

Abstract
The development and progression of colorectal cancer (CRC) is a multi-step process, and the Wnt pathways with its two molecular gladiators adenomatous polyposis coli (APC) and β-catenin plays an important role in transforming a normal tissue into a malignant one. In this study, we aimed to investigate the role of aberrations in the APC and β-catenin genes in the pathogenesis of CRC in the Kashmir valley, and to correlate it with various clinicopathological variables. We examined the paired tumour and normal-tissue specimens of 86 CRC patients for the occurrence of aberrations in the mutation cluster region (MCR) of the APC gene and exon 3 of the β-catenin gene by polymerase chain reaction-single-strand conformation polymorphism (PCR-SSCP) and/or PCR-direct sequencing. Analysis of promoter hypermethylation of the APC gene was also carried out using methylation-specific PCR (MS-PCR). The overall mutation rate of the MCR of the APC gene among 86 CRC cases was 12.8 per cent (11 of 86). Promoter hypermethylation of APC was observed in 54.65 per cent (47 of 86) of cases. Furthermore, we found a significant association between tumour location, tumour grade and node status and the methylation status of the APC gene (p ≤ 0.05). Although the number of mutations in the APC and β-catenin genes in our CRC cases was very low, the study confirms the role of epigenetic gene silencing of the pivotal molecular gladiator, APC, of the Wnt pathway in the development of CRC in the Kashmiri population.
Keywords: Wnt pathway, APC, β-catenin, colorectal cancer, Kashmir, hypermethylation, mutations, Dukes' stage
Introduction
Colorectal cancer (CRC) is a major cause of mortality and morbidity, and the third most common malignancy in the world [1]. The incidence of this malignancy shows considerable variation among racially or ethnically defined populations in multiracial/ethnic countries. It is the fourth most common cancer in men and the third most common in women worldwide [1]. Kashmir has been reported as being a high-incidence area for gastrointestinal (GIT) cancers [2,3]. In the Kashmir valley, CRC represents the third most common GIT cancer after oesophageal and gastric cancer [4,5].
It has been suggested that CRC is a multi-step process which arises from cumulative aberrations of a number of different genes (including tumour suppressor genes, proto-oncogenes, DNA repair genes, the genes encoding growth factors and their receptors, cell cycle checkpoint genes and apoptosis-related genes) or from epigenetic changes in DNA at different stages of development and progression [6,7]. It is believed that mutations in the gene encoding adenomatous polyposis coli (APC) or that encoding β-catenin set the stage for the initiation and transformation of normal colonic epithelial cells. Further accumulation of mutations in other genes then contributes to the progression of cancer through adenoma - carcinoma - metastasis stages. The generally accepted model of CRC tumorigenesis for the majority of tumours has been a stepwise progression, in which mutations in APC are followed by several other mutations, including alterations in the genes encoding Kirsten ras (K-ras) and tumour protein 53 (TP53) [6-8]. During the accumulation of genetic changes, a complex signalling network is established among inactivated and activated cellular pathways [9].
The Wnt pathway regulates cell adhesion, morphology, proliferation, migration and structural remodelling [9,10] and plays an important role in a variety of cellular processes, including proliferation, differentiation, survival, apoptosis and cell motility [11]. Loss of regulation of the Wnt pathway has been implicated in the development of several types of cancers, including colon, lung, breast, thyroid and prostate cancers and leukemia [12-15]. Two of the most important gladiator molecules of the Wnt pathway are APC and β-catenin.
APC is a classical tumour suppressor gene, located on 5q21, containing 21 exons. The APC transcript is 9.0 kilobases (kb) in length and the most common isoform of the APC protein contains 2,843 amino acids, with a molecular weight of 310 kD. Exon 15 of APC is most important, as it comprises > 75 per cent of the coding sequence of APC and hence is the common target for both germline and somatic mutations, which usually span codons 1286-1513 of this exon [16,17]. This region represents the mutation cluster region (MCR), and 68-77 per cent of somatic mutations in APC occur in this region [18]. Mutations in APC are considered to be the earliest genetic aberrations in the initiation and progression of CRC,[7,9,19] and have also been found in ~60-80 per cent of sporadic carcinomas and adenomas [20,21]. Using mutant mouse models, various genetic studies have demonstrated that mutations in APC are responsible for intestinal tumorigenesis [22-24]. Homozygous APC mutations in mice lead to embryonic lethality,[23,25,26] and conditional deletion of the gene in the adult mouse disrupts homeostasis, not only in the intestines but also in other tissues [27-29].
In addition to the mutational inactivation, hypermethylation of the gene promoter is another important mechanism associated with gene silencing [30]. In many tumours the hypermethylation of CpG islands in gene promoters has been found to be a frequent epigenetic change in cancers, and is usually associated with the loss of transcription of APC [31-38]. Hypermethylation of the APC gene promoters has been reported in about 20-48 per cent of human CRCs [32,37,39,40].
The β-catenin gene is located at 3p22-p21.3 and encompasses 23.2 kb of DNA. It contains 16 exons, with a mRNA transcript of about 2343 base pairs (bp), encoding a 781-amino-acid-residue protein with a molecular weight of 92 kD [41]. This gene is mutated in up to 10 per cent of all sporadic CRC by point mutations or in frame deletions of the serine and threonine residues that are phosphorylated by glycogen synthase kinase 3-beta (GSK3β) [42]. These mutations result in the stabilisation of β-catenin and the activation of Wnt signalling. Mutations in the β-catenin gene occur in exclusivity to APC aberrations, as both molecules are components of the same pathway [19].
Based on the hypothesis that CRC carcinogenesis is a multi-step and multi-gene event, we designed this study to elucidate the role of APC and β-catenin in the development and progression of CRC in the Kashmiri population, and to correlate the gene aberrations and hypermethylation with the clinicopathological parameters of CRC cases.
Materials and methods
Patients and specimens
Out of 104 patients who were diagnosed with CRC by clinicians using either sigmoidoscopy or colonoscopy and confirmed by MRI, a total of 86 CRC tissue specimens, comprising tumour tissues and corresponding adjacent normal tissues as controls, were collected for analysis. All samples were surgically resected and collected fresh at the Department of Surgery of the Sher-I-Kashmir Institute of Medical Sciences, Srinagar, Kashmir. Tissue samples were divided into two parts; one part was sent for histopathological diagnosis and the other was snap-frozen at -70°C immediately until needed for further analysis. Only histopathologically confirmed cases were included for molecular analysis. No follow-up of the CRC patients was carried out after the curative surgery. Written informed consent was obtained from all the subjects (and/or their guardians) included in the study, recorded on a prede-signed questionnaire (available on request). The study was carried out in accordance with the principles of the Helsinki Declaration. The study protocol was approved by the Research Ethics Committee of the Sher-I-Kashmir Institute of Medical Sciences, Kashmir.
