

Abstract
As molecular techniques have improved, investigators have attempted to improve pharmaceutical efficacy and safety by making trait associations with genomic, epigenomic, transcriptomic, proteomic, and metabolomic polymorphisms. The ‘omics era has seen screening assays for pharmaceutical efficacy and safety translated into clinical practice. This manuscript will discuss each ‘omic field and the screening assays available to the clinician. While success has been demonstrated in each ‘omic field, many challenges remain. Assays need wider availability, predictive values remain low, and costs remain high. In order for clinicians to realize improved efficacy and safety due ‘omic screens, development of improved techniques, combining of ‘omic assays, and increased clinical utilization is necessary. This is an exciting time for investigators and clinicians that desire improved pharmaceutical therapy.
Introduction
The Human Genome Project (HGP) spawned an eruption of research focused on achieving personalized medicine. Clinicians and researchers hoped that sequencing of the human genome would allow for prediction of disease and personalized pharmaceutical therapy based on an individual’s genetic polymorphisms. Quickly researchers realized the limitations of simple genome associations in clinical medicine. The positive and negative predictive values of genome associations have been poor largely making them ineffective for clinical screening, thus researchers have turned to the downstream products of the genetic sequence. Epigenomics, transcriptomics, proteomics, and metabolomics are fields born out of both the increased knowledge as well as the failures of genomics in clinical medicine. The “omics era” has been characterized by a proliferation in translational research to identify markers of disease and factors associated with pharmaceutical efficacy and toxicity. There are an incredible number of ‘omics investigations relating to a myriad of human diseases; a comprehensive review of the available screens for human disease is impractical. The objective of this manuscript is to review the existing screening tests related to pharmaceutical therapies in each ‘omic category. We will progress through the relevant pharmaceutical screens from the underlying genetic code to ultimate clinical phenotype (Figure 1). Inevitably, there will be reference to ‘omic screening in human disease since disease and therapy are irreconcilably entangled.
Figure 1. Schematic flow from genetic code to drug response.
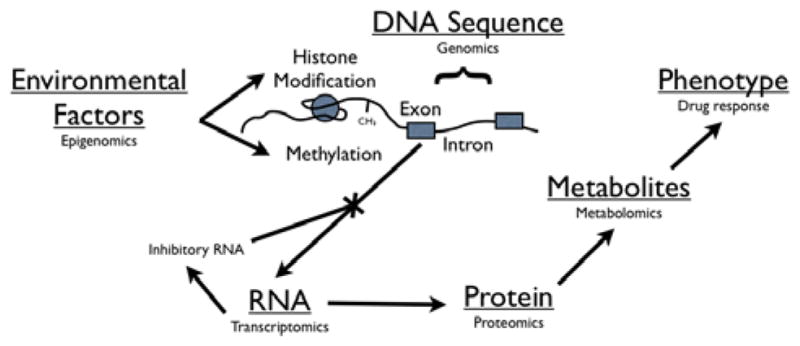
The study of the inherent genetic code is termed “genomics”. The genome can be modified by environmental factors and study of these factors is termed “epigenomics”. Translation of the genome can be modified by RNA factors and study of the interplay and output of these factors is called “transcriptomics”. Characterization of the subsequent protein output is called “proteomics”. “Metabolomics” is the study of the small molecules that are the product of the above biologic functions. Ultimately these biologic interactions result in the phenotypic trait observed as drug response.
Genomics
Genomics is the study of the human genome and the accompanying variability of the contained DNA. Pharmacogenomics, by extension, is the study of how pharmaceutical agents interact with the output of the human genome. In 1956, Arno Motulsky proposed genetic variation as an explanation for individual differences in drug efficacies and adverse reactions. The first pharmacogenomic association was demonstrated with the association of peripheral neuropathy in isoniazid “slow acetylators”in 1960 [1]. Other early genetic associations were largely theoretical until molecular genotyping techniques allowed for a more firm genetic-toxicity linkage. This early neurotoxic-isoniazid association was later confirmed with molecular genotyping techniques. Isoniazid is primarily metabolized by N-acetylation. “Slow acetylators” shunt isoniazid through an alternative CYP isoenzyme to form isonicotinic acid, a neurotoxin that is typically produced in only small amounts by “rapid acetylators” [2].
After the first draft of the HGP was published in 2001 [3], investigators analyzed the genome for common polymorphisms in an attempt to make associations with specific traits within the population. The HapMap Project seeks to catalog common genetic variants describing where the variation occurs within the genome and in which populations worldwide [4,5]. The 1000 Genomes Project seeks to produce an extensive deep catalog of human genetic variation, structural variants, and their haplotype contexts [6]. These projects provide resources for investigators to make large-scale genome level associations. These “genome wide association” (GWA) studies require extremely large sample sizes to reach statistical significance [7]. GWA studies have attempted to provide screening tests for medication-induced toxicity. We have moved from larger gene segment analyses to single base-pair associations using GWA studies. Now single nucleotide polymorphism (SNP) studies seek to characterize functional differences in similar gene outputs putting an even finer point on the genetic polymorphisms within the human population.
Genomic Screens
Microarray assays are the preferred methodology for genotyping analyses because they allow for screening for a large number of polymorphisms with a small sample of blood without the need for multiple DNA extractions and amplification steps, which increase the potential for error. The DNA is isolated, digested, labeled then hybridized within a microarray-well containing the oligonucleotide sequences of interest. Commercial laboratories and pharmaceutical companies now offer United States Food and Drug Association (US FDA) approved screening assays to evaluate for a range of pharmaceutical efficacy and safety (Table 1).
Table 1.
FDA approved/recommended screening assays.
