu M, Yoshimura-Ishikawa N, et al. Novel potent and selective Ca2+ release-activated Ca2+ (CRAC) channel inhibitors. Part 3: synthesis and CRAC channel inhibitory activity of 4′-[(trifluoromethyl)pyrazol-1-yl]carboxanilides. Bioorg Med Chem. 2008;16:9457–9466. doi: 10.1016/j.bmc.2008.09.047. [DOI] [PubMed] [Google Scholar]
- Yuan JP, Kim MS, Zeng W, Shin DM, Huang G, Worley PF, et al. TRPC channels as STIM1-regulated SOCs. Channels (Austin) 2009;3:221–225. doi: 10.4161/chan.3.4.9198. [DOI] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitt C, Strauss B, Schwarz EC, Spaeth N, Rast G, Hatzelmann A, et al. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem. 2004;279:12427–12437. doi: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.