DNA isolation
Genomic DNA was extracted from blood and tissue samples (previously stored at -70°C) from CRC patients using DNA Extraction Kit II (Zymo Research, Orange, CA). The tissue for DNA extraction from the tumour sample was chosen by an experienced pathologist and was ascertained to comprise more than 90 per cent of the tumour cells.
Polymerase chain reaction (PCR)
APC and β-catenin gene analysis was carried out on all of the extracted DNA samples. The MCR region of APC, comprising codons 1260 to 1596, and exon 3 of β-catenin, which encompasses the region for GSK-3β phosphorylation, were amplified using specific oligonucleotide primers (Table 1). PCR was performed in a 50 μl total volume reaction mixture containing 50 ng of genomic DNA, 100 ng of each primer, 100 μM of each deoxynucleotide triphosphate (dNTP), 1.5 mM MgCl2, 10× Taq buffer and 2 U of Taq DNA polymerase (Fermentas Inc, Glen Burnie, MD). The conditions of PCR were as follows: initial denaturation at 95°C for 5 minutes, 35 cycles of denaturation at 95°C, annealing at 52-58°C (see Table 1) and extension at 72°C, for 30 seconds each, and final extension at 72°C for 7 minutes in a Biorad icycler. The PCR products were run on 2-3 per cent agarose gel and analysed under an ultraviolet illuminator.
Table 1.
Primer sequences used for the mutational analysis of β-catenin and APC genes in the Wnt pathway
| Gene | Amplicon | Primer sequence | Amplicon size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|
| β-catenin | Exon 3 | BCat F: 5'-ATG GAA CCA GAC AGA AAA GC-3' BCat R: 5'-GCT ACT TGT TCT TGA GTG AAG-3' |
200 | 58 |
| APC | Exon 15 | APC A F: 5'-CAGACTTATTGTGTAGAAGA- 3' APC A R: 5'-ATCCTGAAGAAAATTCAACA-3' |
295 for codons 1260 to 1359 |
52 |
| APC B F: 5'-AGGGTTCTAGTTTATCTTCA-3' APC B R: 5'-TCTGCTTGGTGGCATGGTTT-3' |
293 for codons 1339 to 1436 |
55 | ||
| APC C F: 5'-GGCATTATAAGCCCCAGTGA-3' APC C R: 5'-AAATGGCTCATCGAGGCTCA-3' |
290 for codons 1417 to 1516 |
55 | ||
| APC D F: 5'-ACTCCAGATGGATTTTCTTG-3' APC D R: 5'-GGCTGGCTTTTTTGCTTTAC-3' |
300 for codons 1497 to 1596 |
55 |
Mutation analysis of the APC gene
Mutation analysis of the APC gene was performed on all cases. Four sets of oligonucleotide primers that have previously been reported were used for fragment-wise amplification (APCA, B, C and D) of codons 1260 to 1596 [43]. All amplicons were 300 bp in length (Figure 1A-D, Table 1), and were then subjected to single-strand conformation polymorphism (SSCP) analysis. For the samples showing an aberrant band in the SSCP analysis, APC BF and APC DR primers were used to amplify the 890 bp target region (Figure 1E) and then were subjected to direct sequencing (Macrogen Inc, Seoul, Korea), including the original 300 bp amplicons; however, studying the aberrations of only the MCR was a limitation of this study.
Figure 1A-E.
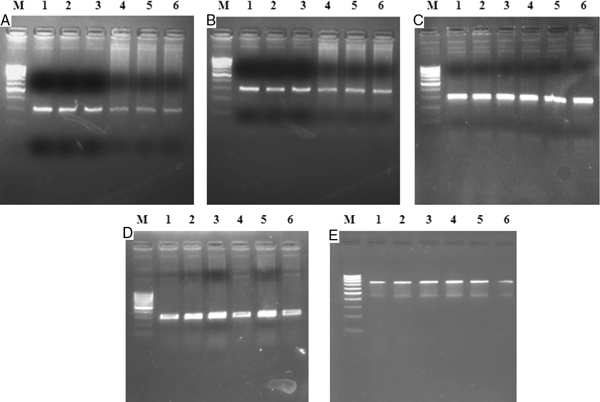
Representative gel picture of mutation cluster regions of APC gene comprising of Exon 15 APCA (295 bp); APCB (293 bp); APCC (290 bp); APCD (295 bp); and APC Full (890 bp) fragments. Lane M: Molecular size marker 100 bp (Middle Prominent Band = 500 bp) Lane 1-6: Amplified product from cancer samples.
Mutation analysis of the β-catenin gene
Genomic DNA from each sample was amplified by PCR using the previously reported primer pair, which amplified a 200-bp amplicon of exon 3 of the β-catenin gene (Figure 2A, Table 1) [43].
Figure 2A.
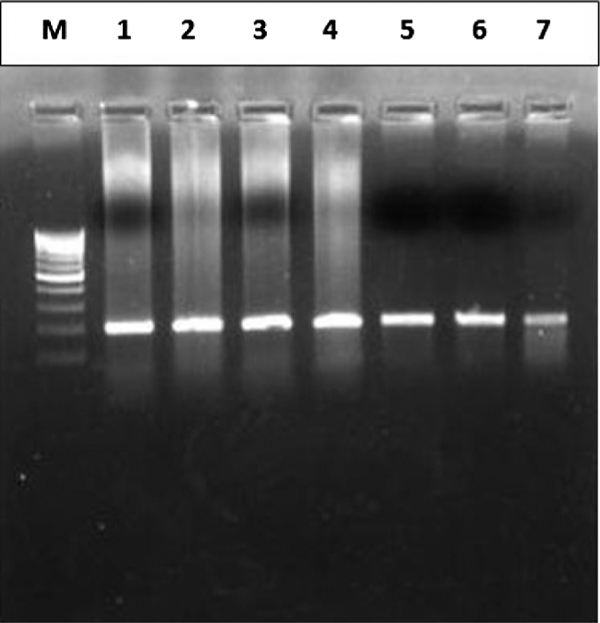
Amplified DNA fragment (200 bp amplicon) of exon 3 of β-catenin gene. Lane M: 100 bp molecular ladder, Lanes 1-7: Amplicons from different tumour tissues.
