


Abstract
Asymmetric dimethylarginine (aDMA) marks are placed on histones and the C-terminal domain (CTD) of RNA Polymerase II (RNAP II) and serve as a signal for recruitment of appropriate transcription and processing factors in coordination with transcription cycle. In contrast to other Tudor domain-containing proteins, Tudor domain-containing protein 3 (TDRD3) associates selectively with the aDMA marks but not with other methylarginine motifs. Here, we report the solution structure of the Tudor domain of TDRD3 bound to the asymmetrically dimethylated CTD. The structure and mutational analysis provide a molecular basis for how TDRD3 recognizes the aDMA mark. The unique aromatic cavity of the TDRD3 Tudor domain with a tyrosine in position 566 creates a selectivity filter for the aDMA residue. Our work contributes to the understanding of substrate selectivity rules of the Tudor aromatic cavity, which is an important structural motif for reading of methylation marks.
INTRODUCTION
Arginine methylation is a frequent post-translational modification of proteins that regulates a variety of cellular processes, including transcriptional regulation, RNA processing, trafficking, signal transduction and DNA repair (1–3). There are three major forms of methylated arginine identified in mammals, monomethylarginine (MMA), asymmetric dimethylarginine (aDMA) and symmetric dimethylarginine (sDMA). These methylation marks are introduced by the protein arginine methyltransferases’ (PRMTs) family, in which, type I PRMTs (PRMT1, 2, 3, 4, 6 and 8) generate MMA and aDMA modifications, whereas type II PRMTs (PRMT5 and 7) produce MMA and sDMA modifications. The MMA modifications introduced by both type I and type II PRMTs are likely generated as ‘intermediates’ on the way to dimethylarginines. PRMTs methylate a large number of protein targets, involved in various aspects of regulation of gene expression (2).
The co-activator-associated arginine methyltransferase 1 (CARM1/PRMT4) deposits an asymmetric dimethylation at the R2 and R17 sites of histone H3 (H3R17 and H3R2) and at the R3 site of histone H4 (H4R3) (4). It also introduces the aDMA mark at R1810 of the mammalian carboxy-terminal domain (CTD) of RNA Polymerase II (RNAP II) (5). The CTD is an important region of RNAP II that undergoes structural remodeling throughout the transcriptional cycle, which allows the association and dissociation of a multitude of effector molecules (6–11). These temporal and spatial interactions couple transcription with most, if not all, pre-mRNA processing steps (12–24). The CTD methylation at R1810 is present on the hyperphosphorylated CTD in vivo, and it facilitates the expression of small nuclear and nucleolar RNAs (snRNAs and snoRNAs) (5).
Pull-down experiments showed that Tudor domain-containing protein 3 (TDRD3) displays a specific interaction with the asymmetrically dimethylated R1810-containing CTD peptide (aDMA-CTD) and histones H3 and H4, but not with unmodified and monomethylated or symmetrically dimethylated peptides (4,5). Consistently, using fluorescence polarization, it was reported that TDRD3 preferentially binds the aDMA marks compared with other methylarginine species (25). Other members of the Tudor family, such as the Survival of Motor Neuron (SMN), survival of motor neuron-related splicing factor 30 (SPF30), staphylococcal nuclease domain-containing protein 1 (SND1) and Tur11 do not discriminate between the aDMA- and sDMA-containing peptides (26–28). They are capable of binding to both dimethylarginine isoforms, with slightly higher affinity to sDMA, which also represents their physiological ligand (29,30). The structures of the Tudor domains of SMN, SPF30, SND1 and Tur11 bound to their ligands have been reported (26–28).
In order to reveal the structural basis of selective recognition of the aDMA marks by TDRD3 that has recently been implicated in transcription activation and RNA processing (4,5), we have determined the solution structure of the Tudor domain of TDRD3 in complex with asymmetrically dimethylated CTD.
