

Abstract
Mammalian cells contain thousands of copies of mitochondrial DNA (mtDNA). At birth, these are thought to be identical in most humans. Here, we use long read length ultra-deep resequencing-by-synthesis to interrogate regions of the mtDNA genome from related and unrelated individuals at unprecedented resolution. We show that very low-level heteroplasmic variance is present in all tested healthy individuals, and is likely to be due to both inherited and somatic single base substitutions. Using this approach, we demonstrate an increase in mtDNA mutations in the skeletal muscle of patients with a proofreading-deficient mtDNA polymerase γ due to POLG mutations. In contrast, we show that OPA1 mutations, which indirectly affect mtDNA maintenance, do not increase point mutation load. The demonstration of universal mtDNA heteroplasmy has fundamental implications for our understanding of mtDNA inheritance and evolution. Ostensibly de novo somatic mtDNA mutations, seen in mtDNA maintenance disorders and neurodegenerative disease and aging, will partly be due to the clonal expansion of low-level inherited variants.
INTRODUCTION
Nucleated mammalian cells contain thousands of copies of the mitochondrial genome. Until recently, it has been generally accepted that, for the vast majority of humans, all mitochondrial DNA (mtDNA) molecules are identical at birth (homoplasmy) (1,2) However, recent work has shown that ∼25% of healthy individuals inherit a mixture of wild-type and variant mtDNA (heteroplasmy), which almost exclusively involves the non-coding mtDNA D-loop (3). Even less frequently, <1 in 200 inherit a potentially pathogenic variant of mtDNA in the coding region (4). Being exclusively maternally inherited, mtDNA undergoes negligible recombination manifested at the population level. As a result, homoplasmic variation of mtDNA has played a key role in determining population migrations on a global scale (5) and in the confident identification of biological samples in forensic medicine (6). Heteroplasmic variants can confound the situation in both circumstances.
MtDNA is highly mutable, with an estimated mutation rate of at least ∼5–15 times that of the nuclear genome. This is partly a result of proximity to the electron transport chain that is the major intracellular source of oxidative free radicals, and partly a result of the relatively limited mtDNA protection and repair mechanisms (7). The high mutation rate contributes to the high levels of mtDNA diversity and also the generation of somatic mtDNA mutations with aging (8). Once present, the intracellular level of both inherited and somatic mtDNA mutations can change during life through the unequal partitioning of mitochondrial genotypes, which can occur during cytokinesis in dividing cells (vegetative segregation) or within a non-dividing cell through the relaxed replication of mtDNA. An increase in heteroplasmy is often referred to as ‘clonal expansion’ that may be enhanced by a replicative advantage for specific mutation types (9). If deleterious mutations exceed a critical threshold level within a cell, they can cause a biochemical defect in the mitochondrial respiratory chain (10). This often involves cytochrome c oxidase (COX), leads to increased free radical production, a defect of ATP synthesis, cell dysfunction and ultimately cell death (1). High percentage levels of inherited mtDNA mutations cause multisystem mitochondrial diseases in ∼1 in 10 000 of the population (11), and high percentage levels of somatic mutations have been described in several age-associated degenerative diseases and possibly contribute to the aging process (12). Understanding the origin of mtDNA variants, therefore, has far-reaching implications, with impact on medicine, human anthropology and forensic science.
Although generally assumed that age-related mtDNA mutations originate during life, it is equally plausible that some ostensibly somatic variants are actually inherited, but fall below the detection threshold of previously applied technologies. With the recent development of massively parallel sequencing, it is now possible to definitively address this issue through the detection of very low-level heteroplasmic variants. Recent studies using whole mtDNA genome sequencing on the Illumina GA/Solexa platforms have suggested that a single heteroplasmic variant can be detected in ∼25% of individuals (3,13–15). However, the relatively conservative detection thresholds (a minor allele frequency of >1.5–10%) used in these studies mean that lower frequency variants were not reported, despite the fact that, from first principles, most somatic mtDNA variants will be expected to fall below this level.
We, therefore, sought to improve the depth of resolution for very low-frequency heteroplasmy by developing an amplicon-based resequencing method on the Roche 454 GS FLX platform. Despite the very great depth of coverage per base position achieved in previous studies using other platforms (3,13–15), the achievable lower limit of resolution for heteroplasmic mtDNA variants is pragmatically limited by the ‘noise’ generated principally during the sequencing reaction. Amplicon resequencing on the 454 GS FLX platform has, thus, become the method of choice for ultra-deep resequencing (UDS) of mutants and quasi-species within other small genomes, for example viruses (16) and bacteria (17), where resolution in the 0.1–1% variant frequency range has been described.
