

Abstract
The construction of mono- and bicyclic phosphate trimesters possessing divalent and multivalent activation and their subsequent use in the production of advanced polyol synthons is presented. The method highlights efforts to employ phosphate tethers as removable, functionally active tethers capable of multipositional activation and their subsequent role as leaving groups in selective cleavage reactions. The development of phosphate tethers represents an integrated platform for a new and versatile tether for natural product synthesis and sheds light on new approaches to the facile construction of small molecules.
Keywords: Phosphate, Tether, Metathesis, Cuprates, Protecting groups
Introduction
The development of new synthetic strategies allowing for efficient asymmetric syntheses of complex biologically active targets, with minimal protecting group manipulations and chemical steps, is an enormous challenge in natural product synthesis. A powerful way of addressing this challenge is through the use of convergent methodologies employing the temporary tethering of two advanced intermediates. Historically, silicon has been the most widely used temporary tether due to its facile installation/cleavage attributes as well as its innate protecting group properties.[1] Moreover, the ability of silicon tethers to undergo myriad functional-group transformations positions them as ideal tethers in the realm of total synthesis.[2] In comparison, phos phorus has the ability to mediate di- and tripodal couplings, provide orthogonal protection and has innate leaving group properties. Although the potential advantages of phosphate tethers are evident, their application in synthesis has until recently largely focused on monovalent activation of a single phosphate ester appendage. Such classical use of phosphates in complex synthesis has focused on nucleophilic displacement reactions of allylic phosphates,[3] cross-coupling/reduction reactions with enol-phosphates,[4] and direct displacements of phosphates in more recent cyclization protocols.[5] Additional uses of phosphate triesters in iodophosphonylation procedures[6] and their role in oligonucleotide synthesis[7] further highlight both nucleophilic properties and facile coupling characteristics innate to phosphates.
While the aforementioned attributes of phosphate triesters is impressive, their reactivity profile is overshadowed by the vast extent to which nature uses the anionic counterparts, organophosphate mono- and dianions, which play a dominant role in a number of key biological processes.[8] A seminal paper in 1987 by Westheimer[8] addresses the issue where he surmises, “while nature capitalizes on the unique features of phosphate monoanions, chemists cannot afford to use compounds as stable as the phosphate anion and with the poor leaving group capabilities of either phosphate anions or dianions.” This concept can be summarized as the phosphate “brake” (Figure 1), which is predicated upon the longer 11-year half-life of the hydrolysis of dimethylsodium phosphate when compared with trimethyl phosphate.
Figure 1.

Hydrolysis half-life of trimethyl- and dimethylsodium phosphate.
The ubiquity of phosphates in nature points to their potential utility in the development of new synthetic methodology. In particular, several attractive features were uncovered in three recent studies illustrating the ability of phosphate triesters to serve as functionally active tethers.[9] These studies showed phosphate tethers capable of coupling two carbinol-containing subunits via ring-closing metathesis (RCM), serving as leaving groups with the ability to undergo selective cleavage reactions and imparting type-III properties in cross-metathesis (CM) reactions with the exocyclic appendant olefin. These studies capitalized on the unique features of the phosphate mono- and dianions as a means of expanding current phosphate methodology to the arena of total synthesis.[9] Reported herein is a summary of efforts to employ phosphate tethers as removable, functionally active tethers capable of multipositional activation and their subsequent role as latent leaving groups in selective cleavage reactions. The development of phosphate tethers (Figure 2) represents an integrated platform for a new and versatile tether for natural product synthesis and sheds light on new approaches to the facile construction of small molecules.
Figure 2.

Monocyclic and bicyclic phosphates.
Divalent Activation in Phosphate Tethers
In 1991, Yamamoto and co-workers reported the superiority of phosphates as allylic leaving groups (Figure 3).[10] In this report, they subjected an allylic phosphate to a copper-mediated anti-SN2′ displacement to afford products with high E:Z selectivity and with excellent chirality transfer. These observations were also reported by Chong and Knochel.[11]
Figure 3.

Seminal report of cuprate addition to allylic phosphates.
More recently, enantioselective allylic displacements via an anti-SN2′ pathway with chiral Schiff base equivalents have been shown to desymmetrize meso-1,3-syn-allylic phosphates [Figure 4, Equation (1)].[12a] Other reagent controlled asymmetric cuprate additions include additional applications of Schiff bases [Equation (2)], BINAP [Equation (3)] and N-heterocyclic carbene ligands [Equation (4)], all of which provide chiral nonracemic products in very good to excellent ee from allylic phosphates.[11b–13]
Figure 4.

Examples of allylic phosphate displacements using cuprates.
In 2004, a phosphate tether was used to construct the pseudo-C2-symmetric monocyclic phosphate 1 en route to desymmetrization studies leading to advanced polyol synthons.[9b] Although the original intent to desymmetrize the C2-symmetric monocyclic phosphate via cuprate additions was met, interesting conformational effects led to further investigations of unsymmetric phosphates, which provided experimental insight into the Corey mechanism of cuprate displacements.[3a]
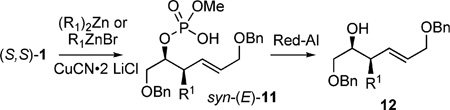
In this method, a three-step protocol was used starting from glycidol ether (S)-5 to generate (S,S)-monocyclic phosphate 1 (Scheme 1) in good yields.[9b] First, treatment of 5 with a sulfur ylide, generated in situ, gave allylic alcohol (S)-6 in excellent yield on multigram scale.[14] After generation of the corresponding alkoxide, condensation with (MeO)POCl2 yielded a phosphate triester, which underwent ring-closing metathesis (RCM) with Grubbs second-generation catalyst cat-B (Figure 5) to afford cyclic phosphate (S,S)-1.[15]
Scheme 1.

Synthesis of monocyclic phosphate 1.
Figure 5.

Olefin metathesis catalysts.
Initial investigations of the reactivity profile of 1 revealed facile cleavage of the phosphate tether when subjected to an excess amount of LiAlH4 (Scheme 2) to yield diol 7.[9b] Removal of the endocyclic olefin was achieved providing cyclic phosphate 8, followed by tether removal to afford diol 9.
Scheme 2.