Base substitutions in codons 32 and 33 were further confirmed by the HinfI restriction endonuclease assay (Fermentas). The 200-bp PCR product for β-catenin contains two HinfI restriction endonuclease sites, yielding 7-bp, 55-bp and 138-bp DNA fragments after digestion of the wild-type allele. β-catenin gene mutations in codons 32 and 33 yield only 62-bp and 138-bp fragments after digestion because of ablation of the first HinfI site. The digested products were run on 10 per cent polyacrylamide gel electrophoresis (PAGE) (Figure 2B) to assess the digested fragments.
Figure 2B.
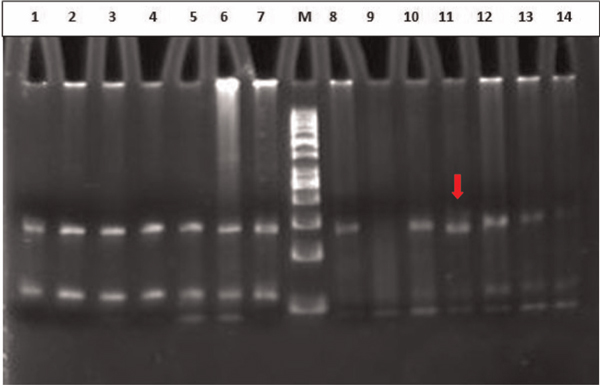
PAGE analysis of Hinf I digest of 200 bp amplicon of exon 3 of the β-catenin gene. Lane M: 100 bp molecular ladder, Lanes 1-14: HinfI digested amplicons of the β-catenin gene. Lane 11 shows the codon 32 mutant.
SSCP analysis
SSCP analysis of PCR products was carried out on 6 per cent non-denaturing polyacrylamide gel (PAG) utilising either non-radioactive silver staining or radioactive procedures, as explained previously [4,44]. In non-radioactive SSCP analysis, PCR products were mixed together in denaturing buffer (95 per cent formamide, 10 mM NaOH, 0.05 per cent xylene-cyanol FF and 0.05 per cent bromophenol blue) in a 1:1 ratio, heat denatured at 95°C for 5 minutes and immediately cooled on ice for 20 minutes. Of the resulting product, 6 μl was loaded on 6 per cent PAG and electrophoresed in 0.5× Tris-borate EDTA buffer at ± 17°C at 4 W constant power for 18-22 hours. Gels were then silver stained. In radioactive SSCP analysis, radiolabelled PCR products (using α32-pCTP) were mixed in a denaturing loading buffer (95 per cent formamide, 20 mM EDTA, 0.05 per cent xylene-cyanol FF and 0.05 per cent bromophenol blue) in a 1:10 ratio and heat denatured at 95°C for 5 minutes. Of the resulting product, 3 μl was loaded on 6 per cent PAG and electrophoresed at 4 W constant power in 0.5× Tris-borate ethylene diamine tetra-acetic acid (EDTA) buffer at ± 17°C for 18-22 hours. The gel was then transferred onto 3 mm Whatman paper, covered with Saran wrap and dried in a vacuum drier at 90°C for 1 hour. The Saran wrap was then replaced by X-ray film and kept at -70°C for 48 hours.
The mobility shift in DNA bands was visualised by developing the X-ray film. Purified PCR products of the samples showing mobility shift on SSCP analysis and randomly chosen samples were used for direct DNA sequencing.
Methylation-specific PCR (MS-PCR) of APC promoters
Both normal and tumour DNAs were subjected to sodium bisulphite modification using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA). About 10 ul of DNA from each sample was modified as described in the protocol. Previously reported primer sets were used for amplification of the two promoters - 1A and 1B of the APC gene (Table 2) [32,37].
Table 2.
Primer sequences used for hypermethylation analysis of the promoter region of the APC gene
| Gene | Amplicon | Primer sequence | Amplicon size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|
| APC | 1A | APC 1A UF: 5'-TGTTTTATTGTGGAGTGTGGGTT-3' | 108 | 60 |
| Promoter | APC 1A UR: 5'-CCAATCAACAAACTCCCAACAA-3' | |||
| APC 1A MF: 5'-TATTGCGGAGTGCGGGTC-3' APC 1A MR: 5'-TCGACGAACTCCCGACGA-3' |
98 | 55 | ||
| APC | 1B | APC 1B UF: 5'-GATAGAATAGTGAATGAGTGTTT-3' | 195 | 55 |
| Promoter | APC 1B UR: 5'-CTTCCAACAACCACACCCCA-3' | |||
| APC 1B MF: 5'-TAGAATAGCGAACGAGTGTTC-3' APC 1B MR: 5'-TCCGACGACCACACCCCG-3' |
190 | 55 |
PCR was performed in a 50 μl total volume reaction mixture containing 10 ng of modified genomic DNA, 100 ng of each primer, 100 μM of each dNTP, 1.5 mM MgCl2, 5 per cent dimethyl sulphoxide (DMSO), 10× Taq buffer and 2 U of Taq DNA polymerase (Fermentas). The reactions were hot started at 97°C for 10 minutes before the addition of 0.75 units of Taq polymerase (Fermentas). The PCR conditions were as follows: 40 cycles of denaturation at 95°C for 40 seconds, annealing at the temperatures specified in Table 1 for 45 seconds, extension at 72°C for 45 seconds and a final extension at 72°C for 10 minutes to complete each PCR. The PCR amplicons were electrophoresed on 2.5 per cent agarose gels and were visualised after staining with ethidium bromide (Figure 3A and 3B).
Figure 3A.
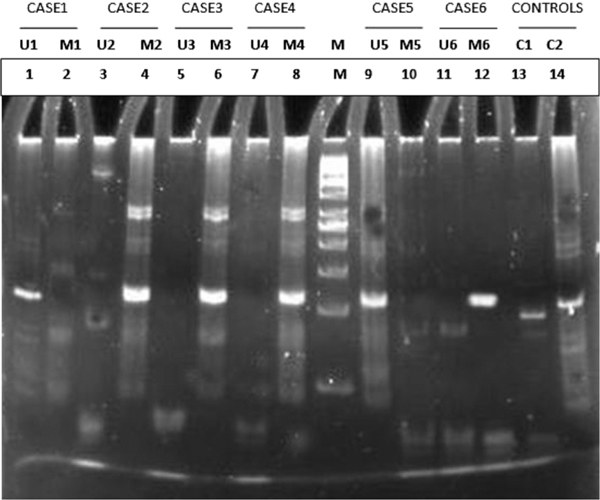
PAGE analysis of APC 1A promoter methylation. Lane M: 50 bp molecular ladder Lanes 1-14: amplicons from paired samples. Case 1 and 5: Unmethylated APC 1A promoter Cases 2, 3, 4 and 6: Methylated APC 1A promoter C1 and C2: Internal unmethylated and methylated human DNA controls.