RESULTS
Developing a highly sensitive and specific method of detecting very low-level mtDNA sequence variants
At the population level, MT-HV2 within the mtDNA non-coding control region is observed to be more polymorphic than the coding region (18). We, therefore, designed two mtDNA amplicons to compare the base substitution rate in two regions in different subjects: one in MT-HV2 (nt 162–455, 294 bp) and one in the coding region MT-CO3 (nt 9307–9591, 285 bp) (see Supplementary Material, Table S1). As nuclear pseudogenes [nuclear mitochondrial sequences (NumtS)] have the potential to confound deep resequencing of mtDNA, we took a three-stage process to exclude the possibility of this affecting our data. Firstly, we compared our amplicon loci with published NumtS to determine the extent to which they nested within these regions and the corresponding degree of sequence identity (19). Only one of the NumtS had a high sequence identity with modern mtDNA (>90%) and was predicted to lie within our MT-CO3 amplicon (Supplementary Material, Table S2). However, this NumtS was still predicted to differ by six nucleotide substitutions from modern mtDNA, which significantly exceeded the number of variants ever seen on single reads within our dataset. Secondly, we used Primer-BLAST analysis of each amplicon primer pair (hg19 build), which showed no predicted non-mtDNA PCR product generation (20). Finally, we attempted to PCR amplify a product using genomic DNA extracted from rho0 cells that contained no mtDNA, following ethidium bromide-induced mtDNA depletion to directly confirm our predictions and to look for unexpected NumtS hits. No product was obtained with either amplicon primer set, despite a strong band with a positive control (nuclear gene primers). We, therefore, concluded that nuclear pseudogene amplification was highly unlikely with the specific primers that we designed to amplify MT-HV2 and MT-CO3. In the 454 assay, the mean depth of coverage using these primers was 8391 reads (range 4158–20 803).
Although commercially available lambda phage controls can be used to estimate the level of background sequencing artifacts, differences in guanine-cytosine content of the cloned templates can influence the outcome. We, therefore, designed several in-house controls to determine the level of background noise using the 454 platform on mtDNA templates: (i) a nuclear DNA amplicon in BRCA2 (NC_000013.10, 32907099–32907295, 197 bp) was designed to compare the intrinsic properties of mtDNA and nuclear genomic DNA, (ii) a cloned nuclear DNA template corresponding exactly to the BRCA2 amplicons was generated from genomic DNA and (iii) we cloned mtDNA templates from exactly the same mtDNA sequences (MT-HV2, MT-CO3). On this basis, any difference in sequence variants between the cloned and genomic DNA templates from the same individual is highly likely to reflect ‘biological’ sequence variants generated in vivo and not technical artifact arising from the sequencing process.
Being based on pyrosequencing, the base-calling errors on the 454 platform are very largely dependent on poly-mononucleotide tracts (21). As our goal was to maximize specificity in calling very low-level variants, we, therefore, excluded these tracts from all of the analysis. We also introduced a further quality-control step, requiring observed variants to be present in both forward and reverse reads at comparable frequencies. A 3-fold difference was permitted to allow for the effects of a binomial sampling distribution at very low variant levels.
Using this highly stringent approach, there were no variants present at >0.2% heteroplasmy in any of the cloned or genomic DNA templates (Fig. 1). This demonstrated the low background noise level using the 454 UDS approach and indicated that variants detected at >0.2% heteroplasmy are highly likely to be generated in vivo and to be of biological origin.
Figure 1.