Preparation of 1,4-diols via phosphate cleavage.
Studies toward desymmetrization of 1 by using a cuprate displacement reaction were investigated in accordance with the previously shown examples of allylic phosphate displacements operating through an anti-SN2′ pathway.[9b] The prerequisite for the aforementioned examples is that the leaving group be orthogonal to the approaching cuprate, requiring coplanar alignment of the σ* and π* orbitals. Thus, cyclic phosphate 1 possesses two possible productive conformations (Scheme 3).
Scheme 3.

Possible modes for cuprate addition.
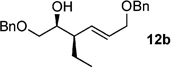
Although four diastereotopic olefin orbitals exist in 1, pseudo-symmetry dictates only two possible products anti-(Z)-10 and syn-(E)-11.[16] Treatment of (S,S)-1 with Et2Zn/CuCN·2LiCl resulted in formation of phosphate acid 11b as a single diastereomer (>20:1), determined by 31P NMR (after acidic workup, no chromatography required). Cleavage of the primary phosphate, in the presence of RedAl®, afforded chiral, nonracemic homoallylic alcohol 12b in a two-step sequence from 1 (Scheme 4).
Scheme 4.

Cuprate addition/phosphate acid cleavage sequence.
Rationalization of the excellent selectivity displayed by the cuprate addition can be explained by using Corey’s proposed concerted, asynchronous mechanism (Figure 6).[3a] The transition state occurs through coordination of the σ* of the phosphate ester leaving group and the π* of the olefin. Since both σ* orbitals in conformers A and B (Scheme 3) are roughly equal in energy, diastereoselectivity is dictated by allylic A1,3-strain from the CH2OBn-side chain, which is more prominent in conformer A when attaining the proper coplanar σ* and π* alignment.[17]
Figure 6.

Corey model for rationalizing stereoselectivity.
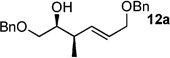
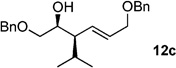
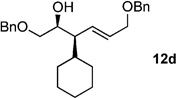
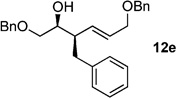
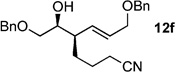
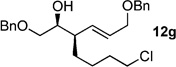
The scope of this reaction was observed with an array of zinc-based organocuprates (Entries 1–3, Table 1).[9b] In all cases, the corresponding homoallylic alcohol 12 was afforded by phosphate cleavage in the presence of RedAl®.
Table 1.
Cuprate addition/phosphate acid cleavage sequence.
 | ||||
|---|---|---|---|---|
| Entry | (R1)2Zn or R1ZnBr |
Phosphate acid - 11 yield (dr) |
Homoallylic alcohol - 12 |
% Yield |
| 1 | Me2Zn[a] | 99% (20:1) 11a |
 |
83 |
| 2 | Et2Zn[a] | 99%(>20:1) 11b |
 |
90 |
| 3 | iPr2Zn[a] | 99%(>20:1) 11c |
 |
95 |
| 4 | cHexZnl[b] | 99% (>20:1) 11d |
 |
70 |
| 5 | BnZnBr[b] | 99% (>20:1) 11e |
 |
84 |
| 6 | CN(CH2)3ZnBr[b] | 99% (>20:1) 11f |
 |
31 |
| 7 | Cl(CH2)4ZnBr[b] | 99% (>20:1) 11g |
 |
71 |
| 8 | CH2=CH(CH2)3ZnBr[b] | 99% (>20:1) 11h |
 |
65 |
Method required 4–5 equiv. of organocuprate.
Method required 8–9 equiv. of organocuprate.
In this study, it was shown that functionally sensitive R groups, e.g. Entries 6–7 in Table 1, gave lower yields after cleavage of the phosphate acid. However, by converting the phosphate acid to the corresponding phosphate ester by in situ methylation (TMSCHN2), better yields of the desired homoallylic alcohol were obtained (Scheme 5).[9b]
Scheme 5.

Methylation/phosphate cleavage.
After investigating activation pathways of pseudo-symmetric 1 in cuprate additions, a number of unsymmetric P-tethered systems were constructed where the electronic energies of the σ* orbitals of the leaving phosphate ester are differentiated by substitution at the reacting carbinol. Cyclic phosphate 2 was synthesized bearing primary and secondary phosphate leaving groups (Scheme 6). From diol 6, phosphorylation and RCM afforded 2 in good yield over the 2-step protocol. Subjecting the monocyclic phosphate 2 to 3.0 equiv. of a diethylzinc-derived organocuprate, followed by phosphate cleavage, furnished homoallylic alcohol 14 as a single stereoisomer. Whereas the stereoselectivity of 1 was based upon conformational preferences, 2 does not encounter the same type of governing A1,3-strain. Instead, the electronic bias of the more labile secondary phosphateleaving group explains the regioselectivity observed in the allylic displacement. Overall, this study contrasted the importance of steric vs. electronic effects in these systems.[9b]
Scheme 6.

Unsymmetric monocyclic phosphates.
To determine the extent to which allylic strain and electronic factors compete, investigations turned toward the unsymmetric phosphate 3 bearing secondary and tertiary allylic phosphate positions (Scheme 7). Subjection of 3 to a diethylzinc-derived organocuprate, followed by phosphate cleavage afforded homoallylic alcohol 16, where displacement of the tertiary allylic phosphate occurred exclusively to provide a single stereoisomer.[9b]
Scheme 7.