| Screening Test | Polymorphisms Represented | Pertinent Pharmaceuticals Represented | Limitations |
|---|---|---|---|
| Roche CYP2D6/2C19 AmpliChip® | 33 CYP2D6 and 2 CYP19 distinct alleles | 25% of known pharmaceuticals[28]. | Only represents 25% of known CYP2D6 and only 5.5% of known CYP2C19 polymorphisms. |
| Affymetrix Drug Metabolizing Enzyme and Transporters (DMET) | 225 genes involved in drug absorption, distribution, metabolism, and elimination. | Virtually all pharmaceuticals may be affected by polymorphisms one of these enzymes. | It is unclear how to clinically interpret this matrix output. |
| HLA-B 5701 | HLA-B*5701 | Presence of this polymorphism is associated with 47% PPV and 99% NPV of Abacavir hypersensitivity[17] | Poor PPV (due to low prevalence of polymorphism) may inappropriately exclude therapy from some patients. |
| Genelex CYP2C9 and VKORC | CYP2C9*1, *2, *3 and VKORC1 -1639 | Warfarin dosing[18] | Screening explains only approx 40% of dosing variation[84]. |
| HLA-B*1502 | HLA-B*1502 | Carbamazepine associated Stevens-Johnson Syndrome or toxic epidermal necrolysis in Asian populations[85]. | Limited to patients of Asian decent. |
| HLA-A*3101 | HLA-A*3101 | Carbamazepine associated hypersensitivity reactions in European populations[78]. | NNT equals 39 European patients, 56 Japanese patients and 83 patients of un-determined decent. |
| HER2 (INFORMR HER2 FISH test, PathVysion® HER2 FISH test, PharmaDX® HER2 FISH test, Spot-Light® HER2 CISH test) | Number HER2 gene copies | Herceptin efficacy in breast[86] and gastric[86] cancer. | Equivocal results should be confirmed by a second screening method. 4 FDA approved genetic methods available. |
| CYP2C19 | CYP2C19 metabolizer status | Poor metabolizers do not form active metabolite of clopidogrel[25]. | Improved outcomes have been demonstrated in Chinese patients[30] only and no major society recommends genotyping prior to therapy to date. |
| G6PD | G6PD mutation by semi-quantitative or spectrophotometric methods[64] | High risk of hemolysis in patients receiving dapsone, methylene blue, nitrofurantoin, phenazopyridine, primaquine, rasburicase, and toluidine blue[87]. | Initial semi-quantitative analysis should be confirmed by spectrophotometric method due to wide variability of enzyme function. |
PPV: positive predictive value
NNT: number needed to treat
Genomic screening has become standard of care for many oncologic therapies. Genomics has been researched and utilized most extensively in this field due to the frequency of patient enrollment in clinical trials and the wide availability of tissue samples for genetic analysis. Tumor genetic polymorphisms have been associated with improved efficacy, identification of resistance, and toxicity of cancer therapeutics. The National Comprehensive Cancer Network (NCCN) now recommends screens for tumor specific genes associated with better chemotherapeutic efficacy [8]. Examples include human epidermal growth recepter 2 (HER2) screening in breast cancer [9,10] and gastric cancer [11] which dictates response to the specific immunologic adjuvant therapy transtuzumab. Optimal testing for HER2 remains controversial and both genomic (fluorescence in situ hybridization [FISH]) and proteomic screens (immunohistochemistry [IHC]) are available though the tests should be used in concert [12]. Cetuximab therapy in metastatic colon cancer is associated with a favorable cost-benefit analysis in tumors with the KRAS wild type gene [13] and KRAS mutations have been associated with a failure of the epidermal growth factor receptor tyrosine kinase inhibitors erotinib and gefitinib in non-small cell lung cancer [14]. BRAF mutations have been associated with failure of therapy in colorectal cancers [15] and improved survival in melanoma patients receiving vemurafenib [16]. See Table 2 for a list of ‘omics screens available for oncology pharmaceuticals.
Table 2.
‘Omics Screens for Cancer Therapeutics.
| Cancer Type | Test | Therapeutic Impact | ‘Omic Category |
|---|---|---|---|
| Breast Cancer | HER2 by FISH | Response to trastuzumab[9,10] | Genomics |
| HER2 by IHC | Response to trastuzumab[9, 10] | Pathology/Proteomics | |
| ER/PR by RT-PCR | Response to hormonal therapies[88–90] | Transcriptomics | |
| DPD by PCR | Fluorouracil toxicity[91] | Genomics | |
| PIK3CA Mutation Analysis | Resistance to trastuzumab in HER2 positive tumors[92] | Genomics | |
| Colorectal Cancer | KRAS mutation analysis | Response to panitumamab, cetuximab[13] | Genomics |
| BRAF mutation analysis | Response to panitumamab, cetuximab[15] | Genomics | |
| EGFR amplication by FISH | Response to cetuximab[93] | Genomics | |
| Thymidylate synthase by IHC | Resistance to fluorouracil or related agents[94] | Pathology/Proteomics | |
| UGT1A1 Molecular Assay | Increased risk of severe irinotecan toxcity[95] | Genomics | |
| DPD by PCR | Fluorouracil toxicity[91, 96] | Genomics | |
| PIK3CA Mutation analysis | Resistance to cetuximab salvage therapy[97] | Genomics | |
| Non-Small Cell Lung Cancer | EGFR Mutation analysis | Response to gefitinib, erlotinib[98] | Genomics |
| KRAS Mutation | Resistance to gefitinib, erlotinib[14] | Genomics | |
| ALK by FISH | Sensitivity to crizotinib[99] | Genomics | |
| ERCC1 by IHC | Resistance to platinum-based chemotherapeutics[100] | Pathology/Proteomics | |
| EGFR Amplication by FISH | Resistance to gefitinib, erlotinib, cetuximab plus paclitaxel, and carboplatin[101] | Genomics | |
| EGFR by IHC | Response to cetuximab plus chemotherapy[102] | Pathology/Proteomics | |
| TS by RT-PCR | Resistance to pemetrexed[103] | Transcriptomics | |
| UGT1A1 Molecular Assay | Irinotecan toxicity[104] | Genomics | |
| ALK by FISH | Response to crizotinib in metastatic disease[105] | Genomics | |
| Gastric Cancer | HER2 by FISH | Response to trastuzumab[11] | Genomics |
| HER2 by IHC | Response to trastuzumab[106,107] | Pathology/Proteomics | |
| ERCC1 by IHC | Resistance to platinum-based chemotherapies[108] | Pathology/Proteomics | |
| Melanoma | BRAF Mutation Analysis | Response to vemurafenib[16] | Genomics |
| Brain Cancer | MGMT methylation | Response to temozolomide[109] | Epigenomics |
| Head and Neck Cancers | DPD status | Fluorouracil toxicity and efficacy[110] | Metabolomics |
| CLL | 17p by FISH | Resistance to fludarabine based regimens | Genomics |
| P53 Mutation analysis | Resistance to fludarabine based regimens[111] | Genomics | |
| CML | BCR-ABL | Recommended imatinib[112] | Genomics |
| BCR-ABL KD Mutation V299L, T315A, F17L/V/I/C | Response to nilotinib rather than dasatinib[41] | Genomics | |
| BCR-ABL KD Mutation Y253H, E255K/V, F359V/C/I | Response to dasatinib rather than nilotinib[41] | Genomics | |
| BCR-ABL transcript mass by RQ-PCR | Response tyrosine kinase inhibitors after therapy ihas been intiated[113] | Transcriptomics | |
| Myelodysplastic Syndrome | Deletion 5q | Response to lenalidomide[114] | Genomics |
| Platelet derived growth factor receptor beta | Indication for imatinib[115,116] | Genomics | |
| Non-Hodgkin’s Lymphoma | MYC translocations | Resistance to retuximab plus cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) in diffuse B-cell lymphoma[117] | Genomics |
HER2: Human epidermal growth receptor 2
FISH: Fluorescence in situ hybridization
IHC: Immunohistochemstry
ER/PR: Estrogen receptor/progesterone receptor
DPD: Dihydropyrimidine Dehydrogenase
PIK3CA: Phosphoinositide-3-kinase, catalytic alpha
EGFR: Epidermal growth factor receptor
TS: thymidylate synthase
ERCC1: Excision repair cross-complementing 1 protein
UGT1A1: Uridine diphosphate glucuronosyltransferase 1A1 gene
ALK: Anaplastic lymphoma receptor tyrosine kinase
MGMT: methylguanine-methyltransferase promoter
One of the most commonly utilized genomic screens in pharmaceutical prescribing is HLA-B*5701 screening for abacavir hypersensitivity. This is likely due to clinical trial data demonstrating improved patient outcomes [17] and the overall prevalence of HIV-1 infection in the population yielding ample opportunity for testing. Interestingly, the positive predictive value (PPV) of this screen was only 47.9% yet the test has become standard of care in Infectious Disease clinics.