MATERIALS AND METHODS
Cloning, expression and purification of Tudor domain of TDRD3 (residues 554–608)
The Tudor domain of TDRD3 (residues 554–608) was cloned into pET22b expression vector to generate C-terminal 6x His-tagged protein. The protein was over-expressed in Escherichia coli BL21-Codon Plus (DE3)-RIPL (Stratagene) overnight at 16°C after induction by 1 mM IPTG and purified by affinity chromatography on Ni-NTA resin (Qiagen) and further purified on Superdex75 gel filtration column. For nuclear magnetic resonance (NMR) experiments, the protein was concentrated into a buffer containing 50 mM Na2HPO4, pH 8.0, 150 mM NaCl and 10 mM β-mercaptoethanol. Mutant constructs were prepared by QuikChange™ Site-directed Mutagenesis Kit with complementary sense (S) and antisense (AS) oligonucleotide primers as follows:
-
Y566F
(S), 5′-cctggagatgaatgttttgcacttttttgggaagacaaca-3′
(AS), 5′-tgttgtcttcccaaaaaagtgcaaaacattcatctccagg-3′;
-
Y566W
(S), 5′-gaaacctggagatgaatgttttgcactttggtgggaagacaacaag-3′
(AS), 5′-cttgttgtcttcccaccaaagtgcaaaacattcatctccaggtttc-3′;
-
W567S
(S), 5′-cctggagatgaatgttttgcactttattcggaagacaacaagtt-3′
(AS), 5′-aacttgttgtcttccgaataaagtgcaaaacattcatctccagg-3′;
-
D569A
(S), 5′-gcactttattgggaagcgaacaagttttaccgggc-3′
(AS) 5′-gcccggtaaaacttgttcgcttcccaataaagtgc-3′.
The mutant proteins were over-expressed in the same E. coli line and purified using the same procedure as described above. Peptides for NMR measurements were purchased from Clonestar peptide service (Brno, CZ).
Nuclear magnetic resonance
All spectra for the backbone and side-chain assignments of ∼1.8 mM uniformly 15N,13C-labeled TDRD3 Tudor in 50 mM sodium phosphate buffer (pH 8.0), 150 mM NaCl, 10 mM β-mercaptoethanol (90% H2O/10% D2O) were recorded on Bruker AVANCE 600 MHz spectrometer equipped with a cryoprobe at a sample temperature of 293.15 K. The spectra were processed using NMRPipe package (31) and the protein resonances were assigned manually, using Sparky software (Goddard T.G. and Kellner D.G., University of California, San Francisco, USA). The 1H, 13C and 15N chemical shifts of TDRD3–aDMA-CTD complex were assigned using standard triple resonance experiments (32). All distance constraints were derived from the 3D 15N- and 13C-separated nuclear Overhauser effect spectroscopy (NOESYs) (with mixing time of 110 ms) collected on a 600 MHz spectrometer. Intermolecular distance constraints were obtained from the 3D F1-13C/15N-filtered NOESY-[13C,1H]-HSQC experiment (33,34), with mixing time of 150 ms on a 600 MHz spectrometer. In the 3D NOESY spectra, intermolecular constraints were semi-quantitatively classified, based on their peak volumes divided by the number of 1H spins involved in nuclear Overhauser effect (NOE) cross-relaxation.
Structure calculation
Structure determination of the TDRD3 Tudor–aDMA-CTD complex was performed with the NOE assignment algorithm implemented in the CYANA program (35). This automated NOE assignment procedure is a re-implementation of the former CANDID algorithm (36) on the basis of a probabilistic treatment of the NOE assignment. CYANA carries out automated assignments, distance calibration of NOE intensities, removal of erroneous restraints, structure calculations with torsion angle dynamics. The resultant NOE cross-peak assignments were subsequently confirmed by visual inspection of the spectra. In the next step, CYANA-generated restraints along with manually assigned TDRD3–aDMA-CTD intermolecular restraints were used for further refinement of 20 preliminary structures with AMBER 11 software (37,38). These calculations employed a modified version (AMBER ff99SB) of the force field described by Cornell et al. (39) and an explicit water solvent. The non-standard aDMA residue was parameterized with Gaussian 09 using restrained electrostatic potential charge approach at HF/6-31G(d) level of theory (40). The compound name for aDMA is DA2. Structural quality was assessed using PROCHECK (41) and WHAT IF (42).
Fluorescence anisotropy
The equilibrium binding of Tudor domain of TDRD3 to the CTD peptides was analyzed by fluorescence anisotropy. The CTD peptides were N-terminally labeled with 5,6-carboxyfluorescein (FAM). The measurements were conducted on a FluoroMax-4 spectrofluorometer (Horiba Jobin-Yvon Edison, NJ, USA). Samples were excited with vertically polarized light at 495 nm, and both vertical and horizontal emissions were recorded at 535 nm. All measurements were conducted at 293.15 K in 50 mM phosphate buffer (pH 8.0) containing 150 mM NaCl. A fixed delay of 30 s was set between each aliquot addition and start of the measurement to allow the reaction to reach equilibrium. This delay was sufficient, as no further change in anisotropy was observed. Each data point is an average of three measurements. The data were analyzed using Gnuplot. The experimental isotherms were fit to a single-site binding model using non-linear least squares regression according to Heyduk and Lee (43).