Secondary vs. tertiary allylic phosphate leaving groups.
An interesting bias of electronic effects over steric effects arises from the observed formation of 16. The preference of conformer F (Scheme 7) reveals severe A1,3 allylic strain between the gem-dimethyl terminus and the olefin. The regioselectivity seen is consistent with the result from allylic displacement of phosphate 2, where the more labile tertiary phosphate-leaving group is preferred. Looking back on the three examples shown, these results are consistent with the asynchronous concerted transition state proposed by Corey, where bond breakage of the lower-energy and more substituted σ* is preferred.[9b]
In summary, the aforementioned phosphate tethers show similar qualities to silicon[1, 2] in terms of facile dipodal coupling, stability, and facile removal. Moreover, the leaving group ability and unique geometry of these heterocycles allow for stereoselective cuprate addition reactions affording a variety of chiral, nonracemic allylic alcohols.[9b]
Multivalent Activation in Phosphate Tethers
The implementation of dipodal coupling to construct cyclic phosphates led further investigations employing tripodal coupling to construct more complex bicyclic phosphates such as (S,S,PR)-4 (Scheme 9). Seminal studies by Burke and co-workers had previously demonstrated utilization of a ketal tether desymmetrization method with C2-symmetric diol 17 (Scheme 8). In this study, ketalization, elimination, and subsequent RCM occurred in the presence of cat-A yielding a single product 19.[18] Overall, the ketal tether allowed for facile differentiation of the homotopic vinyl groups of the starting diol 17 to produce the chiral, nonracemic bicyclic ketal 19.
Scheme 9.

Use of RCM in the construction of the P-chiral, bicy-clo[4.3.1]phosphate (S,S,PR)-4.
Scheme 8.

Burke method of desymmetrization.
It was anticipated that a similar approach could be taken for the assembly of the bicyclic phosphate triester by using desymmetrization of the C2-symmetric 1,3-anti-diol 21 (Scheme 9). Diol 21 was accessed from dichloro-1,3-anti-diol 20,[19] by using a modified Mioskowski-Christie protocol.[14] Condensation with POCl3, followed by coupling with a lithium alkoxide derived from allyl alcohol, gave triene 22 in good yields. Differentiation of the vinyl groups occurs through RCM (cat-B, CH2Cl2, 40°C) where the chair conformer bearing the allyl ester cis to the vinyl group leads to formation of a single chiral, nonracemic bicyclic product (S,S,PR)-4.[9a]
A key feature within 4 is that 7 of the 9 non-oxygen atoms possess electrophilic character, and thus nucleophilic attack can occur at phosphorus or any of the three carbinols [C3, C6, and C8] as well as at allylic phosphate carbons [C4, C5, and C12], respectively (Figure 7).[9a] It was these characteristics that prompted investigations yielding chemo-, regio-, and stereoselective processes.
Figure 7.

Features of P-chiral bicyclo[4.3.1]phosphate 4.
The first of these observations was the notable stability of 4 toward a variety of acidic conditions. Exposure to 10% HCl (aq.)/dioxane and TMSCl afforded clean, unreacted starting material after prolonged reaction times. The enhanced stability appears to be a result of the lack of anti-periplanar (app) lone pairs on the adjacent oxygen atoms to the phosphoryl group, which impedes enhancement of the P=O basicity (Figure 8). This observation is consistent with decreased proton affinities of cyclic phosphites and the inordinate stability of bicyclic phosphates.[20]
Figure 8.

Stability of 4 toward acidic conditions.
As stated earlier, subjecting phosphates to basic hydrolysis conditions are known to stop at the monoanion salt (Figure 1).[8] Taking this fact into consideration, hydrolysis of 4 could likely give rise to three regioisomeric phosphate mono-acid salts (Figure 9).
Figure 9.

Possible hydrolysis products from 4.
Subjecting 4 to LiOH (aq.) in dioxane provided a quantitative and selective cleavage as determined by 31P NMR spectroscopy.[9a] The appearance of a major singlet at −0.06 ppm showed the formation of a major regioisomer formed (rs = 44:1). 13C NMR comparative analysis revealed a diagnostic upfield shift of the resonance corresponding to the C8 carbinol. This indicated regioisomer 23a was formed as the major product (Figure 10). Further evidence of this selective hydrolysis was determined from the loss of C–P coupling at the C8 carbinol. This observation was surprising in the fact that selective phosphate hydrolysis[21] has been considered quite limited[22] in a number of extensive studies[23] done on acidic and basic hydrolysis of phosphate esters.
Figure 10.

13C analysis of basic hydrolysis of 4.
Other nucleophiles were probed showing good regio- and stereoselectivities (Scheme 10). In contrast to hydrolysis, addition of LiSPh gave preferential attack toward the C3 carbinol (rs > 99:1) in nearly quantitative yield to afford linear phosphate 24. As shown earlier (Scheme 2), subjection to excess LiAlH4 resulted in removal of the P-tether furnishing 25 in 65% yield. Consistent with the assembly of syn-(E)-homoallylic alcohols, addition of diethyl-zinc derived organocuprate yielded phosphate acid 26 followed by cleavage with RedAl® to afford diol 27 from allylic displacement at the more sterically accessible exocyclic olefin.[9a]
Scheme 10.

Regio- and diastereoselective reactivity of 4.
Having established the preference of the organocuprate to approach toward the more sterically accessible olefin, investigations were done to observe selectivity on allylic displacement of the endocyclic olefin. Reduction of the exocyclic double bond was carried out under an atmosphere of H2 in presence of Wilkinson’s catalyst to generate 28 (Scheme 11). Treatment with ethyl cuprate resulted in preferential addition to the C5 carbon. Subsequent phosphate cleavage produced 1,3-anti diol 29 as a single diastereomer. The regioselectivity is attributed to proper coplanar alignment of the π* orbital of the olefin and the σ* orbital of the C–OP(O) moiety (Path A, Scheme 11). Facial selectivity was rationalized by the geometry of the bicyclic skeleton, shielding approach of the organocuprate from the concave face. This result was quite exciting from a standpoint of natural product synthesis, since numerous targets bear the anti/anti-1,3,4-stereotriad within 29.[24]
Scheme 11.

Cuprate selectivity on endocyclic olefin of 4.
Another highlight of selective functionalization of 4 was seen in hydroboration (Scheme 12).[9a] Addition of 9-BBN across the exocyclic olefin, followed by mild oxidation of the corresponding borane (NaBO3·4H2O)[25] furnished a primary alcohol,[26] which was protected with TBDPSOTf to give silyl ether 30 in good yield. Further conversion to triol 31[27] was accomplished by using the same cuprate/methylation/cleavage sequence shown previously to afford triol 31 as the sole product (Scheme 5).
Scheme 12.