Significant effort has been invested in genomic screening for warfarin dosing. Several large studies have demonstrated decreased dosing variability [18], decreased hospitalization [19], and earlier time to stable dosing [20]. Demographic and environmental factors have been added to known genotype associations in an effort to further improve efficacy and safety. An unvalidated algorithm including integration of multiple genetic polymorphisms is available to clinicians at www.warfarindosing.org [21]. However, even with incorporation of multiple factors, only 40% of the variability in response to warfarin can be predicted.
It is estimated that more than 90% of known drugs are metabolized by hepatic cytochromes (CYPs) 1A2, 2C9, 2C19, 2D6, 2E1, and 3A4 [22] thus genetic polymorphism in these enzymes is likely to play a large role in drug response. CYP screening is available though application to clinical practice has yet to be regularly employed by clinicians. This is likely due to multiple factors including inherent redundancy in the metabolic function of the CYP enzymes, variable penetrance (expressed phenotype predicted by genotype) [23,24], and lack of clinical data to demonstrate improved patient outcomes. Clinicians, however, should be aware of these screens because polymorphism has been associated with alteration in both clinical efficacy [25,26] and toxicity [27].
The best studied of these enzymes is CYP2D6. It metabolizes approximately 25% of known medications including antidepressants, antipsychotics, anti-arrhythmics, β-blockers, and analgesics. Toxicity and lack of efficacy have been demonstrated due to variable CYP2D6 gene expression [28,29]. However, CYP2D6 screening prior to initiation of therapy is not currently recommended prior to initiation of CYP2D6 dependent medications. CYP2C19 screening has recently been shown to be associated with variable pharmacodynamic responses to clopidogrel [25] and improved outcomes after percutaneous coronary intervention [30]. Again, screening prior to therapy is not currently recommended by major clinical societies though currently underway trials attempting to demonstrate improved outcomes may soon change this. Polymorphisms in other CYP enzymes have been associated with alterations in medication pharmacokinetics and pharmacodynamics subsequently leading to alterations in clinical efficacy and safety [31,32]. However, these associations suffer from the same limitations as other genomic screens, primarily low positive predictive values and failure to demonstrate improved outcomes. While a range of pharmacogenomic screens are available, physicians must realize their limitations in clinical practice.
Limitations of Genomic Screens
Pharmacogenomic screens have become more available with the publication of the human genome sequence and the resulting proliferation of laboratories specialized in characterization of its variation. There are few FDA approved genomic screens available (Table 1) though local laboratories can provide sequencing and analysis in their respective areas of specialty.
Cost remains a major barrier to wide adoption of pharmacogenomic screening. A genomic screen may cost a patient anywhere between $100 to several thousand dollars depending upon the number of polymorphisms sought, and the vendor pricing. Single screens have become less expensive with commercialization, utilization of microarray techniques, and increased clinical demand. For instance the HLA-B*5701 screen for abacavir hypersensitivity can be obtained for approximately $150 in patient cost.
Insurance company re-imbursement remains a limitation to utilization in clinical practice. Perhaps recent trials demonstrating pharmacodynamic associations with genomic polymorphisms [19,20,25] will lead to an investment in screening algorithms that will ultimately decrease clinical costs. However, re-imbursement is unlikely to change until clinical trials demonstrate improved outcomes with antecedent pharmacogenomic screening.
The positive and negative predictive values of currently available screening tests remain the major limitation of these assays. Combining polymorphism screening for CYP2C19 and the vitamin K epoxide reductase complex (VKORC1) only resulted in 41% prediction of the variability in warfarin doses [18]. This combination screen represents the most effective pharmacogenomic screen to date and highlights the shortcomings of utilizing genomics alone. The abacavir and warfarin screens represent the most successful efforts toward “individualized medicine” thus far. The two approaches highlight fundamental differences in screening needs. In one case, toxicity is avoided by identification of one specific polymorphism. In the other case, multiple factors are integrated to improve efficacy. Both situations have yielded less than a 50% success rate for prediction of safety and efficacy [17,18]. This suggests other factors contribute to the ultimately observed phenotype. This has prompted research into the downstream ‘omic fields.
Epigenomics
The term epigenomics refers to environmental factors that regulate gene expression but are not inherently part of the genome. These factors include DNA methylation, variations in histone wrapping, RNA silencing, among other factors. Epigenomic factors are major determinants of when and whether genes are expressed. This field has potential to account for differences in gene expression in different populations and geographic regions affecting drug response from the same genetic code. Numerous methods have been used for analysis of epigenetic factors in basic science laboratories and technique optimization is still underway [33].
Epigenomic Screens
Identification of environmental exposures associated with epigenetic changes leading to cancer [34] is a ripe area of investigation. Expansion into a greater number of human disease processes occurs monthly. The bulk of pharmacoepigenetic work has been done in chemotherapeutic agents. For instance, hypomethylation of the multi-drug resistance gene 1 (MDR-1) dictates increased expression of the gene in the setting of daunorubicin and etoposide treatment [35]. Epigenetic factors have been shown to affect the expression of numerous CYP genes [36] and characterization may account for variation in functionality in patients with the same underlying genetic code. Few other pharmaceutical related epigenetic screens have been investigated.
Limitations of Epigenomic Screens
The major clinical limitation of this field is availability. Ultimately, epigenetic screens validated in one population will be difficult to apply globally due to the inherent differences in environmental factors between populations. This may limit the generalizability of epigenetic screens across populations.
Transcriptomics
Transcriptomics is defined as the study of how the human genome is expressed. Changes in epigenomic, local cellular environment and expression of other genes can alter the human “transcriptome”. The true clinical utility of transcriptomic screens lies in their potential to characterize organ biology through biomarkers in the blood. For example, a medication may modify gene trasnscription in the liver. The mRNA or miRNA profile released from hepatocytes can be measured in serum [37] and the changes can be used to measure the response to treatment. Additionally, peripheral blood cells may change their transcipts in response to a therapy. The measurement of these transcript biomarkers in plasma may provide a means to detect organ specific biologic changes without the need to examine the organ tissue itself, which limits pharmacogenomic and pharmacoepigenomic screens.
Transcriptomic Screens
The most clinically utilized multi-gene transcriptomic screen is the AlloMap (XDx, Inc.), which screens for 11 gene expression levels associated with cardiac transplant rejection. This screen minimizes the need for endomyocardial biopsy in cardiac transplant patients [38] and is now covered by most health insurance companies. This screen is not a direct marker of pharmaceutical efficacy or toxicity but rather, a test for expression of genes associated with rejection that may prompt clinicians to alter pharmaceutical therapy. Two breast cancer multi-gene expression assays are available, the Oncotype DX (Genomic Health, Inc., CA, USA) which screens for tumor expression of 21 genes [39] and the MammaPrint (Agendia, Amsterdam, The Netherlands) which screens for expression of 70 tumor genes [40]. These screens require tissue of resected or biopsied tumors. Several chronic myeloid leukemia (CML) mutations have been associated with varying efficacy of chemotherapeutics [41]. The NCCN now recommends following BCR-ABL kinase transcript levels throughout therapy and consider significant changes an indication to change therapy [42]. Estrogen receptor/progesterone receptor (ER/PR) testing is recommended at diagnosis of all breast cancers to determine appropriate adjuvant chemotherapy. Classically, this testing is performed using immunohistochemistry (IHC) techniques on resected/biopsied tissue though peripheral sampling followed by RT-PCR is becoming more reliable and clinically available [43,44].