NMR binding experiments
1H-15N Heteronuclear single quantum coherence (HSQC) spectra were recorded at same conditions as NOESY experiments. To determine the affinity of different TDRD3 mutants with aDMA- and sDMA-containing peptides, a series of 1H-15N HSQC spectra were recorded. Increasing amounts of the unlabeled dimethylarginine-containing peptides were added to 15N-labeled proteins. Combined chemical shift (CCS) is defined as the normalized length of a vector Ej, whose components are chemical shift differences δji between observed chemical shift and reference experiment (free form). Index j represents the atom type within the primary sequence of the protein
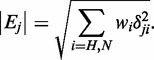 |
Weight factors for each atom type wH = 1 and wN = 0.15 were used. CCS values of three best resolved residues for each titration were used to construct a binding isotherm of 1:1 stoichiometry. Parameters for binding isotherms were found by non-linear least-square regression with Gnuplot. The errors for fitted parameters were calculated within a 95% confidence interval (95% CI).
Quantum chemical calculations
The geometry of aromatic cavity was taken from the deposited NMR structure. The truncated model was made of aDMA and four residues forming the aromatic cavity. The Cα atoms were substituted with methyl groups that were kept frozen during the optimization to account for backbone covalent interactions. The geometry of aDMA and four aromatic cage amino acids was then optimized in vacuo using TPSS-D3/def2-TZVP level of theory with Turbomole 6.3 (44,45). For interaction energy calculations, we used a Hartree–Fock wave function of the monomers as a reference from which, we obtained interaction energy terms in first two orders at symmetry-adapted perturbation theory (SAPT0) approximation using PSI4 suite of codes. Density fitting of 4-index integrals and Laplace transformations of energy denominators were used as described in the following article (46). Reference wavefunction in SAPT analysis was calculated using aug-cc-pVDZ basis set.
RESULTS
Solution structure of TDRD3 in complex with aDMA–CTD peptide
Initial NMR titration experiments suggested that two regions of the Tudor domain of TDRD3 (residues 566–573 and 589–598) were significantly perturbed upon binding to the aDMA-CTD peptide (Figure 1). Analysis of NMR titration data under fast exchange regime allowed to evaluate the binding affinity of the TDRD3 Tudor with the sDMA- and aDMA-containing CTD peptides. TDRD3 binds aDMA-CTD with a KD of 770 ± 30 µM, more than one order of magnitude stronger, compared with the symmetrically methylated isoform (Figure 1D). Similar binding affinity of TDRD3 to fluorescently labeled aDMA-CTD peptide was determined in a quantitative binding assay using fluorescence anisotropy (KD = 900 ± 200 µM; Supplementary Figures S1 and S2).
Figure 1.

Interaction of the TDRD3 Tudor with aDMA-CTD. (A) Sequence of a 13 amino acid peptide with aDMA, used as a mimic of methylated CTD; and domain organization of TDRD3. (B) Scheme of aDMA and sDMA. (C) A 2D 1H-15N HSQC spectra showing four representative steps of NMR titration of 1.8 mM TDRD3 with aDMA-CTD peptide. Trajectories for the three best resolved signals that were used for construction of binding isotherm are highlighted. (D) Estimation of the TDRD3 Tudor–aDMA-CTD (in blue) and TDRD3 Tudor–sDMA-CTD (in red) dissociation constants from NMR titration experiments. CCS values derived from 1H-15N HSQCs are plotted against the CTD peptide concentration. Errors are denoted as 95% CI. (E) Quantification of chemical shift perturbations of the TDRD3 Tudor upon addition of aDMA-CTD. The combined chemical shift perturbations are plotted versus the amino-acid residue number with β-sheets regions shown as blue arrows. Large changes occur in the regions involved in binding of the aDMA-CTD peptide. Green lines represent the residues forming hydrophobic cavity.