Figure 3B.
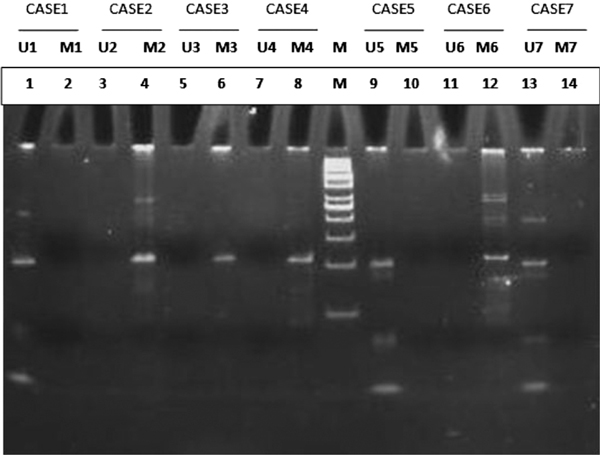
PAGE analysis of APC 1B promoter methylation. Lane M: 50 bp molecular ladder Lanes 1-14: amplicons from paired samples. Case 1, 5 and 7: Unmethylated APC 1B promoter Cases 2, 3, 4 and 6: Methylated APC 1B promoter.
Universal Methylated Human DNA (Zymo Research) was used as positive control for methylated alleles whereas DNA from normal lymphocytes was used as a control for unmethylated alleles. Water was used as a negative PCR control in both reactions.
Direct sequencing
PCR amplicons of the tumour samples and from randomly chosen normal samples were first purified using the DNA Recovery Kit (Zymo Research) and then used for direct DNA sequencing. DNA sequencing was carried out at Macrogen Inc. To minimise the sequencing artefacts by PCR, amplicons from at least two different PCRs were sequenced using forward and reverse primers.
Statistical analysis
All statistical analysis was performed using PASW software, version 18 (IBM, New York, NY). Pearson's chi-square two-proportion test was used to evaluate the hypothesis of equal distribution of molecular alterations with different clinicopathological variables. A Fisher's two-tailed test (p values) of 0.05 or less was considered to be statistically significant.
Results
Of 86 confirmed cases of CRC, 38 were of Dukes' A + B stage and 48 were of C + D stage. All patients presented with constipation and bleeding per rectum as their chief complaint. Furthermore, 81 of these cases were sporadic, four had familial adenomatous polyposis and one had Lynch syndrome. All but one case had adenocarcinoma and only one had squamous cell carcinoma (SCC) of the basal cell type. Thirty-seven patients were female and 49 male; 59 were rural and 27 urban; 36 cases had carcinoma in the colon and 50 in the rectum; and 55 were smokers and 31 non-smokers (Table 3).
Table 3.
Clinico-epidemiological variables of the 86 CRC patients versus 47 hypermethylated phenotypes of APC (1A and 1B promoter) gene
| Variable | Total n = 86 |
Mutantsa n = 11 (12.79%) |
Methylatedb n = 47 (54.65%) |
p
valuec |
|---|---|---|---|---|
| Age group | ||||
| ≤ 60 | 52 (60.5%) | 5 | 20 | < 0.05 |
| > 60 | 34 (39.5%) | 6 | 27 | |
| Gender | ||||
| Female | 37 (43.0%) | 4 | 19 | 0.85 |
| Male | 49 (67.0%) | 7 | 28 | |
| Dwelling | ||||
| Urban | 27 (31.4%) | 5 | 17 | 0.70 |
| Rural | 59 (68.6%) | 6 | 30 | |
| Tumour location | ||||
| Colon | 36 (41.9%) | 8 | 29 | < 0.05 |
| Rectum | 50 (58.1%) | 3 | 18 | |
| Nodal status | ||||
| Involved | 48 (55.8%) | 6 | 38 | < 0.05 |
| Not Involved | 38 (44.2%) | 5 | 9 | |
| Tumour grade | ||||
| A + B | 38 (44.2%) | 5 | 9 | < 0.05 |
| C + D | 48 (55.8%) | 6 | 38 | |
| Smoking status | ||||
| Never | 31 (36.0%) | 4 | 14 | 0.56 |
| Ever | 55 (64.0%) | 7 | 33 | |
| Bleeding PR/Constipation | ||||
| No | 26 (30.2%) | 2 | 16 | 0.69 |
| Yes | 60 (69.8%) | 9 | 31 | |
| Pesticide exposure | ||||
| Never | 33 (38.4%) | 3 | 19 | 0.85 |
| Ever | 53 (61.6%) | 8 | 28 |
aOther than G > A transition at codon 1492 of APC.
bEither 1A or 1B promoter hypermethylation.
cFisher's two-tailed test for hypermethylation status of APC.
Mutation analysis of APC
The overall mutation rate of the MCR of APC among the 86 patients was 12.8 per cent (11 of 86). This is in contrast to other studies that have reported APC as the main gene to undergo aberration in CRC, with a frequency of about 60 per cent. DNA sequencing revealed four missense mutations, three nonsense mutations and four frameshift mutations, including three deletions and one insertion (Table 4, Figure 4). Among the three nonsense mutations, two were Leu > Stop and one was Lys > Stop.
Table 4.
Nature of APC mutation cluster region mutations in 11 CRC patients from the Kashmir valley
| Patient ID | Mutationa | Amino acid change | Affected codon | Effect |
|---|---|---|---|---|
| A6 | TTA > TAA | Leu > Stop | 1277 | NS |
| A8 | ACCAA > ACA | Del CA | 1448/49 | FS |
| A9 | TTA > GTA | Leu > Val | 1489 | MS |
| A22 | AGT > ATT | Ser > Ile | 1494 | MS |
| A25 | AGA > AGT | Arg > Ser | 1336 | MS |
| A27 | AGT > ATT | Ser > Ile | 1494 | MS |
| A28 | TTA > TAA | Leu > Stop | 1277 | NS |
| A31 | TAAAAGAAAAG > TAAAAGA | Del AAAAG | 1307/08/09 | FS |
| A33 | TAAAAG > TAAG | Del AA | 1307/08 | FS |
| A37 | AAG > TAG | Lys > Stop | 1449 | NS |
| A77 | ATG > ATAG | Ins A | 1525 | FS |
| XXb | ACG > ACA | Thr > Thr | 1492 | S |
aMutated, deleted or inserted nucleotide underlined.
bXX refers to any general tumour sample.