Limitations of Transcriptomic Screens
Transcriptomic analyses have largely failed to move into the clinical arena to date. This field remains entrenched in basic science laboratories with few efforts to move the technique into translational studies. The fundamental limitation of using transciptomic assays is that mRNAs are intermediate products of disease that fail to adequately predict the clinical effect. Polymorphism of downstream proteins including post-translational changes, changes in second-messenger systems, and interaction with downstream metabolic enzymes leads to persistent variability of the observed clinical effect. This is compounded by short mRNA half-lives resulting in difficulty in quantitative interpretation [45]. Circulating cancer cells may represent a biologically distinct cell type when compared to the primary tumor transcriptome. This discordance has been observed in circulating breast cancer transcription of ER/PR [46]. Biomarkers have been studied in numerous animal models and when applied in humans, they have not performed better than a coin-flip for prediction of disease/toxicity. Perhaps continued work that characterizes the kinetics of mRNAs/transcripts and further enhancement of sensitivity of their detection will lead to a larger role of transcriptomic analysis in pharmacotherapy.
Proteomics
In the same Nature issue that published the first draft of the HGP [3], The Human Proteome Organization (HUPO) announced their efforts to increase resources for characterization of the proteins encoded by the HGP [47]. Proteomics is defined as the study of proteins and their variability in human disease states. The proteome may vary between different individuals, different organs, or even between cells in the same organ in an individual. Most efforts have focused upon characterizing protein mediators and predictors of disease rather than the protein targets of pharmacotherapy.
Proteomic Screens
The most prominently available proteomic screens are utilized in tumor specific cancer screening. For instance, evaluation of estrogen receptor (ER), and progesterone receptor (PR), is recommended in addition to genomic screening of HER2 for evaluation of newly diagnosed breast cancers. These evaluations are pathology based and are typically performed using IHC assays. IHC may technically be considered a proteomic approach because it quantifies a human protein associated with disease though many would consider it a more traditional pathologic technique because it does not characterize the inherent protein variability. Newer ultrasensitive proteomic techniques use a combination of high performance chromatography and mass spectrometry to characterize thousands of proteins from as little as a microgram of tissue [48]. Identification of a variable proteomic fingerprint has been associated with aspirin resistance and the use of clopidogrel in this population is recommended [49]. Proteomic measurements of inflammatory markers, such as tumor necrosis factor alpha (TNF-a) and haptoglobin alpha-2-polypeptide has been associated with increased manifestations of atherosclerotic disease in patients with systemic lupus erythematosus (SLE) [50]. Inflammatory markers can be measured by commercial laboratories [51,52] and some have suggested targeting therapies, such as infliximab, in SLE patients with elevated markers [53]. While these tests have face validity, there are no data to demonstrate that therapy modification based on the testing improves patient outcomes.
Limitations of Proteomic Screens
The major limitation of proteomic screens is that they are tissue specific and therefore require tissue to characterize protein variability. Unlike blood-based targets or resected tumor tissue, from which the pertinent biologic matrix is practical to obtain, tissue samples from organs such as lung, kidney, heart, or brain are not easily obtained for proteomic screens. Most proteomic screens are not yet commercially available to clinicians. While a subset of patients may be able to finance tests such as TNF-α, which costs only a few hundred dollars, no proteomic screens are reimbursed by insurance companies in the United States.
Metabolomics
Metabolomics is an emerging field that quantitates endogenous and exogenous metabolic products to measure metabolic response or predict disease. Advances in metabolomic techniques have yielded a sensitive approach that may be altered by minute-to-minute physiologic changes and account for upstream variations in an individual’s genome, transcriptome, and proteome. Multiple techniques are employed to measure these small molecule metabolites; a combination of ultra performance liquid chromatography with nuclear magnetic resonance (NMR), orthogonal quadrupole time-of-flight (QTOF), or mass spectroscopy time-of-flight (MALDI TOF) have become the most common and preferred methodologies [54,55], though optimization is still occurring [56]. This approach has been applied to multiple disease processes [57–62]. This approach may ultimately prove to be the most sensitive screening modality for pharmaceutical efficacy and safety in the individual patient because it accounts for upstream genomic, epigenomic, transcriptomic and proteomic factors[63].
Metabolomic Screens
The classic metabolomic screen for toxicity is for glucose-6-phosphate defficiency (G6PD). G6PD has been associated with xenobiotic-induced hemolysis. The screen can be performed either qualitatively or quantitatively via spectrophotometric metabolite quantification [64]. G6PD testing is used prior to prescription of many anti-malarial therapies and prior to dapsone prescription. G6PD testing is recommended by the Association of Infectious Diseases Society of America for all HIV patients [65]. G6PD status is tested in all US military recruits.
To date, only one clinical trial has demonstrated an ultrasensative metabolomic relationship between a medication and drug induced liver injury (DILI). The SPORTIF trial examined ximelagatran anticoagulation for stroke prevention in patients with atrial fibrillation. Blood sampling before and after ximelagatran administration revealed that lower levels of serum pyruvate before dosing predicted DILI [66].
Winnike examined the metabolomic profiles of subjects taking therapeutic doses of acetaminophen using using NMR spectroscopy and was able to identify evidence of hepatotoxicity suggesting the utility of “early-intervention pharmacometabonomics” [67]. However these evoked metabolomic profiles can’t truly be considered screening tests. This same group demonstrated alterations in acetaminophen urinary metabolite profile predicted by predose p-cresol urinary concentrations [68]. While this study did not directly demonstrate toxicity, it demonstrated proof that screening metabolomic profiles can predict variation in individual metabolic activity, and likely toxicity. Markers of hepatotoxicity have been observed in children taking valproic acid utilizing organic acid metabolism as the metabolic matrix [69]. A similar association has been demonstrated with statin induced liver/muscle toxicity [70]. None of these assays are validated for screening prior to therapy. However, these association studies represent with groundwork required for metabolomic toxicity screening.
Metabolomic profiles have been utilized in drug discovery using small compounds to target specific metabolic changes in specific cell lines [71–73]. This technique suggests that screening of individual metabolomes could predict subsequent pharmaceutical response and may yield more specific therapies.
Limitations of Metabolomic Screens
At this time, high sensitivity metabolomic screens for pharmacotherapy have not left research laboratories and thus are of little utility to the practicing clinician. The human metabolome needs to be further characterized and subsequently therapeutic associations can be made. In addition, these highly specific techniques raise issues with reproducibility [74]. It is unclear how clinicians will use this sensitive data since even small changes in physiology, such as food ingestion [75] or going up a flight of stairs, can have significant impact on the metabolome [55]. Combining pharmacometabolomics with pharmacogenomics can lead to identification of more clinically relevant associations [76,77]. If clinicians initially focus on larger stable metabolic signals, such as the output of CYP systems, hopefully we can minimize being misled by transient metabolic associations that do not represent causation.