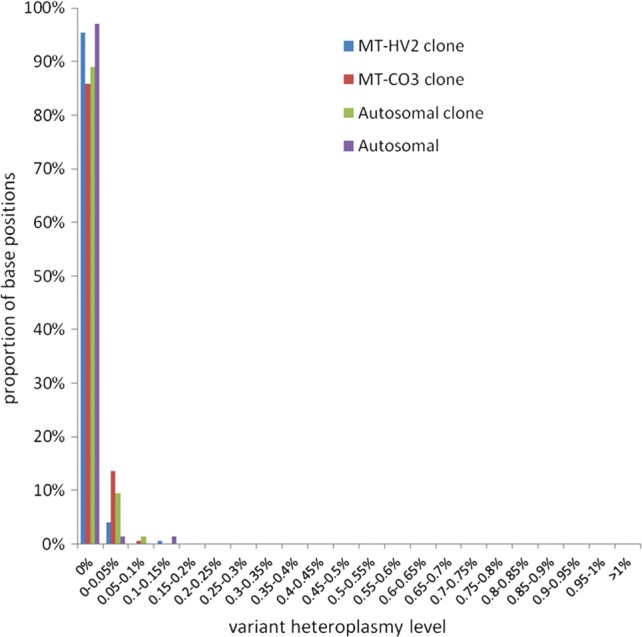
Resolution of ultra-deep sequencing-by-synthesis assay. Demonstration of very low levels of noise in negative controls after quality-control filtering for poly-mononucleotide tracts and bidirectional validation of variants. An amplicon was produced from cloned DNA for each mtDNA amplicon (MT-HV2, MT-CO3) along with an autosomal amplicon (BRCA2) and a clone. All negative controls showed minimal numbers of base positions with any variants, and none at >0.2% heteroplasmy level, with no inherent differences between the different DNA templates.
Further validation of this approach came from repeated experiments performed using the same 454 platform, but different chemistry (Titanium) and different amplicon generation primers. Duplicate experiments were performed on four samples of genomic DNA. There was 100% consistency of variant detection between runs, but 6% of the variants present in the first run fell below the 0.2% threshold in the second run. The heteroplasmy level range for these variants in the second run was 0.12–0.19%, demonstrating that these fell just outside our defined threshold for detection, but were indeed present.
Frequency of very low-level mtDNA sequence variants in healthy control subjects
Next, we studied genomic DNA extracted from whole blood and the skeletal muscle from unrelated and related healthy individuals at different ages. Demographic and genotypic details of the subjects are shown in Supplementary Material, Table S3. Detected mtDNA sequence variants are shown in Supplementary Material, Table S4.
All of the healthy control subjects showed variants at >0.2% heteroplasmy in both the blood and skeletal muscle DNA samples at one or more base positions. These data indicate that very low-level single-base heteroplasmy appears to be a universal finding among different control subjects (Fig. 2A and B). The total number of variant base positions was the same in the blood and skeletal muscle samples, but variants at higher percentage heteroplasmy levels (>2%) were present only in the skeletal muscle of some individuals (three of seven healthy subjects) and never seen in the blood. Higher level variants (>2%) were only found in the MT-HV2 amplicon.
Figure 2.

Very low-level heteroplasmic mtDNA variance was detected by ultra-deep amplicon resequencing. (A) Comparison of variants detected in the skeletal muscle (skm) DNA within the MT-HV2 amplicon in different patient groups shows no difference between healthy control (n = 7) and OPA1 (n = 8) subjects, but a significant excess of variance at all heteroplasmy levels in POLG subjects (n = 8). (B) Comparison of variants detected in the blood DNA in healthy control (n = 7), POLG (n = 4) and OPA1 (n = 7) subjects shows that variants are present, but less common than in the skeletal muscle DNA with the absence of higher level variants (>2% heteroplasmy) and no difference between patient groups.
Overall, the number of variants per base position was significantly more common in the skeletal muscle than in the blood (P = 0.001), and this difference was greater for MT-HV2 than MT-CO3 (P < 0.001). In addition, the number of variants per base position was significantly greater in MT-HV2 than MT-CO3 (P < 0.001). In summary, the greatest frequency of very low-frequency variants was seen in the MT-HV2 region, and both the greatest number of different variants and the highest heteroplasmy levels were seen in the skeletal muscle.
The frequency of very low-level variants in disorders of mtDNA maintenance
We studied patients with inherited defects of POLG encoding the sole mtDNA polymerase γ, predicted to have error-prone mtDNA replication ((22–24). For comparison, we also studied subjects with defects of OPA1. Skeletal muscle biopsies from these individuals show an excess of cells containing clonally amplified mtDNA somatic mutations; however, unlike for POLG defects, the mechanism is believed to be indirect, mediated through defects in mitochondrial fission and fusion, rather than by any direct effect on the polymerase (25–27). Clinical details and pedigrees are shown in Supplementary Material, Table S3 and Supplementary Material, Figure S1.