Hydroboration/cuprate/cleavage sequence of 4.
The utility of the phosphate tether for the rapid generation of advanced polyol subunits is highlighted in with chiral allylic alcohol 32, successful RCM gave the complex phosphate 33. Hydrolysis of the phosphate yielded 35 as the phosphate lithio-salt in 3 steps from 21. Exhaustive hydrogenation[28] of both olefins (H2, 500 psi) and quantitative phosphate cleavage (LiAlH4) afforded the polyol subunit 36 in an efficient 4-step protocol from 21.[9a]
Overall, a number of selective nucleophilic additions to 4 have been demonstrated. These observations highlight the ability of multivalent activation in phosphate triesters to rapidly access complex, differentiated polyol subunits, which are applicable to natural product synthesis.
Phosphate Tethers in Cross Metathesis
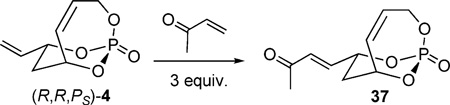
Interest in further manipulation of the bicyclic phosphate through olefin cross-metathesis (CM) led additional studies. Olefin CM has established itself as a powerful technique for accessing highly substituted olefins in a rapid, mild, and selective manner.[29] It was thought that the exocyclic olefin in 4 could be utilized as a cross partner to further functionalize the phosphate scaffold. Initially studies sought to acquire a compatible metathesis catalyst by using excess amounts of methyl vinyl ketone in refluxing CH2Cl2 (Table 2).[9c] Neither Grubbs first- or second-generation catalysts (cat-A and cat-B)[30] were efficient in obtaining CM adduct 37. However, subjecting (R,R,PS)-4 to Hoveyda–Grubbs second-generation catalyst cat-C[31] gave improved yields. The conditions employed in Entries 4 and 5 were developed by Blechert and co-workers, which are compatible with electron-deficient systems.[32] Excellent olefin selectivity was observed (E:Z = 44:1 for Entry 4 and 15:1 for Entry 5), where more dilute conditions promoted a more efficient CM.
Table 2.
Catalyst screening for CM of 4 and methyl vinyl ketone.
 | |||
|---|---|---|---|
| Entry | Catalyst (10 mol-%) | Cone./ solvent | % Yield |
| 1 | cat-A | 0.05 m/CH2Cl2 | <5 [a] |
| 2 | cat-B | 0.05 m/CH2Cl2 | 25 [a] |
| 3 | cat-B | 0.1 m/CH2Cl2 | 26 [a] |
| 4 | cat-C | 0.05 m/CH2Cl2 | 75 [b] |
| 5 | cat-C | 0.1 m/CH2Cl2 | 71 [b,c] |
Conversion determined by 31P NMR spectroscopy.
Yields determined by isolated, purified products.
Used 4.0 equiv. of MVK.
With this result in hand, a series of CM were carried out with various Type I (characterized by rapid homodimerization) and Type II (slow homodimerization) olefins.[33] Using 1.1 equiv. of allyl alcohol and TBS-protected alcohol gave excellent yields and selectivity in presence of 10 mol-% of cat-C (Entries 1–2 in Table 3). When switching to a Bocprotected allylamine (Entry 3), yield of the corresponding CM product decreased, however selectivity was maintained. Implementing allyloxy dimethoxyphosphate (Entry 4) afforded 37 in good yield, albeit in low E:Z selectivity.[9c]
Table 3.
CM of 4 with type I, II, and III olefins. All entries reported used with cat-C catalyst.
| Entry | CM partner |
Product | % Yield | E/Z | Type |
|---|---|---|---|---|---|
| 1 | 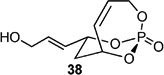 |
86 | >99:1 | I | |
| 2 | 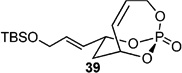 |
87 | >99:1 | I | |
| 3 | 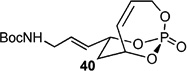 |
69 | >99:1 | I | |
| 4 | 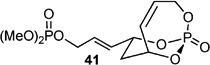 |
80 | 2:1 | I | |
| 5 | 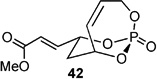 |
78 | 8:1 | II | |
| 6 | 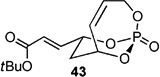 |
60 | 5:1 | II | |
| 7 | 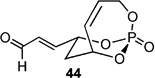 |
78 | >99:1 | II | |
| 8 | 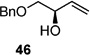 |
 |
72 | >99:1 | II |
| 9 |  |
 |
<5[a] | n/a | III |
| 10 | 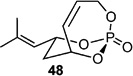 |
<5[a] | n/a | III | |
| 11 | <5[b] | n/a | III | ||
| 12 | 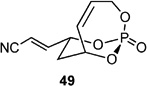 |
n.r.[b] | n/a | III |
Used 4.0 equiv. of CM partner.
Reaction attempted neat.
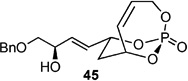
Screening electron-deficient Type II olefins revealed 4–5 equiv. of cross-partner, and 10–12 mol-% of cat-C were required for optimum results. Treatment with methyl acrylate and tert-butyl acrylate (Entries 5–6) gave good selectivity. When switching to acrolein, selectivity increased greatly. Entry 8 shows an attractive extension of this chemistry in which more elaborate cross partners can be exploited to achieve complex subunits. In this example, treatment of 4 with (R)-1-(benzyloxy)but-3-en-2-ol 46 provided smooth conversion to phosphate 45 in good yield and excellent selectivity. Subsequent facile removal of the phosphatetether was achieved in the presence of LiAlH4 (Scheme 14) to construct 50 in a concise five-step sequence.[9c]
Scheme 14.