Abbreviations: MS, missense mutation; NS, nonsense mutation; S, silent mutation; FS, Frameshift mutation.
Figure 4.
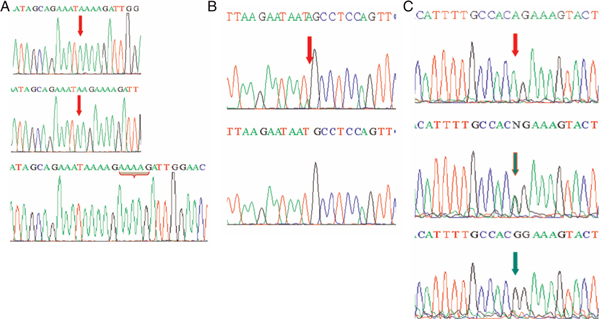
Partial electropherograms representing the mutant (above) and normal (below) forms (shown by arrows) of the mutation cluster region (MCR) of APC. (A) Deletion of the AAAAG pentamer at codons 1307/08/09 in the MCR region of APC and deletion of the AA dimer at codons 1307/08 in the MCR region of APC. (B) Insertion of A at codon 1525 in the MCR region of APC. (C) Polymorphism of codon 1492 in the MCR region of APC.
We also found a novel single nucleotide polymorphism (SNP) in our study, a G > A polymorphism in codon 1492 of APC. The polymorphism changes ACG to ACA, without changing the resulting amino-acid residue. We found, that among 86 CRC cases, only 14 (16.3 per cent) had the homozygous wild-type (GG) genotype, while 53 (61.6 per cent) had the homozygous variant (AA) genotype and 19 (22.1 per cent) had the heterozygous (GA) genotype (Table 5, Figures 3A and 3B). In addition, 72 cases had G > A variants, 67 were variants for G > A at codon 1492 only, while five also had mutations at other sites.
Table 5.
Codon 1492 status of APC gene in 86 colorectal carcinoma cases in Kashmiri population
| APC codon 1492 status | Cases (n = 86) |
|---|---|
| Variant | 53 (61.6%) |
| Heterozygous | 19 (22.1%) |
| Wild-type | 14 (16.3%) |
Mutation analysis of the β-catenin gene
The overall mutation rate of the β-catenin gene was 8.1 per cent (seven of 86). Of these seven mutations, three affected codon 32, three affected codon 49 and one affected codon 45 (Figure 5, Table 6); five were missense and two were nonsense mutations. Both nonsense mutations affected codon 49, changing lysine to a stop codon leading to truncation of the protein. In addition, five of the seven patients with β-catenin mutations had higher-grade tumours (C + D). One also had a mutation in APC, but six β-catenin had an intact APC gene.
Figure 5.
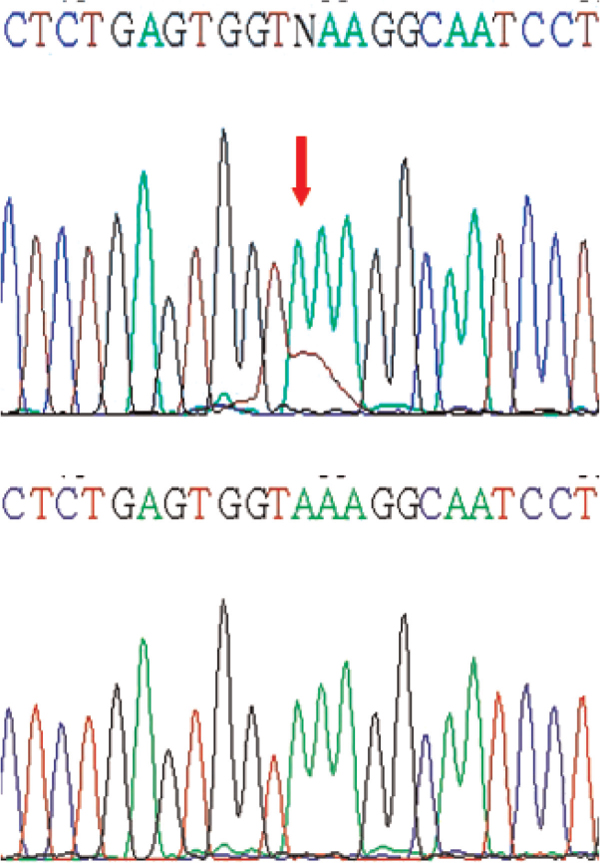
Partial electropherograms representing the mutant (above) and normal (below) forms (shown by arrows) of exon 3 of the β-catenin gene (AAA > TAA).
Table 6.
Nature of β-catenin gene in seven colorectal cancer patients from the Kashmir valley
| Patient ID | Mutationa | Amino acid change | Affected codon | Effect |
|---|---|---|---|---|
| A1 | GAC > GGC | Asp > Gly | 32 | MS |
| A5 | AAA > TAA | Lys > Stop | 49 | NS |
| A9 | AAA > TTA | Lys > Leu | 49 | MS |
| A10 | GAC > GGC | Asp > Gly | 32 | MS |
| A62 | AAA > TAA | Lys > Stop | 49 | NS |
| A74 | GAC > GGC | Asp > Gly | 32 | MS |
| A82 | TCT > TTT | Ser > Phe | 45 | MS |
aMutated, deleted or inserted nucleotide underlined.
Abbreviations: MS, missense mutation; NS, nonsense mutation.
Table 7.