First, we studied eight patients with a variety of POLG mutations (A467T/A467T, A467T/G848S, A467T/W748S, R627Q/W748S and R627Q/R1096H). In skeletal muscle biopsies, the overall burden of mtDNA variants was greater in patients with POLG mutations when compared with healthy control subjects, but only for MT-HV2, where the majority of variants were seen (P = 0.004) (Fig. 2A). The excess of mtDNA variants was present across all levels of heteroplasmy, and increased significantly with age (r = 0.86, P = 0.006), suggestive of a time-dependent accumulation of low-level somatic mutations. There was no significant correlation of mutation burden with age in healthy controls (r = 0.62, P = 0.14). Given that the mutational burden was significantly greater at lower heteroplasmy levels in POLG when compared with control subjects (for <1% heteroplasmy level variants, P = 0.019), these findings imply that that the higher mutational burden is primarily due to de novo somatic mutation in POLG patients.
We, then, studied multiple tissues from patients with mutations in OPA1 (M1fsX208, S545R, V294fsX667, R905X and intron 8 and intron 15 splicing defects). In contrast to the POLG patients, there was no significant increase in mutational burden in OPA1 patients when compared with healthy controls in either the blood or skeletal muscle for the MT-HV2 or MT-CO3 amplicons (Fig. 2A and B). These findings argue against an increased rate of de novo mutagenesis in patients with OPA1 mutations, consistent with there being no direct effect on the polymerase γ and in keeping with previous suggestions that accelerated clonal expansion of existing somatic mutations is responsible for the cellular cytochrome c oxidase defects (28). We studied the mutational burden in the skeletal muscle of one subject on two occasions 10 years apart. Six heteroplasmic variants were detected in the first muscle sample of this individual. All six variants were also detectable at comparable levels in the follow-up muscle sample taken 10 years later, although two variants in the second sample fell below the 0.2% threshold (0.17%, 0.06%). Overall, five variants showed a decrease in heteroplasmy level and one showed an increase over the 10 year period; however, the absolute changes were small (Supplementary Material, Table S5).
Determining whether very low-level variants are maternally inherited
Having identified low-level heteroplasmic mtDNA variants in the different pedigrees, we, then, compared related and unrelated individuals to determine the extent to which the mtDNA variants arose through maternal transmission or somatic mutation. We focused on families with known nuclear genetic defects associated with mtDNA maintenance because of the larger number of variants available for scrutiny (i.e. increased signal strength).
We compared 13 pairs of samples from 12 maternal first-degree relatives, including one subject with repeated blood and muscle samples taken 10 years apart. To determine the background level of similarity between unrelated individuals, we compared the same tissue between 79 different random pairs of unrelated subjects. Out of 485 variants, 59 variants were common in unrelated subjects (12%). In contrast, when the analysis was restricted to first-degree maternal relatives, 25/63 variants were shared (40%, P < 0.001, Table 1). We also compared the frequency of sharing between 13 pairs of related individuals and 13 random pairs of unrelated controls. This also repeatedly revealed a highly significant difference (P < 0.001), adding further weight to our conclusions. On average, 39% of variants in a given individual were shared with their maternal relative. Interestingly, shared variants in related subjects were principally seen in the skeletal muscle mtDNA, where 17/24 variants were shared (71%), compared with 50/395 variants shared in unrelated subjects (13%, P < 0.001). In contrast, variant sharing in related subjects was rather uncommon in the blood mtDNA, where 8/39 variants were shared (21%) when compared with 9/90 variants in unrelated subjects (10%, P = 0.10).
Table 1.
Extent of sharing of variants in 13 pairs of maternally related and 79 pairs of unrelated samples
| Maternally related samples |
Unrelated samples |
P-value | |||||
|---|---|---|---|---|---|---|---|
| Shared variants (n) | Unshared variants (n) | Shared variants (%) | Shared variants (n) | Unshared variants (n) | Shared variants (%) | ||
| Skeletal muscle | 17 | 7 | 70.8 | 50 | 345 | 12.7 | <0.0001 |
| Blood | 8 | 31 | 20.5 | 9 | 81 | 10.0 | 0.10 |
| All samples | 25 | 38 | 39.7 | 59 | 426 | 12.2 | <0.0001 |
These findings have two implications. First, some of the more common MT-HV2 variants are present in the background population at very low heteroplasmy levels. Second, 4-fold more variants are family specific and are transmitted between first-degree maternal relatives. Although it is not possible, without longitudinal study, to state exactly what proportion of variants are due to somatic mutation, the data clearly show that a large proportion appear to be inherited at very low heteroplasmy levels.