Five-step protocol from (R,R)-21 to polyol 50.
When attempting CM with various Type-III olefins (no homodimerization),[33] poor reactivity was observed. Treatment with methyl methacrylate for 12 h resulted in clean recovery of the cross partner (Table 3, Entry 9). Trace amounts of product were seen when using varying equivalents of isobutylene (Table 3, Entries 10 and 11). Use of acrylonitrile also resulted in no reaction with the electron-deficient cross partner (Table 3, Entry 12).[34] Throughout these studies, no homodimerization was observed even when subjecting 4 to cat-C for 24 h, leading to the conclusion that the exocyclic olefin behaves as a Type-III partner. A simple sequence to the complex polyol 50 starting from (R,R)-21 highlights the power of this method (Scheme 14).
Overall, this method has been utilized for the synthesis of complex polyketide structures, namely 55, which possesses a key stereotriad found in a number of natural products.[24] Compound 55 was accessed through incorporating a geminal dimethyl group to construct 51, which underwent successful CM with homoallyl alcohol (Scheme 15). Regioselective hydrogenation of the exocyclic olefin was achieved,[35] followed by subsequent PMB-protection of the primary alcohol to afford 52. Consistent with earlier results seen in cuprate additions to these cyclic phosphates, the same three-step protocol towards the anti/anti stereotriad, in compound 55 was achieved in 65% yield.[9c]
Scheme 15.

CM/hydrogenation/cuprate sequence.
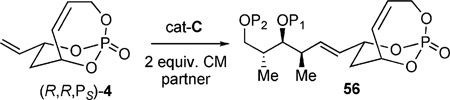
Investigations were carried out to find a cross partner bearing the syn/anti stereotriad contained within dolabelides A–D.[24a, 24b] A noticeable steric effect was observed when changing the protecting group on the primary alcohol of the cross partner (Table 4).[36]
Table 4.
Remote steric effects of cross partners in CM with 4.
 | ||||
|---|---|---|---|---|
| CM partner | cat-C loading | Solvent | Temp. (°C) | Yield (%) |
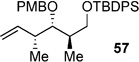 |
6 mol-% | PhCH3 | 60 | N.A. |
| 6 mol-% | PhCH3 | 90 | 28 | |
| 12 mol-% | PhCH3 | 90 | 31 | |
 |
6 mol-% | CH2Cl2 | 50 | 42 |
| 6 mol-% | PhCH3 | 90 | 45 | |
| 12 mol-% | PhCH3 | 90 | 60 | |
| 6 mol-% | DCE | 90 | 72 | |
 |
6 mol-% | CH2Cl2 | 50 | 32 |
| 6 mol-% | PhCH3 | 90 | 63 | |
| 6 mol-% | DCE | 90 | 73 | |
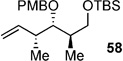
Using the bulky tert-butyldiphenylsilyl (TBDPS, 57 in Table 4) as a protecting group gave poor results, showing the poor reactivity of the cross partner. Switching to the less sterically demanding tert-butyldimethylsilyl (TBS, 58) protecting group gave much better conversion. Optimal conditions were found to utilize 6 mol-% of Hoveyda–Grubbs second-generation catalyst cat-C at elevated temperatures (90 °C) in 1,2-dichloroethane (DCE). Alternatively, use of a PMP-acetal 59 gave similar results to that of 58.[36]
Phosphate Tethers in Natural Product Synthesis
Given the observations from the use of diastereo- and chemoselective cuprate additions, regioselective hydrogenation, cross-metathesis, and facile tether removal in presence of a hydride source, the next step was applying this method in natural product synthesis, namely aimed at the total synthesis of dolabelide C. Fragments 50 and 55 (Scheme 16) were attractive subunits in obtaining the requisite C1–C14 and C15–C30 subunits bearing the necessary stereochemistry required in the total synthesis of the macrolactone dolabelide C.
Scheme 16.

Differentiated polyol subunits accessed from 4.
Dolabelides A–D were isolated from a sea hare, Dolabella auricularia.[24a, 24b] To date, Leighton’s total synthesis of Dolabelide D stands as the lone synthesis of any member in this family.[37] Retrosynthetic analysis revealed two subunits (61 and 62, Scheme 17) bearing stereochemistry, which can be accessed by using the developed P-tether technology. The C1–C14 subunit could be accessed by using chemistry similar to that of fragment 50, and the C15–C30 subunit could be accessed by using chemistry similar to that of fragment 55 (Scheme 9).
Scheme 17.

Retrosynthetic analysis of dolabelide C.
Employing the previously developed CM methodology for the union of bicyclic phosphate (S,S,PR)-4 and the readily prepared subunit 58 resulted in clean reaction to generate the advanced homoallylic PMB-ether 63 in 72% yield. Subsequent regioselective diimide hydrogenation[38] of the exocyclic olefin gave 64. A Pd-formate reduction was next utilized to transpose the C10–C11 olefin to the C11–C12 position for necessary alkene oxidation to introduce the C13–C14 fragment as well as to obtain the C11 carbinol.[36] In this protocol, Pd-formate reduction[39] gave excellent regio-selectivity (preferred internal addition into π-allyl complex 65) of allylic phosphate displacement to afford 66 after methylation of the phosphate acid. Removal of the phosphate ester in presence of LiAlH4 completed a phosphate-mediated sequence constructing the C1–C11 subunit (Scheme 18).
Scheme 18.

Phosphate-mediated construction of C1–C11 subunit.
Final steps to completing the C1–C14 framework included acetonide protection, followed by ozonolysis of the terminal C11–C12 olefin and Grignard addition (derived from 1-iodo-3-methyl-3-butene) to produce 68 (Scheme 19). Oxidation of the C11 carbinol (Dess–Martin periodinane) and removal of the acetonide by using CeCl3·7H2O set the stage for a stereoselective reduction of 69 by using Evan’s syn-reduction[40] conditions (20:1 ds of desired C11 epimer), affording all of the necessary stereocenters (70) in 13 steps from phosphate (S,S,PR)-4.[36]
Scheme 19.

Final steps to C1–C14 subunit.
The strategy toward the C15–C30 subunit employed phosphate-mediated synthesis of aldehyde 71 (Scheme 20), which was then to be subjected toward a coupling with vinyl iodide 72 to afford desired intermediate 62.[41] Utilizing the aforementioned regioselective hydroboration, starting from (R,R,PS)-4, the resultant primary alcohol was protected by using PMB-imidate. Intermediate 73 was subjected to cuprate conditions (Me2Zn, CuCN·2LiCl), yielding a single diastereomer at C22 (Scheme 21). Subsequent methylation gave phosphate ester 74, followed by tether removal, afforded diol 75. Acetonide protection of the 1,3-diol or a two-step sequence to obtain orthogonal protection yielded intermediates 76a and 76b, respectively. Both species underwent successful oxidation of their terminal olefins under ozonolysis conditions to yield both aldehydes (77a and 77b) by a phosphate-mediated approach.
Scheme 20.