APC gene methylation status in 86 CRC cases in the Kashmiri population
| APC promoter status | Cases (n = 86) |
|---|---|
| Either methylated (1A and/or 1B) | 47 (54.65%) |
| Only 1A methylated | 9/47 (19.1%) |
| Only 1B methylated | 15/47 (31.9%) |
| Both methylated | 23/47 (48.9%) |
| Neither methylated | 39 (45.35%) |
Hypermethylation of APC promoters
Methylation analysis of APC carried out on two promoters, 1A and 1B, revealed a high methylation status of these two promoters. Forty-seven (54.65 per cent) of the tumours were methylated at either one of the two promoter regions, while 39 (45.35 per cent) of the tumours were not methylated at any of the promoters (Tables 3 and 7). Among the tumours which were methylated, only nine (19.1 per cent) were found to be methylated at the APC 1A promoter exclusively, 15 (31.9 per cent) were found to be methylated at the APC 1B promoter exclusively, while 23 (48.9 per cent) tumours were methylated at both promoters. Furthermore, we also found that only four tumours were mutated as well as hypermethylated for the APC gene (Table 8). Statistical analysis showed a significant association between APC methylation status and the age group, tumour location (colon) and tumour grade (C + D) of the patients (Table 3).
Table 8.
Correlation of APC mutation status versus APC methylation status
| APC status | OR; 95% CI; p value | ||
|---|---|---|---|
|
Wild-type W = 75 |
Mutanta M = 11 |
||
| APC promoter methylationb | |||
| Unmethylated; n = 39 | 32 (42.7%) | 7 (63.6%) | 2.35; 0.63-8.72; 0.22 |
| Methylated; n = 47 | 43 (57.3%) | 4 (36.4%) | |
aOther than G > A transition at codon 1492.
bEither 1A or 1B promoter hypermethylation.
Abbreviations: OR, odds ratio; CI, confidence interval.
Discussion
The Kashmir valley, located in the northern division of India, has a unique ethnic population, living in temperate environmental conditions and with distinctive food habits, which, along with genetic factors, play a large role in the development of GIT cancers [3-5,45]. As previously reported, the aetiology and incidence of various GIT cancers in this population has been attributed to a probable exposure to nitroso compounds, amines and nitrates reported to be present in local foodstuffs such as hoakhe suen (sun-dried vegetables), pharei and hoggade (sundried and/or smoked fish and meat), hakh (a leafy vegetable of the Brassica family), hot noon chai (salted tea), dried and pickled vegetables and red chilli, and also through smoking hukka (a water pipe) [2-5,46].
According to the multi-step model of colorectal tumorigenesis,[7] the most common and principal causes of APC inactivation are gene aberrations. A somatic mutation in APC leads to a truncated protein in most sporadic CRCs [15,36]. Hypermethylation of APC at the promoter region constitutes an alternative mechanism for APC inactivation in breast, lung and GIT cancers, especially CRCs [30,33,34,38,40,47]. Combined with these two mechanisms of APC inactivation and the aberrations in the β-catenin gene, the Wnt pathway molecules play an important role in CRC development and progression [48,49].
The present study involved the mutational analysis of exon 15 (MCR) of APC and exon 3 of the β-catenin gene and also the hypermethylation analysis of two promoters of APC. Although being the important genetic molecule of the Wnt pathway, and implicated in almost 60 per cent of sporadic CRCs, we found APC gene to be aberrant in only 12.79 per cent of CRCs, which was considerably lower than the previously reported frequencies [9,19,35,50-53]. This low frequency suggests that APC may not be the foremost gene to be implicated in the development of CRCs in this population.
Furthermore, we found an SNP (G > A) at codon 1492 in 72 (83.7 per cent) CRC cases. Out of 72 cases, 53 were homozygous variants. This was a novel finding, as it has not been reported previously.
We also found a low frequency (8.1 per cent; seven of 86) of β-catenin mutations in CRC. These results were in line with those in the published literature [54-57]. Exon 3 of β-catenin contains a regulatory domain which is the hotspot for genetic aberrations. Mutations in this exon have been reported in various tumours, resulting in its nuclear accumulation and leading to progression of the tumour [9,43,58]. The mutations in the hotspot codons - 32, 33, 41, 45 and 49 - in exon 3 of β-catenin result in an amino acid change at the GSK-3β phosphorylation sites, which in turn affect the phosphorylation mechanism and result in the decreased sequestration of β-catenin by APC [58]. Furthermore, the mutation affecting codon 45 (TCT > TTT; Ser > Phe) was present in a Lynch syndrome patient, as has been reported previously [55,56]. Also, six of seven tumour samples which harboured β-catenin gene mutations were wild-type for APC (MCR only), which further corroborated findings in the literature that mutations of the genes encoding these two Wnt pathway molecules are mutually exclusive [9,54,57,59,60]. Only one case (A9) had mutations in both genes (Tables 4 and 6). Overall, mutations in Wnt pathway molecule genes were found to be present in 20.9 per cent (18 of 86) of CRC cases. Thus, our observation identifies this pathway as being important in determining the development and progression of CRC but is less important than in other populations, where the mutational frequency of these Wnt gladiators is higher.
CpG island hypermethylation is one of the important mechanisms of gene inactivation. Cancer cell lines have in general demonstrated an increased frequency of hypermethylation by comparison with primary tumours [61]. Inactivation of tumour suppressor genes by promoter hypermethylation has been recognised to be as common as gene disruption by mutation in tumorigenesis [36,37,62,63]. A number of studies on CRC around the globe have demonstrated the role of promoter hypermethylation of a number of different genes in the development and progression of CRC [32,64,65]. Promoter hypermethylation of APC, similarly to that of other genes, plays a pivotal role in the inactivation of APC, which in turn enhances tumour development [30,33].
In the present study, we found hypermethylation in 54.65 per cent (47/86) of CRC cases, which is consistent with the results found in some other major studies, although markedly higher than reported in others [30,32,33]. However, only 26.7 per cent (23/86) of the tumours were hypermethylated at both the 1A and 1B promoters. This may be due to the fact that there is less mutational inactivation of APC in this population and also because this population is exposed to a special set of environmental challenges, such as extreme temperature, high altitude, special food habits and exposure to agricultural by-products such as pesticides and nitrosamines [5,46]. As has been revealed in previous studies, promoter hypermethylation constitutes an alternative hit in the inactivation of APC in cancers,[39,66] and we have identified the same phenomenon as the major cause of APC inactivation in our population. Various studies have shown transcriptional repression of APC by hypermethylation in tumours as well as cell lines [34,67].
Arnold et al. demonstrated the loss of protein expression due to the promoter hypermethylation of APC [39]. In addition, 42.8 per cent (three of seven) of patients with a mutation in β-catenin were also found to have hypermethylation of APC. We found the methylation status of the APC promoter to be associated with age (> 60), tumour location (colon) and nodal status/tumour grade (C + D) in CRC.