DISCUSSION
Using amplicon-based UDS at unprecedented depth, we have shown that detectable very low-frequency mtDNA variants (0.2–2% heteroplasmy) are present in all tested healthy subjects.
It is highly unlikely that nuclear pseudogene (NumtS) contamination influenced our results for the following reasons: Primer-BLAST analysis (hg19 build) failed to identify any predicted non-mtDNA PCR products using our specific MT-HV2 and MT-CO3 primers; we were unable to generate a product from rho0 genomic DNA using these primers. Moreover, further evidence came from the post hoc analysis of individual sequence reads and scrutiny of all predicted nuclear pseudogenes (Supplementary Material, Table S2) (19). Based on this analysis, a minimum of six changes from the mtDNA consensus sequence would be expected in a read from a pseudogene with our MT-CO3 primers and ∼48 changes for the MT-HV2 primers. Read-by-read analysis never identified this number of variants, so pseudogene contamination is extremely unlikely in our dataset. We conclude, therefore, that heteroplasmy is a universal finding in humans. Although our approach does not allow us to state with absolute certainty whether any one variant is inherited or a somatic mutation, a comparison of maternal relatives shows substantial sharing between first-degree relatives, and our findings, therefore, show that inherited variants make a significant contribution to the overall mutation load in any one individual.
None of the heteroplasmic variants detected were haplogroup defining or haplogroup specific. Twelve of 40 base positions containing heteroplasmic variants are reported to be polymorphic (>1% of reported sequences with variants) at the population level (http://www.mtdb.igp.uu.se/). None of the variants detected have been ascribed definite pathogenic potential, although a single mutation at position m.9544 has previously been associated with optic neuropathy. We also examined whether variants detected had previously been reported as somatic mutations. In our data, 21 of 31 control region (MT-HV2) heteroplasmic variants were detected, and 2 of 9 coding region (MT-CO3) variants have previous been reported as somatic variants (http://www.mitomap.org/MITOMAP) (Supplementary Material, Table S4).
Using this approach, we also show that a disrupted mtDNA polymerase γ due to a POLG defect leads to increased levels of mutations, and that these increase during life, mirroring observations in POLG-deficient mice (29,30). In contrast, we did not observe a correlation between age and mutation burden in control subjects, whether in the blood or muscle. This suggests that de novo mutagenesis throughout life is unlikely to contribute significantly to the cellular COX defects observed in healthy aged individuals. Rather, age-related COX defects are more likely to be the result of clonal expansions of mutations occurring during early life (31). This notion has recently been demonstrated in aged colonic crypt cells, where multimethod measurement of mtDNA mutation load failed to demonstrate an age-dependent increase (32). However, our observation of very low-frequency heteroplasmy transmission indicates that that many of these mutations will be inherited down the maternal line. As a result, all clonally expanded species need not be the result of somatic mutation events in early life, and some could have arisen from low-level inherited variants.
The changes in low-level heteroplasmy value between each mother and offspring (Supplementary Material, Figure S1 and Supplementary Material, Table S4) are relatively small when compared with the shifts observed in patient pedigrees carrying higher levels of pathogenic mutations (33). However, this is consistent with the neutral drift theory (34) which predicts that the variance in the offspring heteroplasmy due to the mtDNA bottleneck decreases as the mother's mutation level decreases. These are the first data to show the inheritance of such low-level mtDNA mutations. The high rate of inheritance of the low-level mutation in muscle (71%) may be surprising, considering our expectations of the effect of the mtDNA inheritance bottleneck; however, two points should be considered here. First, that muscle data is from only two sibling pairs [subjects A-B and P-Q (Supplementary Material, Figure S1 and Supplementary Material, Table S4)]. Second, as noted above, the neutral drift theory predicts that very low-level mutations will have low variance in the offspring, making them more likely to be preserved through the mtDNA inheritance bottleneck.