The Future ‘Omics in Pharmacotherapy
‘Omic modalities have utility in screening for efficacy and safety of pharmaceutical therapy. Screening must ultimately fulfill practical criteria in order to be clinically useful. For screening assays that seek to improve efficacy, the positive predictive value (PPV) must be high or patients that may have benefit from the therapy will be inappropriately excluded. Likewise, if a screen is meant to eliminate high morbidity toxicity, such as Stevens-Johnson Syndrome, then the PPV is less important and the negative predictive value (NPV) is more pertinent because false negative screens put patients at significant risk. Predictive values are preferred for clinical screening assays because they account for disease prevalence in the population, which sensitivity and specificity do not. Ideally, the screen would have both high PPV and NPV. However this is unlikely in a single screen due to the limitations listed in the respective ‘omic sections. Genomic screens are perfectly suited for rare, high morbidity condition, such as HLA-B*1502 associated Stevens-Johnson Syndrome in carbamazepine therapy [78]. More common and less severe toxicities, such as statin-rhabdomyolysis, may be best screened by a combination or syllogistic approach. A biologic systems panel (BSP) [79] has the potential to account for several common genetic polymorphisms, proteomic, and metabolomic variables improving the overall predictive values of the panel. More common toxicities may be cost-effectively screened with an initial genomic screen, followed by a functional screen, if positive on the initial screen, so that patients are not denied therapies due to false positive tests.
The complexity of human-medication interactions are further complicated by idiosyncrasies of disease. For instance, HER2 status of breast cancer may change during therapy [80]. The chemotherapeutic agent tamoxifen is metabolically activated by CYP2D6 and despite numerous pharmacokinetic studies demonstrating a lower metabolite serum concentrations [81, 82], no increased rate of breast cancer recurrence in patients taking CYP2D6 inhibitors when compared to controls [83]. This suggests that lower serum concentrations of tamoxifen’s active metabolite are sufficient for treatment, or other factors such as tumor specific changes diminish the pharmacokinetic association. Associating metabolic traits with GWA identified genotypes has been shown to improve the predictive value for observed clinical effect for common medical conditions [76,77]. This work highlights the power of combining ‘omic methodologies. Again, integration of the ‘omics fields into panels may improve the predictive efficacy and safety of prescribing practices.
Conclusions
‘Omics pharmaceutical screens are slowly entering clinical medicine. Continued basic research is necessary to establish a robust matrix of ‘omics data. As associations are identified, researchers should determine if pre- therapy screening improves patient outcomes. Insurance companies must be pushed to cover screening assays that enhance outcomes. Integration of numerous ‘omics techniques into panels will yield screens with higher positive and negative predictive values. ‘Omics panels will prove more clinically useful to physicians than single ‘omic screens.
Acknowledgments
Funding
The NIH grants include: EY11490 and EY17963 (both NIH). The 3rd grant is for Andrew Monte, Emergency Medicine Foundation Research Training Grant: 2012–2014.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Evans DA, Manley KA, McKusick VA. Genetic control of isoniazid metabolism in man. Br Med J. 1960;2:485–491. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crettol S, Petrovic N, Murray M. Pharmacogenetics of phase I and phase II drug metabolism. Curr Pharm Des. 2010;16:204–219. doi: 10.2174/138161210790112674. [DOI] [PubMed] [Google Scholar]
- 3.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 4.International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 5.International hapmap project.
- 6.1000 genomes-a deep catalog of human genetic variation.
- 7.Bernstein JA, Zhang G, Jin L, Abbott C, Nebert DW. Olfactory receptor gene polymorphisms and nonallergic vasomotor rhinitis. J Asthma. 2008;45:287–292. doi: 10.1080/02770900701867579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.NCCN Guidelines. The National Comprehensive Cancer Network; [Google Scholar]
- 9.Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- 10.Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007;369:29–36. doi: 10.1016/S0140-6736(07)60028-2. [DOI] [PubMed] [Google Scholar]
- 11.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 12.Carlson RW, Moench SJ, Hammond ME, Perez EA, Burstein HJ, et al. HER2 testing in breast cancer: NCCN Task Force report and recommendations. J Natl Compr Canc Netw. 2006;3 [PubMed] [Google Scholar]
- 13.Mittmann N, Au HJ, Tu D, O’Callaghan CJ, Isogai PK, et al. Prospective cost-effectiveness analysis of cetuximab in metastatic colorectal cancer: evaluation of National Cancer Institute of Canada Clinical Trials Group CO.17 trial. J Natl Cancer Inst. 2009;101:1182–1192. doi: 10.1093/jnci/djp232. [DOI] [PubMed] [Google Scholar]
- 14.Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13:2890–2896. doi: 10.1158/1078-0432.CCR-06-3043. [DOI] [PubMed] [Google Scholar]
- 15.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 16.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mallal S, Phillips E, Carosi G, Molina JM, Workman C, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–579. doi: 10.1056/NEJMoa0706135. [DOI] [PubMed] [Google Scholar]
- 18.Namazi S, Azarpira N, Hendijani F, Khorshid MB, Vessal G, et al. The impact of genetic polymorphisms and patient characteristics on warfarin dose requirements: a cross-sectional study in Iran. Clin Ther. 2010;32:1050–1060. doi: 10.1016/j.clinthera.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Epstein RS, Moyer TP, Aubert RE, O Kane DJ, Xia F, et al. Warfarin genotyping reduces hospitalization rates results from the MM-WES (Medco-Mayo Warfarin Effectiveness study) J Am Coll Cardiol. 2010;55:2804–2812. doi: 10.1016/j.jacc.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 20.Caraco Y, Blotnick S, Muszkat M. CYP2C9 genotype-guided warfarin prescribing enhances the efficacy and safety of anticoagulation: a prospective randomized controlled study. Clin Pharmacol Ther. 2008;83:460–470. doi: 10.1038/sj.clpt.6100316. [DOI] [PubMed] [Google Scholar]
- 21.Gage BF. IsoDynamic. Warfarin dosing. [Google Scholar]
- 22.Wang JF, Zhang CC, Chou KC, Wei DQ. Structure of cytochrome p450s and personalized drug. Curr Med Chem. 2009;16:232–244. doi: 10.2174/092986709787002727. [DOI] [PubMed] [Google Scholar]
- 23.Gaedigk A, Bradford LD, Marcucci KA, Leeder JS. Unique CYP2D6 activity distribution and genotype-phenotype discordance in black Americans. Clin Pharmacol Ther. 2002;72:76–89. doi: 10.1067/mcp.2002.125783. [DOI] [PubMed] [Google Scholar]