Conclusion
We conclude that, in the Kashmir valley population, although mutational aberration of the genes encoding two pivotal molecules of the Wnt pathway - APC and β-catenin - occurs at a low frequency in CRC cases, the high level of epigenetic silencing of APC plays a pivotal role in the initial tumorigenesis and also enhances the chances of tumour development and progression to advanced stages.
References
- Center MM, Jemal A, Smith RA, Ward E. Worldwide variations in colorectal cancer. CA Cancer J Clin. 2009;59:366–378. doi: 10.3322/caac.20038. [DOI] [PubMed] [Google Scholar]
- Mir MM, Dar NA, Gochhait S, Zargar SA. et al. p53 Mutation profile of squamous cell carcinomas of the esophagus in Kashmir (India): A high-incidence area. Int J Cancer. 2005;116:62–68. doi: 10.1002/ijc.21002. [DOI] [PubMed] [Google Scholar]
- Murtaza I, Mushtaq D, Margoob MA, Dutt A. et al. A study on pp53 gene alterations in esophageal squamous cell carcinoma and their correlation to common dietary risk factors among population of the Kashmir valley. World J Gastroenterol. 2006;12:4033–4037. doi: 10.3748/wjg.v12.i25.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sameer AS, Chowdri NA, Syeed N, Banday MZ. et al. SMAD4 -- Molecular gladiator of the TGF-β signaling is trampled upon by mutational insufficiency in colorectal carcinoma of Kashmiri population: An analysis with relation to KRAS proto-oncogene. BMC Cancer. 2010;10:300. doi: 10.1186/1471-2407-10-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sameer AS, Shah ZA, Syeed N, Banday MZ. et al. TP53 Pro47Ser and Arg72Pro polymorphisms and colorectal cancer predisposition in an ethnic Kashmiri population. Genet Mol Res. 2010;9:651–660. doi: 10.4238/vol9-2gmr751. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE. et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- Samowitz WS, Slattery ML, Sweeney C, Herrick J. et al. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res. 2007;5:165–170. doi: 10.1158/1541-7786.MCR-06-0398. [DOI] [PubMed] [Google Scholar]
- Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer. 2003;2:41. doi: 10.1186/1476-4598-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widelitz R. Wnt signaling through canonical and non-canonical pathways: Recent progress. Growth Factors. 2005;23:111–116. doi: 10.1080/08977190500125746. [DOI] [PubMed] [Google Scholar]
- Willert K, Jones KA. Wnt signaling: is the party in the nucleus? Genes Dev. 2006;20:1394–1404. doi: 10.1101/gad.1424006. [DOI] [PubMed] [Google Scholar]
- Mikesch JH, Steffen B, Berdel WE, Serve H. et al. The emerging role of Wnt signaling in the pathogenesis of acute myeloid leukemia. Leukemia. 2007;21:1638–1647. doi: 10.1038/sj.leu.2404732. [DOI] [PubMed] [Google Scholar]
- Turashvili G, Bouchal J, Burkadze G, Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology. 2006;73:213–223. doi: 10.1159/000098207. [DOI] [PubMed] [Google Scholar]
- Yardy GW, Brewster SF. Wnt signaling and prostate cancer. Prostate Cancer Prostatic Dis. 2005;8:119–126. doi: 10.1038/sj.pcan.4500794. [DOI] [PubMed] [Google Scholar]
- Jass JR, Barker M, Fraser L, Walsh MD. et al. APC mutation and tumour budding in colorectal cancer. J Clin Pathol. 2003;56:69–73. doi: 10.1136/jcp.56.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim Biophys Acta. 1997;1332:127–147. doi: 10.1016/s0304-419x(97)00008-5. [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Nagase H, Ando H, Horii A. et al. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- Beroud C, Soussi T. APC gene: Database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1996;24:121–124. doi: 10.1093/nar/24.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J. The role of the Wnt signalling pathway in colorectal tumorigenesis. Biochem Soc Trans. 2005;33:672–676. doi: 10.1042/BST0330672. [DOI] [PubMed] [Google Scholar]
- Aoki K, Taketo MM. Adenomatous polyposis coli (APC): A multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–3335. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM. et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- Fodde R, Kuipers J, Rosenberg C, Smits R. et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M. et al. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC. et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Ishikawa TO, Tamai Y, Li Q, Oshima M. et al. Requirement for tumor suppressor Apc in the morphogenesis of anterior and ventral mouse embryo. Dev Biol. 2003;253:230–246. doi: 10.1016/S0012-1606(02)00020-9. [DOI] [PubMed] [Google Scholar]
- Moser AR, Shoemaker AR, Connelly CS, Clipson L. et al. Homozygosity for the Min allele of Apc results in disruption of mouse development prior to gastrulation. Dev Dyn. 1995;203:422–433. doi: 10.1002/aja.1002030405. [DOI] [PubMed] [Google Scholar]
- Andreu P, Colnot S, Godard C, Gad S. et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development. 2005;132:1443–1451. doi: 10.1242/dev.01700. [DOI] [PubMed] [Google Scholar]
- Gounari F, Chang R, Cowan J, Guo Z. et al. Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat Immunol. 2005;6:800–809. doi: 10.1038/ni1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H. et al. Loss of Apc in vivo immediately perturbs Wnt signalling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Röcken C, Lofton-Day C, Schulz HU. et al. Molecular analysis of APC promoter methylation and protein expression in colorectal cancer metastasis. Carcinogenesis. 2005;26:37–43. doi: 10.1093/carcin/bgh280. [DOI] [PubMed] [Google Scholar]
- Zare M, Jazii FR, Alivand MR, Nasseri NK. et al. Qualitative analysis of adenomatous polyposis coli promoter: Hypermethylation, engagement and effects on survival of patients with esophageal cancer in a high risk region of the world, a potential molecular marker. BMC Cancer. 2009;9:24. doi: 10.1186/1471-2407-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind GE, Thorstensen L, Løvig T, Meling GI. et al. A CpG island hypermethylation profile of primary colorectal carcinomas and colon cancer cell lines. Mol Cancer. 2004;3:28. doi: 10.1186/1476-4598-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang GH, Lee S, Kim JS, Jung HY. Profile of aberrant CpG island methylation along the multistep pathway of gastric carcinogenesis. Lab Invest. 2003;83:635–641. doi: 10.1097/01.lab.0000067481.08984.3f. [DOI] [PubMed] [Google Scholar]