The different patterns of mutation observed in the blood and skeletal muscle are, at least in part, likely to be a consequence of different rates of cell and mtDNA turnover between dividing (blood) and post mitotic tissues. Rapid turnover of blood cells can lead to the loss of mtDNA mutations during life, either through selection against a particular mutation (35) or simply by genetic drift (36). On the other hand, the loss of mutations is much less likely in a post mitotic tissue such as the skeletal muscle, where the replication of mtDNA can lead to an increase in mutation load during life within individual cells and the tissue as a whole, even from very low levels of heteroplasmy (31). Thus, it is plausible that an inherited mutation is lost from the blood, but detected in the muscle, explaining why some inherited mutations are more likely to be detected in the muscle than in the blood.
It is intriguing that the frequency of low-level variants in MT-HV2 is significantly greater than in MT-CO3 in healthy control subjects (OR 3.3). Why should this be the case, given that our UDS of cloned mtDNA showed no intrinsic sequence-specific difference between the two templates? One possible explanation is that much of the non-coding D-loop heteroplasmy is actually inherited at a very low level. Being a non-coding region, these substitutions may be tolerated during transmission, unlike coding region variants that undergo strong negative selection during transmission (37).
Perhaps most importantly, we show here that next generation sequencing has the potential to reliably detect very low levels of heteroplasmy, when a very stringent analytical approach is employed. It is important to note that we did not perform a head-to-head comparison of different next generation sequencing platforms, so it would be premature to conclude that the 454 approach is superior to other platforms. However, using this method, we have gained novel insight into mtDNA within individuals and within pedigrees. Prospective studies in larger family-based cohorts will substantiate these findings. However, given that mtDNA heteroplasmy levels can change dramatically during life, and during maternal transmission, the finding of universal mtDNA heteroplasmy has significant implications for our understanding of mtDNA at the population, family and individual level. If deleterious mutations are inherited, these have the potential to accumulate within single cells during life and thus contribute to neurodegenerative disease. Or, if they segregate rapidly through the mtDNA bottleneck, they could lead to a maternally inherited mitochondrial disorder. This places greater emphasis on the importance of developing techniques to prevent the transmission of mtDNA heteroplasmy and preventing the clonal expansion of pre-existing mtDNA mutations.
MATERIALS AND METHODS
Patient and samples
All subjects gave informed consent for participation in research. Characteristics of patient and control samples are shown in Supplementary Material, Table S3 and Supplementary Material, Figure S1.
Molecular analyses
Amplicon resequencing was performed on the Roche 454 GS FLX platform. Two mtDNA amplicons were used: MT-HV2 (NC_012920, nt.162–455) and MT-CO3 (nt.9307–9591). Confirmatory experiments on a subset of samples used the following amplicon positions: MT-HV2 (nt.109–483) and MT-CO3 (nt.9304–9653). Full primer sequences are shown in Supplementary Material, Table S1. Owing to the potential of nuclear pseudogenes to confound deep resequencing studies of mtDNA, we used BLAST to ensure that our primers avoided these areas and confirmed this by lack of amplification from mtDNA deplete rho0 DNA. Amplicon-specific mtDNA clones comprised the following inserts in a pGEM-T-easy vector (Promega): MT-HV2 clone (nt.16548–771) and MT-CO3 clone (nt.9127–9661). Autosomal negative control comprised amplicon BRCA2 (NC_000013.10, 32907099–32907295) and BRCA2 clone (32906828–32907480). Amplicon generation PCR reactions were performed in a 50 µl volume comprising: 1x buffer for KOD Hot Start DNA Polymerase (Novagen), 1.5 mm MgSO4, 0.2 mm dNTPs, 0.3 μm primers, 1 U KOD Hot Start DNA Polymerase (Novagen) and 100 ng DNA. Cycling conditions were 95°C for 2 min followed by 30 cycles of 95°C for 20 s, 60°C for 10 s and 70°C for 4 s. EmPCR and bidirectional sequencing were performed according to the manufacturer's instructions (Roche 454).
Bioinformatic analyses
Base calling and alignment were performed using the algorithm of PyroBayes and Mosaik. Subsequent analysis of variants was performed using the custom R library flowgram. To maximize specificity for very low-level variants, base positions within or immediately adjacent to poly-mononucleotide tracts were excluded from analysis. Variant calls were then validated by comparison of read directions.
Primer-BLAST (20) analysis to confirm complete specificity of amplicon generation primer pairs was performed using standard stringent parameters: human genome assembly hg19, blast E value 30 000and unintended targets with <6 mismatches were considered.