To understand the structural basis for molecular recognition of aDMA-containing CTD by TDRD3, we determined the solution structure of a reconstituted complex consisting of the Tudor domain of TDRD3 (residues 555–610) and a 13-mer peptide aDMA-CTD [YSPSSP(aDMA)YTPQSP; Figure 1A] (Figure 2A and B; Supplementary Figure S2B and C; Supplementary Table S1). The Tudor domain of TDRD3 forms a four-stranded β-barrel fold and is similar to the structure of TDRD3 in free form (Supplementary Figure S2B) (25).
Figure 2.

NMR structure of the TDRD3 Tudor–aDMA-CTD complex. (A) Overlay of the 20 lowest energy structures of the TDRD3 Tudor–aDMA-CTD complex shown in stereo view. Backbone and side-chains of the aromatic cavity (Y566, Y573, F591 and Y594) are shown in wire representation. For clarity, only aDMA residue of the bound peptide is shown in wire representation. The TDRD3 Tudor domain and aDMA are shown in black and red, respectively. (B) Structure of the human TDRD3 Tudor domain bound to the aDMA-CTD peptide. The aDMA-CTD peptide is represented as a magenta ribbon with the aDMA residue in sticks (only methyl protons are shown) and the protein is shown as a blue ribbon model. Residues forming the aromatic cavity (Y566, Y573, F591 and Y594) are shown in green sticks and the cube-shaped cavity is highlighted by a square. (C) aDMA recognition by the TDRD3 Tudor domains. The hydrogen bond and aDMA interactions with the aromatic cavity are shown with yellow dotted lines. Only aDMA and the side-chain of amino acids that form, or interact with the aromatic cavity are shown (starting from Cα atoms).
The β1–β2 and β3–β4 loops of the Tudor domain contain aromatic residues (Y566, Y573, F591 and Y594). These residues form a partially exposed aromatic cavity of rectangular cuboid shape that is side-walled by N596 (Figure 2C). Upon binding of TDRD3 to the CTD peptide, the aromatic cavity accommodates aDMA that is placed parallel between two tyrosines (Y566 and Y594). This binding results from cation–π and stacking interactions between positively charged guanidinium group of aDMA and aromatic rings of the two parallel tyrosine residues. Furthermore, CH–π interactions between the methyl groups of aDMA and two remaining aromatic residues (Y573 and F591) stabilize the interaction. One guanidinomethyl group faces Y573, whereas the second guanidinomethyl group faces F591 at the back wall of the cavity (Figure 2C). Interestingly, conserved N596 does not form a H-bond to aDMA-CTD, akin to the structures of s/aDMA bound to the canonical Tudor domains of SPF30 and SMN (26). This conserved asparagine in the structures of extended Tudor domains forms an H-bond to sDMA, altering its role in this Tudor subfamily (27,28).
Our NMR data show no intermolecular contacts between the aromatic cavity and neighboring residues of aDMA (Supplementary Figure S2C). This suggests that those residues are flexible and do not interact with the TDRD3 Tudor domain, and that the CTD peptide with the aDMA mark is recognized in a sequence-independent manner. Furthermore, phosphorylations of aDMA-CTD (at Ser2 and Ser5) showed no effect on the binding affinity to TDRD3.
TDRD3 Y566 mutants loose specificity to aDMA-CTD peptides
It was shown that substitution of any of the four aromatic residues of the cavity with a non-aromatic amino acid, abrogates dimethylarginine binding (26). In the present study, we have investigated the effect of aromatic substitutions of the least conserved residue within the aromatic cavity (Figures 2C and 3A). In TDRD3, Y566 is a unique residue, whereas SMN, SPF30, SND1 and Tur11 contain tryptophan or phenylalanine in this position. Y566F substitution diminishes binding to aDMA-CTD and it does not increase binding affinity to sDMA-CTD (Figure 3B and Supplementary Figure S5). On the other hand, Y566W substitution promotes complex formation with sDMA-CTD, yet it has similar binding affinity to aDMA-CTD as the wild-type protein. This indicates that both phenylalanine and tryptophan substitutions at position Y566 abrogate TDRD3 selectivity for aDMA- against sDMA-CTD peptides. Residue Y566 is thus the key element, which determines the specificity of TDRD3 toward aDMA-containing peptides.
Figure 3.