- 24.Ieiri I, Yamada S, Seto K, Morita T, Kaneda T, et al. A CYP2D6 phenotype-genotype mismatch in Japanese psychiatric patients. Pharmacopsychiatry. 2003;36:192–196. doi: 10.1055/s-2003-43049. [DOI] [PubMed] [Google Scholar]
- 25.Mega JL, Hochholzer W, Frelinger AL, 3rd, Kluk MJ, Angiolillo DJ, et al. Dosing clopidogrel based on CYP2C19 genotype and the effect on platelet reactivity in patients with stable cardiovascular disease. JAMA. 2011;306:2221–2228. doi: 10.1001/jama.2011.1703. [DOI] [PubMed] [Google Scholar]
- 26.Lobello KW, Preskorn SH, Guico-Pabia CJ, Jiang Q, Paul J, et al. Cytochrome P450 2D6 phenotype predicts antidepressant efficacy of venlafaxine: a secondary analysis of 4 studies in major depressive disorder. J Clin Psychiatry. 2010;71:1482–1487. doi: 10.4088/JCP.08m04773blu. [DOI] [PubMed] [Google Scholar]
- 27.Gasche Y, Daali Y, Fathi M, Chiappe A, Cottini S, et al. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N Engl J Med. 2004;351:2827–2831. doi: 10.1056/NEJMoa041888. [DOI] [PubMed] [Google Scholar]
- 28.De Gregori M, Allegri M, De Gregori S, Garbin G, Tinelli C, et al. How and why to screen for CYP2D6 interindividual variability in patients under pharmacological treatments. Curr Drug Metab. 2010;11:276–282. doi: 10.2174/138920010791196274. [DOI] [PubMed] [Google Scholar]
- 29.Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5:6–13. doi: 10.1038/sj.tpj.6500285. [DOI] [PubMed] [Google Scholar]
- 30.Luo Y, Zhao YT, Verdo A, Qi WG, Zhang DF, et al. Relationship between cytochrome P450 2C19*2 polymorphism and stent thrombosis following percutaneous coronary intervention in Chinese patients receiving clopidogrel. J Int Med Res. 2011;39:2012–2019. doi: 10.1177/147323001103900548. [DOI] [PubMed] [Google Scholar]
- 31.Hellmann A, Rule S, Walewski J, Shpilberg O, Feng H, et al. Effect of cytochrome P450 3A4 inducers on the pharmacokinetic, pharmacodynamic and safety profiles of bortezomib in patients with multiple myeloma or non-Hodgkin’s lymphoma. Clin Pharmacokinet. 2011;50:781–791. doi: 10.2165/11594410-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 32.Callaghan JT, Bergstrom RF, Ptak LR, Beasley CM. Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin Pharmacokinet. 1999;37:177–193. doi: 10.2165/00003088-199937030-00001. [DOI] [PubMed] [Google Scholar]
- 33.Ho SM, Tang WY. Techniques used in studies of epigenome dysregulation due to aberrant DNA methylation: an emphasis on fetal-based adult diseases. Reprod Toxicol. 2007;23:267–282. doi: 10.1016/j.reprotox.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herceg Z, Vaissière T. Epigenetic mechanisms and cancer: an interface between the environment and the genome. Epigenetics. 2011;6:804–819. doi: 10.4161/epi.6.7.16262. [DOI] [PubMed] [Google Scholar]
- 35.Baker EK, Johnstone RW, Zalcberg JR, El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene. 2005;24:8061–8075. doi: 10.1038/sj.onc.1208955. [DOI] [PubMed] [Google Scholar]
- 36.Baer-Dubowska W, Majchrzak-CeliÅ„ska A, Cichocki M. Pharmocoepigenetics: a new approach to predicting individual drug responses and targeting new drugs. Pharmacol Rep. 2011;63:293–304. doi: 10.1016/s1734-1140(11)70498-4. [DOI] [PubMed] [Google Scholar]
- 37.Powell EE, Kroon PA. Low density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme A reductase gene expression in human mononuclear leukocytes is regulated coordinately and parallels gene expression in human liver. J Clin Invest. 1994;93:2168–2174. doi: 10.1172/JCI117213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pham MX, Teuteberg JJ, Kfoury AG, Starling RC, Deng MC, et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med. 2010;362:1890–1900. doi: 10.1056/NEJMoa0912965. [DOI] [PubMed] [Google Scholar]
- 39.Paik S, Shak S, Tang G, Kim C, Baker J, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 40.Kunz G. Use of a genomic test (MammaPrintâ„¢) in daily clinical practice to assist in risk stratification of young breast cancer patients. Arch Gynecol Obstet. 2011;283:597–602. doi: 10.1007/s00404-010-1454-9. [DOI] [PubMed] [Google Scholar]
- 41.Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118:1208–1215. doi: 10.1182/blood-2010-12-326405. [DOI] [PubMed] [Google Scholar]
- 42.O’Brien S, Abboud C, Akhtari M, Altman J, Berman E, et al. NCCN Guidelines TM. National Comprehensive Cancer Network; 2012. [Google Scholar]
- 43.Andreopoulou E, Yang LY, Rangel KM, Reuben JM, Hsu L, et al. Comparison of assay methods for detection of circulating tumor cells in metastatic breast cancer: AdnaGen AdnaTest BreastCancer Select/Detectâ„¢ versus Veridex CellSearchâ„¢ system. Int J Cancer. 2012;130:1590–1597. doi: 10.1002/ijc.26111. [DOI] [PubMed] [Google Scholar]
- 44.Oda M, Arihiro K, Kataoka T, Osaki A, Asahara T, et al. Comparison of immunohistochemistry assays and real-time reverse transcription-polymerase chain reaction for analyzing hormone receptor status in human breast carcinoma. Pathol Int. 2010;60:305–315. doi: 10.1111/j.1440-1827.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 45.Mendrick DL. Transcriptional profiling to identify biomarkers of disease and drug response. Pharmacogenomics. 2011;12:235–249. doi: 10.2217/pgs.10.184. [DOI] [PubMed] [Google Scholar]
- 46.Aktas B, Müller V, Tewes M, Zeitz J, Kasimir-Bauer S, et al. Comparison of estrogen and progesterone receptor status of circulating tumor cells and the primary tumor in metastatic breast cancer patients. Gynecol Oncol. 2011;122:356–360. doi: 10.1016/j.ygyno.2011.04.039. [DOI] [PubMed] [Google Scholar]
- 47.Abbott A. And now for the proteome. Nature. 2001;409:747. doi: 10.1038/35057460. [DOI] [PubMed] [Google Scholar]
- 48.Altelaar AM, Heck AJ. Trends in ultrasensitive proteomics. Curr Opin Chem Biol. 2012 doi: 10.1016/j.cbpa.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Mateos-Cáceres PJ, Macaya C, Azcona L, Modrego J, Mahillo E, et al. Different expression of proteins in platelets from aspirin-resistant and aspirin-sensitive patients. Thromb Haemost. 2010;103:160–170. doi: 10.1160/TH09-05-0290. [DOI] [PubMed] [Google Scholar]
- 50.Pavón EJ, Muñoz P, Lario A, Longobardo V, Carrascal M, et al. Proteomic analysis of plasma from patients with systemic lupus erythematosus: increased presence of haptoglobin alpha2 polypeptide chains over the alpha1 isoforms. Proteomics. 2006;6(1):282–292. doi: 10.1002/pmic.200500404. [DOI] [PubMed] [Google Scholar]
- 51.Cytokine panel. Life Extension. [Google Scholar]
- 52.Tumor necrosis factor alpha panel. LabCorp; [Google Scholar]
- 53.López-Pedrera C, Aguirre MÁ, Barbarroja N, Cuadrado MJ. Accelerated atherosclerosis in systemic lupus erythematosus: role of proinflammatory cytokines and therapeutic approaches. J Biomed Biotechnol. 2010 doi: 10.1155/2010/607084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson CH, Patterson AD, Idle JR, Gonzalez FJ. Xenobiotic metabolomics: major impact on the metabolome. Annu Rev Pharmacol Toxicol. 2012;52:37–56. doi: 10.1146/annurev-pharmtox-010611-134748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson CH, Gonzalez FJ. Challenges and opportunities of metabolomics. J Cell Physiol. 2011 doi: 10.1002/jcp.24002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang A, Sun H, Wang P, Han Y, Wang X. Modern analytical techniques in metabolomics analysis. Analyst. 2012;137:293–300. doi: 10.1039/c1an15605e. [DOI] [PubMed] [Google Scholar]