Statistical analyses
Comparison of proportions of base positions between groups was done using the chi-squared test.
SUPPLEMENTARY MATERIAL
FUNDING
Funding to pay Open Access publication for this article was provided by the Wellcome Trust.
Supplementary Material
Acknowledgements
B.P. is a M. R. C. Clinical Research Training Fellow. P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science and an NIHR Senior Investigator who also receives funding from the Medical Research Council (UK) and the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust. R.H. was supported by the MRC (UK). P.Y.W.M. is an MRC Clinician Scientist. D.C.S. was supported by the National Institutes of Health through grant GM073744. R.W.T. was supported by the Wellcome Trust (906919).
Conflict of interest statement. All authors confirm that they have no conflict of interest to declare.
References
- 1.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parsons T.J., Muniec D.S., Sullivan K., Woodyatt N., Alliston-Greiner R., Wilson M.R., Berry D.L., Holland K.A., Weedn V.W., Gill P., Holland M.M. A high observed substitution rate in the human mitochondrial DNA control region. Nat. Genet. 1997;15:363–368. doi: 10.1038/ng0497-363. [DOI] [PubMed] [Google Scholar]
- 3.Li M., Schonberg A., Schaefer M., Schroeder R., Nasidze I., Stoneking M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 2010;87:237–249. doi: 10.1016/j.ajhg.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torroni A., Achilli A., Macaulay V., Richards M., Bandelt H.J. Harvesting the fruit of the human mtDNA tree. Trends Genet. 2006;22:339–345. doi: 10.1016/j.tig.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Just R.S., Leney M.D., Barritt S.M., Los C.W., Smith B.C., Holland T.D., Parsons T.J. The use of mitochondrial DNA single nucleotide polymorphisms to assist in the resolution of three challenging forensic cases. J. Forensic Sci. 2009;54:887–891. doi: 10.1111/j.1556-4029.2009.01069.x. [DOI] [PubMed] [Google Scholar]
- 7.Wallace D.C. Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man. Proc. Natl. Acad. Sci. USA, 2011;107(suppl. 2):8947–8953. doi: 10.1073/pnas.0914635107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- 9.Birky C.W. Relaxed and stringent genomes: why cytoplasmic genes don't obey Mendel's laws. J. Heredity. 1994;85:355–365. [Google Scholar]
- 10.Schon E.A., Bonilla E., DiMauro S. Mitochondrial DNA mutations and pathogenesis. J. Bioenerget. Biomembr. 1997;29:131–149. doi: 10.1023/a:1022685929755. [DOI] [PubMed] [Google Scholar]
- 11.Schaefer A.M., McFarland R., Blakely E.L., He L., Whittaker R.G., Taylor R.W., Chinnery P.F., Turnbull D.M. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 12.Park C.B., Larsson N.G. Mitochondrial DNA mutations in disease and aging. J. Cell. Biol. 2011;193:809–818. doi: 10.1083/jcb.201010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Y., Wu J., Dressman D.C., Iacobuzio-Donahue C., Markowitz S.D., Velculescu V.E., Diaz L.A., Jr, Kinzler K.W., Vogelstein B., Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goto H., Dickins B., Afgan E., Paul I.M., Taylor J., Makova K.D., Nekrutenko A. Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome Biol. 2011;12:R59. doi: 10.1186/gb-2011-12-6-r59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang S., Huang T. Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques. 2010;48: 287–296. doi: 10.2144/000113389. [DOI] [PubMed] [Google Scholar]
- 16.Daly G.M., Bexfield N., Heaney J., Stubbs S., Mayer A.P., Palser A., Kellam P., Drou N., Caccamo M., Tiley L., et al. A viral discovery methodology for clinical biopsy samples utilising massively parallel next generation sequencing. PLoS One. 2011;6:e28879. doi: 10.1371/journal.pone.0028879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soares A.R., Pereira P.M., Santos M.A. Next-generation sequencing of miRNAs with Roche 454 GS-FLX technology: steps for a successful application. Methods Mol. Biol. 2011;822:189–204. doi: 10.1007/978-1-61779-427-8_13. [DOI] [PubMed] [Google Scholar]