Recognition of aDMA by the TDRD3 Tudor. (A) Sequence alignment of the human TDRD3 Tudor domain with other dimethylarginine binding Tudor domains of SMN (Homo sapiens), SPF30 (H. sapiens), SND1 (H. sapiens), and the 11th Tudor domain of Tud (Drosophila melanogaster; Tud11). Residues forming an aromatic cavity are highlighted in green squares, variable residues are shown in red. The β-sheet regions (β1, β2, β3 and β4) of human TDRD3 Tudor are shown with blue arrows. (B) Bar plot of the NMR-derived association constants (Ka) of various TDRD3 mutants with the aDMA-CTD peptide (blue) and sDMA-CTD peptide (red) in a logarithmic scale. Asterisk indicates that the binding constant could not be determined. (C) Bar plot showing decomposed interactions’ energies between the aromatic cavity and aDMA calculated by SAPT. The exchange-repulsion (blue bar) accounts for an interaction caused by tunneling of the electrons between interacting systems and electron−electron repulsion due to the Pauli exclusion principle. The induction interaction (yellow bar) is a second-order energy contribution, which originates from mutual distortion of electron density distribution of interacting molecules. The dispersion interaction (green) arises from the correlated electron fluctuations in the interacting molecules (48). (D) Pyramidalization of the aDMA amino group as predicted by DFT-D theory. The hydrogen bond (2.7 Å) that is responsible for aDMA recognition is shown with yellow dotted line. Only heavy atoms and non-polar hydrogens are shown.
Comparison of the TDRD3–aDMA-CTD complex with previously determined structures of SPF30/SMN–a/sDMA complexes show variations in accommodation of the substrates in the aromatic cavities (Supplementary Figure S3A). The sDMA substrates are inserted at a different angle than aDMA, so that they maximize stacking interactions with the tryptophan aromatic residue in the position 566 (TDRD3 numbering). This underlines the importance of the aromatic residue type (Y/W/F; Figure 3A) in this position for dimethylarginine recognition.
Pyramidalization of aDMA amino group promotes hydrogen bond formation with TDRD3
Force field approximations along with limited resolution of experimental data often imperfectly describe molecular interactions. In our structure of the TDRD3–aDMA-CTD complex, we identified a possibility of H-bond between the aDMA amino group and the hydroxyl group of Y566. Such H-bond would require a distortion of the aDMA amino group planarity. Studies of high-resolution x-ray structures have shown amino group pyramidalization allowing hydrogen bond formation in nucleic acids (47). However, empirical force fields used for the structural calculations enforce the planarity of the amino group; therefore, we had to resort to other methods. To probe the existence of this H-bond, we performed a dispersion-corrected density functional theory (DFT-D) study of aDMA bound to the aromatic cavity of TDRD3 (for details, see ‘Materials and Methods’ section). Geometry of the DFT-D optimized model is very close to the NMR structure with heavy atom RMSD of 0.9 Å (Supplementary Figure S3B). Most importantly, the planarity of aDMA amino group was slightly distorted (due to partial sp3 hybridization), allowing the H-bond formation with the hydroxyl group of Y566 (Figure 3D). As the cavity is formed by hydrophobic residues and its interior is not accessible to solvents, it is likely that this H-bond has significant energy. Y566F substitution in TDRD3 or the lack of amino group in sDMA prevents formation of this H-bond (Figures 1D and 3B). This hydrogen bond explains why, during titration experiments, Y566F substitution abrogated TDRD3 binding with aDMA and why sDMA did not bind to wild-type TDRD3.
Dispersion interactions play a major role in complex formation
To understand the forces contributing to the complex formation, we calculated the interaction energy of previously optimized DFT-D model of TDRD3–aDMA complex using SAPT calculations (48). In this perturbation approach, the interaction energy is expressed as a sum of physically well defined parts. Electrostatic and exchange-repulsion terms represent the first order contributions, whereas the second order terms are represented by induction and dispersion energy. Calculated interaction energy terms are summarized in Figure 3C.