- 57.Serkova NJ, Standiford TJ, Stringer KA. The emerging fieldof quantitative blood metabolomics for biomarker discovery in critical illnesses. Am J Respir Crit Care Med. 2011;184:647–655. doi: 10.1164/rccm.201103-0474CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stringer KA, Serkova NJ, Karnovsky A, Guire K, Paine R, 3rd, et al. Metabolic consequences of sepsis-induced acute lung injury revealed by plasma ¹H-nuclear magnetic resonance quantitative metabolomics and computational analysis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L4–4L11. doi: 10.1152/ajplung.00231.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cohen MJ, Serkova NJ, Wiener-Kronish J, Pittet JF, Niemann CU. 1H-NMR-based metabolic signatures of clinical outcomes in trauma patients--beyond lactate and base deficit. J Trauma. 2010;69:31–40. doi: 10.1097/TA.0b013e3181e043fe. [DOI] [PubMed] [Google Scholar]
- 60.Chorell E, Svensson MB, Moritz T, Antti H. Physical fitness level is reflected by alterations in the human plasma metabolome. Mol Biosyst. 2012;8:1187–1196. doi: 10.1039/c2mb05428k. [DOI] [PubMed] [Google Scholar]
- 61.van der Kloet FM, Tempels FW, Ismail N, van der Heijden R, Kasper PT, et al. Discovery of early-stage biomarkers for diabetic kidney disease using ms-based metabolomics (FinnDiane study) Metabolomics. 2012;8:109–119. doi: 10.1007/s11306-011-0291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mihalik SJ, Michaliszyn SF, de Las Heras J, Bacha F, Lee S, et al. Metabolomic Profiling of Fatty Acid and Amino Acid Metabolism in Youth With Obesity and Type 2 Diabetes: Evidence for enhanced mitochondrial oxidation. Diabetes Care. 2012;35:605–611. doi: 10.2337/DC11-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mikami T, Aoki M, Kimura T. The application of mass spectrometry to proteomics and metabolomics in biomarker discovery and drug development. Curr Mol Pharmacol. 2011 doi: 10.2174/1874467211205020301. [DOI] [PubMed] [Google Scholar]
- 64.Minucci A, Giardina B, Zuppi C, Capoluongo E. Glucose-6-phosphate dehydrogenase laboratory assay: How, when, and why? IUBMB Life. 2009;61:27–34. doi: 10.1002/iub.137. [DOI] [PubMed] [Google Scholar]
- 65.Aberg JA, Kaplan JE, Libman H, Emmanuel P, Anderson JR, et al. Primary care guidelines for the management of persons infected with human immunodeficiency virus: 2009 update by the HIV medicine Association of the Infectious Diseases Society of America. Clin Infect Dis. 2009;49:651–681. doi: 10.1086/605292. [DOI] [PubMed] [Google Scholar]
- 66.Olsson SB Executive Steering Committee of the SPORTIF III Investigators. Stroke prevention with the oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with non-valvular atrial fibrillation (SPORTIF III): randomised controlled trial. Lancet. 2003;362:1691–1698. doi: 10.1016/s0140-6736(03)14841-6. [DOI] [PubMed] [Google Scholar]
- 67.Winnike JH, Li Z, Wright FA, Macdonald JM, O’Connell TM, et al. Use of pharmaco-metabonomics for early prediction of acetaminophen-induced hepatotoxicity in humans. Clin Pharmacol Ther. 2010;88:45–51. doi: 10.1038/clpt.2009.240. [DOI] [PubMed] [Google Scholar]
- 68.Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification -microbiome significant host of metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Price KE, Pearce RE, Garg UC, Heese BA, Smith LD, et al. Effects of valproic acid on organic acid metabolism in children: a metabolic profiling study. Clin Pharmacol Ther. 2011;89:867–874. doi: 10.1038/clpt.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang HJ, Choi MJ, Wen H, Kwon HN, Jung KH, et al. An effective assessment of simvastatin-induced toxicity with NMR-based metabonomics approach. PLoS One. 2011;6:e16641. doi: 10.1371/journal.pone.0016641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Okada T, Afendi FM, Altaf-Ul-Amin M, Takahashi H, Nakamura K, et al. Metabolomics of medicinal plants: the importance of multivariate analysis of analytical chemistry data. Curr Comput Aided Drug Des. 2010;6:179–196. doi: 10.2174/157340910791760055. [DOI] [PubMed] [Google Scholar]
- 72.Fan TW, Lorkiewicz PK, Sellers K, Moseley HN, Higashi RM, et al. Stable isotope-resolved metabolomics and applications for drug development. Pharmacol Ther. 2012;133:366–391. doi: 10.1016/j.pharmthera.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiaradonna F, Moresco RM, Airoldi C, Gaglio D, Palorini R, et al. From cancer metabolism to new biomarkers and drug targets. Biotechnol Adv. 2012;30:30–51. doi: 10.1016/j.biotechadv.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 74.Simó C, Ibáñez C, Gómez-Martínez A, Ferragut JA, Cifuentes A. Is metabolomics reachable? Different purification strategies of human colon cancer cells provide different CE-MS metabolite profiles. Electrophoresis. 2011;32:1765–1777. doi: 10.1002/elps.201100019. [DOI] [PubMed] [Google Scholar]
- 75.Fujimura Y, Kurihara K, Ida M, Kosaka R, Miura D, et al. Metabolomics-driven nutraceutical evaluation of diverse green tea cultivars. PLoS One. 2011;6:e23426. doi: 10.1371/journal.pone.0023426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abo R, Hebbring S, Ji Y, Zhu H, Zeng ZB, et al. Merging pharmacometabolomics with pharmacogenomics using ‘1000 Genomes’ single-nucleotide polymorphism imputation: selective serotonin reuptake inhibitor response pharmacogenomics. Pharmacogenet Genomics. 2012 doi: 10.1097/FPC.0b013e32835001c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477:54–60. doi: 10.1038/nature10354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCormack M, Alfirevic A, Bourgeois S, Farrell JJ, Kasperavičiūtė D, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364:1134–1143. doi: 10.1056/NEJMoa1013297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Monte AA, Heard KJ, Vasiliou V. Prediction of drug response and safety in clinical practice. J Med Toxicol. 2012;8:43–51. doi: 10.1007/s13181-011-0198-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fabi A, Di Benedetto A, Metro G, Perracchio L, Nisticò C, et al. HER2 protein and gene variation between primary and metastatic breast cancer: significance and impact on patient care. Clin Cancer Res. 2011;17:2055–2064. doi: 10.1158/1078-0432.CCR-10-1920. [DOI] [PubMed] [Google Scholar]
- 81.Stearns V, Johnson MD, Rae JM, Morocho A, Novielli A, et al. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J Natl Cancer Inst. 2003;95:1758–1764. doi: 10.1093/jnci/djg108. [DOI] [PubMed] [Google Scholar]
- 82.Jin Y, Desta Z, Stearns V, Ward B, Ho H, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97:30–39. doi: 10.1093/jnci/dji005. [DOI] [PubMed] [Google Scholar]
- 83.Azoulay L, Dell’Aniello S, Huiart L, du Fort GG, Suissa S. Concurrent use of tamoxifen with CYP2D6 inhibitors and the risk of breast cancer recurrence. Breast Cancer Res Treat. 2011;126:695–703. doi: 10.1007/s10549-010-1162-y. [DOI] [PubMed] [Google Scholar]