- 18.Macaulay V., Richards M., Hickey E., Vega E., Cruciani F., Guida V., Scozzari R., Bonne-Tamir B., Sykes B., Torroni A. The emerging tree of West Eurasian mtDNAs: a synthesis of control-region sequences and RFLPs. Am. J. Hum. Genet. 1999;64:232–249. doi: 10.1086/302204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simone D., Calabrese F.M., Lang M., Gasparre G., Attimonelli M. The reference human nuclear mitochondrial sequences compilation validated and implemented on the UCSC genome browser. BMC Genomics. 2011;12:517. doi: 10.1186/1471-2164-12-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quince C., Lanzen A., Curtis T.P., Davenport R.J., Hall N., Head I.M., Read L.F., Sloan W.T. Accurate determination of microbial diversity from 454 pyrosequencing data. Nat. Methods. 2009;6:639–641. doi: 10.1038/nmeth.1361. [DOI] [PubMed] [Google Scholar]
- 22.Van Goethem G., Dermaut B., Lofgren A., Martin J.-J., Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat. Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- 23.Hakonen A.H., Heiskanen S., Juvonen V., Lappalainen I., Luoma P.T., Rantamaki M., Goethem G.V., Lofgren A., Hackman P., Paetau A., et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am. J. Hum. Genet. 2005;77:430–441. doi: 10.1086/444548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naviaux R.K., Nguyen K.V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004;55:706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 25.Alexander C., Votruba M., Pesch U.E., Thiselton D.L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 26.Hudson G., Amati-Bonneau P., Blakely E.L., Stewart J.D., He L., Schaefer A.M., Griffiths P.G., Ahlqvist K., Suomalainen A., Reynier P., et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–337. doi: 10.1093/brain/awm272. [DOI] [PubMed] [Google Scholar]
- 27.Amati-Bonneau P., Valentino M.L., Reynier P., Gallardo M.E., Bornstein B., Boissiere A., Campos Y., Rivera H., de la Aleja J.G., Carroccia R., et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- 28.Yu-Wai-Man P., Griffiths P.G., Gorman G.S., Lourenco C.M., Wright A.F., Auer-Grumbach M., Toscano A., Musumeci O., Valentino M.L., Caporali L., et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. 2010;133:771–786. doi: 10.1093/brain/awq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J.N., Rovio A.T., Bruder C.E., Bohlooly Y.M., Gidlof S., Oldfors A., Wibom R., et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 30.Kujoth G.C., Hiona A., Pugh T.D., Someya S., Panzer K., Wohlgemuth S.E., Hofer T., Seo A.Y., Sullivan R., Jobling W.A., et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 31.Elson J.L., Samuels D.C., Turnbull D.M., Chinnery P.F. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am. J. Hum. Genet. 2001;68:802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greaves L.C., Barron M.J., Plusa S., Kirkwood T.B., Mathers J.C., Taylor R.W., Turnbull D.M. Defects in multiple complexes of the respiratory chain are present in ageing human colonic crypts. Exp. Gerontol. 2010;45:573–579. doi: 10.1016/j.exger.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chinnery P.F., Thorburn D.R., Samuels D.C., White S.L., Dahl H.H., Turnbull D.M., Lightowlers R.N., Howell N. The inheritance of mtDNA heteroplasmy: random drift, selection or both? Trends Genet. 2000;16:500–505. doi: 10.1016/s0168-9525(00)02120-x. [DOI] [PubMed] [Google Scholar]
- 34.Wonnapinij P., Chinnery P.F., Samuels D.C. Previous estimates of mitochondrial DNA mutation level variance did not account for sampling error: comparing the mtDNA genetic bottleneck in mice and humans. Am. J. Hum. Genet. 2010;86:540–550. doi: 10.1016/j.ajhg.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pyle A., Taylor R.W., Durham S.E., Deschauer M., Schaefer A.M., Samuels D.C., Chinnery P.F. Depletion of mitochondrial DNA in leucocytes harbouring the 3243A->G mtDNA mutation. J. Med. Genet. 2007;44:69–74. doi: 10.1136/jmg.2006.043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chinnery P.F., Samuels D.C. Relaxed replication of mtDNA: a model with implications for the expression of disease. Am. J. Hum. Genet. 1999;64:1158–1165. doi: 10.1086/302311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart J.B., Freyer C., Elson J.L., Wredenberg A., Cansu Z., Trifunovic A., Larsson N.G. Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 2008;6:e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.