Retrosynthetic analysis of the C15–C30 subunit.
Scheme 21.

Phosphate-mediated approach to aldehydes 76a and 76b.
Alternatively, a CM-selective hydrogenation approach (as previously shown in Scheme 15), was also employed to append the C15–C18 framework. The two-step protocol smoothly converted (S,S,PR)-4 to 79, followed by cuprate addition and tether removal to afford diol 81. Orthogonal protection and ozonolysis furnished aldehyde 83 over a three-step sequence (Scheme 22).[41]
Scheme 22.

Alternative approach to C15–C30 subunit.
Aldehydes 77b and 83 were coupled together with a vinyl-lithiate generated from treating vinyl iodide 84 with tert-butyllithium to give a 1:1 ratio of C23 epimers for both cases (Scheme 23). Protection of the C23 carbinol with MOMCl gave fully protected polyols 87a and 87b, which were converted to a C15–C30 subunit of dolabelide C 88, via different pathways.
Scheme 23.

Final steps to the C15–C30 subunit of dolabelide C.
Conclusions
Phosphate tethers are effective tools in constructing advanced polyol synthons rapidly from simple glycidyl ether 6 or C2-symmetric diol 20. Utilization of divalent activation from monocyclic tethers 1–3 to construct homoallylic alcohols by regio- and stereoselective cuprate additions further display the effectiveness of SN2′ displacements of allylic phosphates. In addition, multivalent activation from the bicyclo[4.3.1]phosphate 4 has been expanded by the aforementioned cuprate chemistry, along with other selective nucleophilic additions, including hydride, hydroxide, and sulfide. Furthermore, regioselective hydrogenation and hydroboration, in conjunction with Type-III behavior in cross-metathesis reactions position this method for use in advanced polyol synthesis. Overall, synthons accessed by this methodology have been applied to natural product synthesis. The potential for further application of P-tethers in total synthesis will be reported in due course.
Scheme 13.

Construction of complex differentiated polyol subunits from 4.
Acknowledgments
The authors would like to thank the National Science Foundation (NSF) (NSF CHE-0503875) and the National Institute of General Medical Sciences (NIH RO1 GM077309) for their kind and generous support of our program. The authors also kindly acknowledge Daiso Co., Ltd., Fine Chemical Department for donating each antipode of both benzyl- and trityl-protected glycidols and Materia, Inc. for supplying metathesis catalysts and helpful suggestions.
Biographies

Christopher D. Thomas was born in Kansas City, MO (U.S.A.) in 1981. He completed his B.S. degree in 2005 from Northwest Missouri State University in Maryville, MO (U.S.A.), where he completed two summers of undergraduate research under the direction of Professor Barrett E. Eichler. Since 2005, he has been working under the supervision of Professor Paul R. Hanson at the University of Kansas in Lawrence, KS (U.S.A.), where his efforts are focused on the use of phosphate tethers in the total synthesis of complex natural products.

James P. McParland, born in Topeka, KS, obtained his B.S. degree from Loyola University-Chicago in 2003 with a major in chemistry under the direction of James H. Babler. He then matriculated to the University of Kansas, Department of Chemistry, subsequently entering graduate work under the guidance of Paul R. Hanson. His research has involved regioand stereoselective displacement of phosphates to generate advanced polyol synthons and the utilization of phosphate tethered systems in natural product synthesis.