- 84.Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011;364:1144–1153. doi: 10.1056/NEJMra1010600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen P, Lin JJ, Lu CS, Ong CT, Hsieh PF, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med. 2011;364:1126–1133. doi: 10.1056/NEJMoa1009717. [DOI] [PubMed] [Google Scholar]
- 86.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 87.Youngster I, Arcavi L, Schechmaster R, Akayzen Y, Popliski H, et al. Medications and glucose-6-phosphate dehydrogenase deficiency:an evidence-based review. Drug Saf. 2010;33:713–726. doi: 10.2165/11536520-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 88.Iverson AA, Gillett C, Cane P, Santini CD, Vess TM, et al. A single-tube quantitative assay for mRNA levels of hormonal and growth factor receptors in breast cancer specimens. J Mol Diagn. 2009;11:117–130. doi: 10.2353/jmoldx.2009.080070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Müller BM, Kronenwett R, Hennig G, Euting H, Weber K, et al. Quantitative determination of estrogen receptor, progesterone receptor, and HER2 mRNA in formalin-fixed paraffin-embedded tissue--a new option for predictive biomarker assessment in breast cancer. Diagn Mol Pathol. 2011;20:1–10. doi: 10.1097/PDM.0b013e3181e3630c. [DOI] [PubMed] [Google Scholar]
- 90.Sun B, Zhang F, Wu SK, Guo X, Zhang LL, et al. Gene expression profiling for breast cancer prognosis in Chinese populations. Breast J. 2011;17:172–179. doi: 10.1111/j.1524-4741.2010.01049.x. [DOI] [PubMed] [Google Scholar]
- 91.Lee A, Ezzeldin H, Fourie J, Diasio R. Dihydropyrimidine dehydrogenase of pharmacogenetics on 5-fluorouracil therapy. Clin Adv deficiency: Hematol Oncol. 2004;2:527–532. [PubMed] [Google Scholar]
- 92.Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, et al. A functional genetic approach identifiesthe PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 93.Cappuzzo F, Finocchiaro G, Rossi E, Jänne PA, Carnaghi C, et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol. 2008;19:717–723. doi: 10.1093/annonc/mdm492. [DOI] [PubMed] [Google Scholar]
- 94.Cascinu S, Aschele C, Barni S, Debernardis D, Baldo C, et al. Thymidylate synthase protein expression in advanced colon cancer: correlation with the site of metastasis and the clinical response to leucovorin-modulated bolus 5-fluorouracil. Clin Cancer Res. 1999;5:1996–1999. [PubMed] [Google Scholar]
- 95.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 96.Ciaparrone M, Quirino M, Schinzari G, Zannoni G, Corsi DC, et al. Predictive role of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer patients receiving adjuvant 5-fluorouracil. Oncology. 2006;70:366–377. doi: 10.1159/000098110. [DOI] [PubMed] [Google Scholar]
- 97.Souglakos J, Philips J, Wang R, Marwah S, Silver M, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer. 2009;101:465–472. doi: 10.1038/sj.bjc.6605164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martínez-Navarro EM, Rebollo J, González-Manzano R, Sureda M, Evgenyeva E, et al. Epidermal growth factor receptor (EGFR) mutations in a series of non-small-cell lung cancer (NSCLC) patients and response rate to EGFR-specific tyrosine kinase inhibitors (TKIs) Clin Transl Oncol. 2011;13:812–818. doi: 10.1007/s12094-011-0739-1. [DOI] [PubMed] [Google Scholar]
- 99.Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Olaussen KA, Dunant A, Fouret P, Brambilla E, André F, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006;355:983–991. doi: 10.1056/NEJMoa060570. [DOI] [PubMed] [Google Scholar]
- 101.Gaughan EM, Costa DB. Genotype-driven therapies for non-small cell lung cancer: focus on EGFR, KRAS and ALK gene abnormalities. Ther Adv Med Oncol. 2011;3:113–125. doi: 10.1177/1758834010397569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gridelli C, Maione P, Ferrara ML, Rossi A. Cetuximab and other anti-epidermal growth factor receptor monoclonal antibodies in the treatment of non-small cell lung cancer. Oncologist. 2009;14:601–611. doi: 10.1634/theoncologist.2008-0153. [DOI] [PubMed] [Google Scholar]
- 103.Hanauske AR, Eismann U, Oberschmidt O, Pospisil H, Hoffmann S, et al. In vitro chemosensitivity of freshly explanted tumor cells to pemetrexed is correlated with target gene expression. Invest New Drugs. 2007;25:417–423. doi: 10.1007/s10637-007-9060-9. [DOI] [PubMed] [Google Scholar]
- 104.Shulman K, Cohen I, Barnett-Griness O, Kuten A, Gruber SB, et al. Clinical implications of UGT1A1*28 genotype testing in colorectal cancer patients. Cancer. 2011;117:3156–3162. doi: 10.1002/cncr.25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shaw AT, Solomon B, Kenudson MM. Crizotinib and testing for ALK. J Natl Compr Canc Netw. 2011;9:1335–1341. doi: 10.6004/jnccn.2011.0115. [DOI] [PubMed] [Google Scholar]
- 106.Croxtall JD, McKeage K. Trastuzumab: in HER2-positive metastatic gastric cancer. Drugs. 2010;70:2259–2267. doi: 10.2165/11205900-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 107.Herceptin: summary of product characteristics.
- 108.Kwon HC, Roh MS, Oh SY, Kim SH, Kim MC, et al. Prognostic value of expression of ERCC1, thymidylate synthase, and glutathione S-transferase P1 for 5-fluorouracil/oxaliplatin chemotherapy in advanced gastric cancer. Ann Oncol. 2007;18:504–509. doi: 10.1093/annonc/mdl430. [DOI] [PubMed] [Google Scholar]
- 109.Motomura K, Natsume A, Kishida Y, Higashi H, Kondo Y, et al. Benefits of interferon-β and temozolomide combination therapy for newly diagnosed primary glioblastoma with the unmethylated MGMT promoter: A multicenter study. Cancer. 2011;117:1721–1730. doi: 10.1002/cncr.25637. [DOI] [PubMed] [Google Scholar]
- 110.Yang CG, Ciccolini J, Blesius A, Dahan L, Bagarry-Liegey D, et al. DPD-based adaptive dosing of 5-FU in patients with head and neck cancer: impact on treatment efficacy and toxicity. Cancer Chemother Pharmacol. 2011;67:49–56. doi: 10.1007/s00280-010-1282-4. [DOI] [PubMed] [Google Scholar]
- 111.Döhner H, Fischer K, Bentz M, Hansen K, Benner A, et al. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood. 1995;85:1580–1589. [PubMed] [Google Scholar]
- 112.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 113.Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108:28–37. doi: 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.List A, Dewald G, Bennett J, Giagounidis A, Raza A, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456–1465. doi: 10.1056/NEJMoa061292. [DOI] [PubMed] [Google Scholar]
- 115.David M, Cross NC, Burgstaller S, Chase A, Curtis C, et al. Durable responses to imatinib in patients with PDGFRB fusion gene-positive and BCR-ABL-negative chronic myeloproliferative disorders. Blood. 2007;109:61–64. doi: 10.1182/blood-2006-05-024828. [DOI] [PubMed] [Google Scholar]
- 116.Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med. 2002;347:481–487. doi: 10.1056/NEJMoa020150. [DOI] [PubMed] [Google Scholar]
- 117.Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood. 2009;114:3533–3537. doi: 10.1182/blood-2009-05-220095. [DOI] [PubMed] [Google Scholar]