- Virmani AK, Rathi A, Sathyanarayana UG, Padar A. et al. Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin Cancer Res. 2001;7:1998–2004. [PubMed] [Google Scholar]
- Rowan AJ, Lamlum H, Ilyas M, Wheeler J. et al. APC mutations in sporadic colorectal tumors: A mutational "hotspot" and interdependence of the "two hits". Proc Natl Acad Sci USA. 2000;97:3352–3357. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Herman JG. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J Pathol. 2002;196:1–7. doi: 10.1002/path.1024. [DOI] [PubMed] [Google Scholar]
- Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M. et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60:4366–4371. [PubMed] [Google Scholar]
- Tsuchiya T, Tamura G, Sato K, Endoh Y. et al. Distinct methylation patterns of two APC gene promoters in normal and cancerous gastric epithelia. Oncogene. 2000;19:3642–3646. doi: 10.1038/sj.onc.1203704. [DOI] [PubMed] [Google Scholar]
- Arnold CN, Goel A, Niedzwiecki D, Dowell JM. APC promoter hypermethylation contributes to the loss of APC expression in colorectal cancers with allelic loss on 5q. Cancer Biol Ther. 2004;3:960–964. doi: 10.4161/cbt.3.10.1113. [DOI] [PubMed] [Google Scholar]
- Hiltunen MO, Alhonen L, Koistinaho J, Myöhänen S. et al. Hypermethylation of the APC (adenomatous polyposis coli) gene promoter region in human colorectal carcinoma. Int J Cancer. 1997;70:644–648. doi: 10.1002/(SICI)1097-0215(19970317)70:6<644::AID-IJC3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Debuire B, Lemoine A, Saffroy R. CTNNB1 (Catenin, beta-1) Atlas of Genetics and Cytogenetics in Oncology and Haematology. 2002. http://AtlasGeneticsOncology.org/Genes/CTNNB1ID71.html
- Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Abraham SC, Wu TT, Klimstra DS, Finn LS. et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: Frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol. 2001;159:1619–1627. doi: 10.1016/S0002-9440(10)63008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain I, ul Rehman S, Afroze D, Zahoor L. et al. Mutational spectrum of conserved regions of TP53 and PTEN genes in Kangri cancer (of the skin) in the Kashmiri population. Mutat Res. 2009;676:5–10. doi: 10.1016/j.mrgentox.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Salam I, Hussain S, Mir MM, Dar NA. et al. Aberrant promoter methylation and reduced expression of p16 gene in esophageal squamous cell carcinoma from Kashmir valley: A high-risk area. Mol Cell Biochem. 2009;332:51–58. doi: 10.1007/s11010-009-0173-7. [DOI] [PubMed] [Google Scholar]
- Siddiqi M, Kumar R, Fazili Z, Spiegelhalder B. et al. Increased exposure to dietary amines and nitrate in a population at high risk of esophageal and gastric cancer in Kashmir (India) Carcinogenesis. 1992;13:1331–1335. doi: 10.1093/carcin/13.8.1331. [DOI] [PubMed] [Google Scholar]
- Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22:4632–4642. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- Schneikert J, Behrens J. The canonical Wnt signaling pathway and its APC partner in colon cancer development. Gut. 2007;56:417–425. doi: 10.1136/gut.2006.093310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim RZ, Tse GMK, Putti TC, Scolyer RA. et al. The significance of the Wnt pathway in the pathology of human cancers. Pathology. 2004;36:120–128. doi: 10.1080/00313020410001671957. [DOI] [PubMed] [Google Scholar]
- Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- Luchtenborg M, Weijenberg MP, Roemen GMJM, de Bruine AP. et al. APC mutations in sporadic colorectal carcinomas from The Netherlands Cohort Study. Carcinogenesis. 2004;25:1219–1226. doi: 10.1093/carcin/bgh117. [DOI] [PubMed] [Google Scholar]
- Jeon C, Lee H, Shin IH, Park J. Genetic alterations of APC, K-ras, p53, MSI, and MAGE in Korean colorectal cancer patients. Int J Colorectal Dis. 2008;23:29–35. doi: 10.1007/s00384-007-0373-0. [DOI] [PubMed] [Google Scholar]
- Smith G, Carey FA, Beattie J, Wilkie MJV. et al. Mutations in APC, Kirsten-ras, and p53 -- Alternative genetic pathways to colorectal cancer. Proc Nat Acad Sci USA. 2002;99:9433–9438. doi: 10.1073/pnas.122612899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samowitz WS, Powers MD, Spirio LN, Nollet F. et al. β-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res. 1999;59:1442–1444. [PubMed] [Google Scholar]
- Miyaki M, Iijima T, Kimura J, Yasuno M. et al. Frequent mutation of beta-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res. 1999;59:4506–4509. [PubMed] [Google Scholar]
- Johnson V, Volikos E, Halford SE, Eftekhar Sadat ET. et al. Exon 3 β-catenin mutations are specifically associated with colorectal carcinomas in hereditary non-polyposis colorectal cancer syndrome. Gut. 2005;54:264–267. doi: 10.1136/gut.2004.048132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwao K, Nakamori S, Kameyama M, Imaoka S. et al. Activation of the β-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res. 1998;58:1021–1026. [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B. et al. Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N. et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Paz MF, Fraga MF, Avila S, Guo M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003;63:1114–1121. [PubMed] [Google Scholar]
- Karpiñski P, Sasiadek MM, Blin N. Aberrant epigenetic patterns in the etiology of gastrointestinal cancers. J Appl Genet. 2008;49:1–10. doi: 10.1007/BF03195243. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Lee S, Hwang KS, Lee HJ, Kim JS. et al. Aberrant CpG island hypermethylation of multiple genes in colorectal neoplasia. Lab Invest. 2004;84:884–893. doi: 10.1038/labinvest.3700108. [DOI] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG. et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Arnold CN, Boland CR. Multistep progression of colorectal cancer in the setting of microsatellite instability: New details and novel insights. Gastroenterology. 2001;121:1497–1502. doi: 10.1053/gast.2001.29978. [DOI] [PubMed] [Google Scholar]
- Moreno-Bueno G, Hardisson D, Sanchez C, Sarrio D. et al. Abnormalities of the APC/β-catenin pathway in endometrial cancer. Oncogene. 2002;21:7981–7990. doi: 10.1038/sj.onc.1205924. [DOI] [PubMed] [Google Scholar]