Paul R. Hanson was born in 1963 in Mason City, IA, and graduated from Hampton High School in Hampton, IA, in 1981. He received his B. S. degree in 1985 from Luther College (Decorah, IA) with a major in chemistry under the direction of Adrian Docken. In 1993, he obtained his Ph. D. in chemistry from the University of Minnesota under the mentorship of Thomas R. Hoye. From 1993 to 1996, he served as an NIH postdoctoral fellow at Stanford University under the guidance of Barry M. Trost. His independent research career began in 1996 at the University of Kansas where he was hired as an assistant professor of chemistry. He was promoted to associate professor in 2001 and full professor in 2005. His current interests lie in the development of new approaches to the synthesis of phosphorus and sulfur heterocyclic compounds with biological and synthetic utility, natural product synthesis, chemical methods for library development, and the design of high-load oligomers with tunable properties for use in facilitated synthesis.
References
- 1.a) Fensterbank L, Malacria M, Sieburth S. Synthesis. 1997:813–854. [Google Scholar]; b) Gauthier DR, Zandi KS, Shea KJ. Tetrahedron. 1998;54:2289–2338. [Google Scholar]
- 2.a) White JD, Carter RG. In: Science of Synthesis: Houben-Weyl, Methods of Molecular Transformations. Fleming I, editor. vol. 4. Stuttgart, New York: Thieme; 2001. pp. 371–412. and references cited therein. [Google Scholar]; b) Evans PA, Cui J, Gharpure SJ, Polosukhin A, Zhang HR. J. Am. Chem. Soc. 2003;125:14702–14703. doi: 10.1021/ja0384734. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cox LR, Ley SV. In: Templated Organic Synthesis. Diederich F, Stang PJ, editors. Weinheim: Wiley-VCH; 2000. pp. 275–395. [Google Scholar]
- 3.a) Corey EJ, Boaz NW. Tetrahedron Lett. 1984;25:3063–3066. [Google Scholar]; b) Yanagisawa A, Noritake Y, Yamamoto N, Nomura H. Synlett. 1991:251–253. [Google Scholar]; c) Lipshutz B. Synlett. 1990:119–128. [Google Scholar]; d) Magid RM. Tetrahedron. 1980;36:1901–1930. [Google Scholar]
- 4.a) Takai K, Oshima K, Nozaki H. Tetrahedron Lett. 1980;21:2531–2534. [Google Scholar]; b) Sato M, Takai K, Oshima K, Nozaki H. Tetrahedron Lett. 1981;22:1609–1612. [Google Scholar]; c) Fugami K, Oshima K, Utimoto K. Chem. Lett. 1987:2203–2206. [Google Scholar]; d) Nicolaou KC, Namoto K. Chem. Commun. 1998:1757–1758. doi: 10.1039/b104949f. [DOI] [PubMed] [Google Scholar]; e) Lepifre F, Clavier S, Bouyssou P, Coudert G. Tetrahedron. 2001;57:6969–6975. [Google Scholar]; f) Kadota I, Takamura H, Sato K, Yamamoto Y. Tetrahedron Lett. 2001;42:4729–4731. [Google Scholar]
- 5.Morin MD, Rychnovsky SD. Org. Lett. 2006;8:2051–2053. doi: 10.1021/ol050567q. [DOI] [PubMed] [Google Scholar]
- 6.a) Bartlett PA, Jernstedt KK. J. Am. Chem. Soc. 1977;99:4829–4830. [Google Scholar]; b) Bartlett PA, Jernstedt KK. Tetrahedron Lett. 1980;21:1607–1610. [Google Scholar]
- 7.a) Letsinger RL, Finnan JL, Heavner GA, Lunsford WB. J. Am. Chem. Soc. 1975;97:3278–3279. doi: 10.1021/ja00844a090. [DOI] [PubMed] [Google Scholar]; b) Beaucage SL, Caruthers MH. In: Bioorganic Chemistry: Nucleic Acids. Hecht SM, editor. New York: Oxford University Press; 1996. pp. 36–74. chapter 2. [Google Scholar]
- 8.Westheimer FH. Science. 1987;235:1173–1178. doi: 10.1126/science.2434996. [DOI] [PubMed] [Google Scholar]
- 9.a) Whitehead A, McReynolds MD, Moore JD, Hanson PR. Org. Lett. 2005;7:3375–3378. doi: 10.1021/ol0512886. [DOI] [PubMed] [Google Scholar]; b) Whitehead A, McParland JP, Hanson PR. Org. Lett. 2006;8:5025–5028. doi: 10.1021/ol061756r. [DOI] [PubMed] [Google Scholar]; c) Waetzig JD, Hanson PR. Org. Lett. 2006;8:1673–1676. doi: 10.1021/ol0602809. [DOI] [PubMed] [Google Scholar]
- 10.Yanagisawa A, Noritake Y, Nomura N, Yamamoto H. Synlett. 1991;4:251–253. [Google Scholar]
- 11.a) Belelie JL, Chong M. J. Org. Chem. 2001;66:5552–5555. doi: 10.1021/jo0104431. [DOI] [PubMed] [Google Scholar]; b) Calaza MI, Hupe E, Knochel P. Org. Lett. 2003;5:1059–1061. doi: 10.1021/ol0340742. [DOI] [PubMed] [Google Scholar]
- 12.a) Piarulli U, Daubos P, Claverie C, Roux M, Gennari C. Angew. Chem. Int. Ed. 2003;42:234–236. doi: 10.1002/anie.200390088. [DOI] [PubMed] [Google Scholar]; b) Piarulli U, Claverie C, Daubos C, Gennari C, Minnaard AJ, Feringa BL. Org. Lett. 2003;5:4493–4496. doi: 10.1021/ol035807l. [DOI] [PubMed] [Google Scholar]
- 13.a) Kacprznski MA, Hoveyda AH. J. Am. Chem. Soc. 2004;126:10676–10681. doi: 10.1021/ja0478779. [DOI] [PubMed] [Google Scholar]; b) Larsen AO, Leu W, Oberhuber CN, Campbell JE, Hoveyda AH. J. Am. Chem. Soc. 2004;126:11130–11131. doi: 10.1021/ja046245j. [DOI] [PubMed] [Google Scholar]
- 14.Davoille RJ, Rutherford DT, Christie SDR. Tetrahedron Lett. 2000;41:1255–1259. [Google Scholar]
- 15.Nosse B, Schall A, Jeong WB, Reiser O. Adv. Synth. Catal. 2005;347:1869–1874. [Google Scholar]
- 16.Cuprate addition leads to loss of the prochiral phosphorus center, thereby providing only two potential products via four diastereomeric pathways. These pathways correspond to addition syn- or anti- to the P=O or OMe groups attached to the prochiral phosphorus atom. The other two possible diastereomers, anti-(E)-7 and syn-(Z)-8 can not be accessed from (S,S)-1 via an anti-SN2′-allylic displacement mechanism. Use of meso-1 would be required to access these two diastereomers.
- 17. Hoffmann RW. Chem. Rev. 1989;89:1841–1860. b) For A1,3-strain in acyclic allylic phosphates, see: Harrington-Frost NA, Leuser H, Calaza MI, Kneisel FF, Knochel P. Org. Lett. 2003;5:2111–2114. doi: 10.1021/ol034525i.
- 18.a) Burke SD, Beaudry N, Muller CM. Org. Lett. 1999;1:1827–1829. doi: 10.1021/ol9910971. [DOI] [PubMed] [Google Scholar]; b) Burke SD, Voight EA. Org. Lett. 2001;3:237–240. doi: 10.1021/ol006887l. [DOI] [PubMed] [Google Scholar]; c) Lambert WT, Burke SD. Org. Lett. 2003;5:515–518. doi: 10.1021/ol027389a. [DOI] [PubMed] [Google Scholar]
- 19.Rychnovsky SD, Griesgraber G, Powers JP. Org. Synth. 2000;77:1–11. [Google Scholar]
- 20.It is known that when oxygen lone pairs occupy a trans-app position to an adjacent polar bond (P-OR), they donate electron density from their lone pair orbital, n, to the antibonding O–P σ*, thus weakening the P=O bond and increasing the basicity at O. Conversely, lack of app lone pairs imparts orthogonal stability to hydrolysis with acid; Deslongchamps P. Stereoelectronic Effects in Organic Chemistry. Oxford: Pergamon Press; 1983. Vande Griend LJ, Verkade JG, Pennings JFM, Buck HM. J. Am. Chem. Soc. 1977;99:2459–2463.
- 21.Thatcher GRJ, Kluger R. Adv. Phys. Org. Chem. vol. 25. Academic Press, Inc.; 1989. Mechansim and Catalysis of Nucleophilic Substitution in Phosphate Esters. [Google Scholar]
- 22.The most prominent synthetic case is a recent report by Imanishi and coworkers, see: Miyashita K, Ikejiri M, Kawasaki H, Maemura S, Imanishi T. J. Am. Chem. Soc. 2003;125:8238–8243. doi: 10.1021/ja030133v.
- 23.a) Cox JR, Ramsay JOB., Jr Chem. Rev. 1964;64:317–352. [Google Scholar]; b) Bunton CA. Acc. Chem. Res. 1970;3:257–265. [Google Scholar]; c) Westheimer F. Acc. Chem. Res. 1968;1:70–78. [Google Scholar]; d) Gorenstein DG, Rowell R, Taira K. Stereoelectronic Effects in Phosphate Esters, ACS Symposium Series. 1981;171:69–75. [Google Scholar]; e) Fanni T, Taira K, Gorenstein DG, Vaidyanathaswamy R, Verkade JG. J. Am. Chem. Soc. 1986;108:6311–6314. and references cited therein. [Google Scholar]
- 24.Dolabelides A–D: Ojika M, Nagoya T, Yamada K. Tetrahedron Lett. 1995;36:7491–7494. Suenaga K, Nagoya T, Shibata T, Kigoshi H, Yamada K. J. Nat. Prod. 1997;60:155–157. Salicylihalamides: Wu Y, Seguil OR, De Brabander JK. Org. Lett. 2000;2:4241–4244. doi: 10.1021/ol0068086. Holloway GA, Hugel HM, Rizzacasa MA. J. Org. Chem. 2003;68:2200–2204. doi: 10.1021/jo026798h. Snider B, Song F. Org. Lett. 2001;3:1817–1820. doi: 10.1021/ol015822v. Bitungolides A-F: Sirirath S, TanakaI J, Ohtani II, Ichiba T, Rachmat R, Ueda K, Usui T, Osada H, Higa T. J. Nat. Prod. 2002;65:1820–1823. doi: 10.1021/np0200865. Rhizoxin D: Lafontaine JA, Provencal DP, Gardelli C, Leahy JW. Tetrahedron Lett. 1999;40:4145–4148. (+)-Discodermolide: Smith AB, III, Kaufman MD, Beauchamp TJ, LaMarche MJ, Arimoto H. Org. Lett. 1999;1:1823–1826. doi: 10.1021/ol9910870.
- 25.Lucas BS, Luther LM, Burke SD. Org. Lett. 2004;6:2965–2968. doi: 10.1021/ol0488800. [DOI] [PubMed] [Google Scholar]
- 26.The yield of the hydroboration reaction was optimized later in ref.[41]
- 27.TBDPS was unexpectedly removed during the 3-step protocol.
- 28.Louie J, Bielawski CH, Grubbs RH. J. Am. Chem. Soc. 2001;123:11312. doi: 10.1021/ja016431e. [DOI] [PubMed] [Google Scholar]
- 29.a) Grubbs RH. Handbook of Metathesis. Weinheim, Germany: Wiley-VCH; 2003. [Google Scholar]; b) Connon SJ, Blechert S. Angew. Chem. Int. Ed. 2003;42:1900. doi: 10.1002/anie.200200556. [DOI] [PubMed] [Google Scholar]
- 30.a) Schwab P, Grubbs RH, Ziller JW. J. Am. Chem. Soc. 1996;118:100–110. [Google Scholar]; b) Schwab P, France MB, Ziller JW, Grubbs RH. Angew. Chem. Int. Ed. Engl. 1995;34:2039–2041. [Google Scholar]
- 31.For a review see: Hoveyda AH, Gillingham DG, Van Veldhuizen JJ, Kataoka O, Garber SB, Kingsbury JS, Harrity JPA. Org. Biol. Chem. 2004;2:8–23. doi: 10.1039/b311496c. and references cited therein. b) A study of CM reaction using Hoveyda-Grubbs catalyst was reported by: Cossy J, BouzBouz S, Hoveyda AH. J. Organomet. Chem. 2001;624:327–332.
- 32.Dewi P, Randl S, Blechert S. Tetrahedron Lett. 2005;46:577–580. [Google Scholar]
- 33.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH. J. Am. Chem. Soc. 2003;125:11360–11370. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 34.Hoyveda HR, Vezina M. Org. Lett. 2005;7:2113. doi: 10.1021/ol050387g. [DOI] [PubMed] [Google Scholar]
- 35.a) Drouin SD, Zamanian F, Fogg DE. Organometallics. 2001;20:5495–5497. [Google Scholar]; b) Bielawski CW, Louie J, Grubbs RH. J. Am. Chem. Soc. 2000;122:12872–12873. [Google Scholar]
- 36.Waetzig JD, Hanson PR. Org. Lett. 2008;10:109–112. doi: 10.1021/ol7025944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park PK, O’Malley SJ, Schmidt DR, Leighton JL. J. Am. Chem. Soc. 2006;128:2796–2797. doi: 10.1021/ja058692k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myers AG, Zheng B, Movassaghi M. J. Org. Chem. 1997;62:7507. doi: 10.1021/jo9710137. [DOI] [PubMed] [Google Scholar]
- 39.a) Hughes G, Lautens M, Wen C. Org. Lett. 2000;2:107–110. doi: 10.1021/ol991170n. [DOI] [PubMed] [Google Scholar]; b) Chau A, Paquin J-F, Lautens M. J. Org. Chem. 2006;71:1924–1933. doi: 10.1021/jo052267s. [DOI] [PubMed] [Google Scholar]
- 40.a) Chen K-M, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155–158. [Google Scholar]; b) Evans DA, Chapman KT, Carreira EM. J. Am. Chem. Soc. 1988;110:3560–3578. [Google Scholar]
- 41.Whitehead A, Waetzig JD, Thomas CD, Hanson PR. Org. Lett. 2008;10:1421–1424. doi: 10.1021/ol8001865. [DOI] [PMC free article] [PubMed] [Google Scholar]