The ‘electrostatic’ term is stabilizing. This is consistent with our picture that  hydrogen bond and cation–π interactions contribute to complex formation. However, the ‘exchange-repulsion’ over-compensates the electrostatic attraction, as many electron pairs come close when aDMA and aromatic residues form the complex. Therefore, the first order contributions are repulsive. The second order contributions showed that ‘induction’ term plays a minor role for the overall binding energy. In general, cation–π interactions are dominated by electrostatic forces and cation-induced polarization of π systems (49). This suggests that induction mainly represents an interaction between a positive guanidinium group of aDMA and π orbitals of parallel tyrosines. The ‘dispersion’ interaction is the most significant stabilizing term. It should be noted that all non-covalent interactions contain some degree of a dispersion-type component (49). However, the role of dispersion in cation–π and hydrogen bonds is modest (50). As the dispersion energy is almost as large as the total interaction energy, we conclude that the dispersion originates predominantly from stacking interactions between aDMA and two parallel tyrosines (Y566 and Y594).
hydrogen bond and cation–π interactions contribute to complex formation. However, the ‘exchange-repulsion’ over-compensates the electrostatic attraction, as many electron pairs come close when aDMA and aromatic residues form the complex. Therefore, the first order contributions are repulsive. The second order contributions showed that ‘induction’ term plays a minor role for the overall binding energy. In general, cation–π interactions are dominated by electrostatic forces and cation-induced polarization of π systems (49). This suggests that induction mainly represents an interaction between a positive guanidinium group of aDMA and π orbitals of parallel tyrosines. The ‘dispersion’ interaction is the most significant stabilizing term. It should be noted that all non-covalent interactions contain some degree of a dispersion-type component (49). However, the role of dispersion in cation–π and hydrogen bonds is modest (50). As the dispersion energy is almost as large as the total interaction energy, we conclude that the dispersion originates predominantly from stacking interactions between aDMA and two parallel tyrosines (Y566 and Y594).
Surrounding amino acids stabilize the aromatic cavity
The structure of the TDRD3–aDMA-CTD complex reveals that D569 of the β1–β2 loop forms the H-bond with the hydroxyl group of Y573 and thus, stabilizes the geometry of the aromatic cavity (Figure 2C). If the H-bond formation is impaired by D569A substitution, the [1H,15N]-HSQC spectrum is significantly altered when compared with that of the wild-type (Supplementary Figure S4A). The residues of the aromatic cavity and their nearest neighbors display large chemical shift perturbations, suggesting that the geometry of the cavity is disturbed (Supplementary Figure S4B). Furthermore, D569A substitution abrogates binding to the aDMA-CTD peptide, indicating the importance of D569 for the stabilization of the aromatic cavity (Supplementary Figure S4B).
Conserved W567 residue stacks with Y566, stabilizing the geometry of aromatic cavity (Figure 2C and Supplementary Figure S2A). W567S substitution of TDRD3 Tudor yields lower affinity to aDMA-CTD (Figure 3B). These results demonstrate the importance of residues surrounding the cavity for the recognition of aDMA by TDRD3.
DISCUSSION
Available structures of the SMN, SPF30, SND1 and Tud11 Tudor domains in complex with their corresponding dimethylarginine ligands (26–28), together with the structure of the TDRD3 Tudor domain bound to aDMA-CTD, define structural determinants for the recognition of sDMA- and aDMA-containing peptides. The extended Tudor domains (SND1 and Tur11) contain an additional α-helix and two β-strands at aminoterminal and several α-helices and β-strands at the carboxy-terminal to the canonical Tudor core. They preferably interact with sDMA-containing peptides, because of hydrogen bond formation between sDMA and asparagine residue (Supplementary Figure S6). In contrast, the structures of the canonical Tudor domains bound to dimethylarginine ligands lack this hydrogen bond (Supplementary Figure S7). They utilize a different strategy for the dimethylarginine recognition. The SMN and SPF30 canonical Tudor domains contain tryptophan at position 566 (Figure 3A). The presence of the fused-ring heterocyclic amino acid alters the stacking interactions between aromatic cavity and ligand. As a result, both SMN and SPF30 preferably bind to sDMA but also possess a comparable affinity to aDMA.
The presence of tyrosine at position 566 in the canonical Tudor domain of TDRD3, creates unique binding properties of the cavity as it selectively binds the aDMA mark but not the sDMA mark. The TDRD3–aDMA complex formation is driven by a combination of cation–π, CH–π and stacking interactions between aromatic cavity of Tudor domain and aDMA ligand. The complex is further stabilized by the 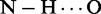 hydrogen bond between the aDMA amino group and the hydroxyl group of Y566. Y566F variant of TDRD3, which causes the loss of this hydrogen bond, abrogates the interaction between TDRD3 and aDMA. Interestingly, Y566W variant allows TDRD3 to form a complex with sDMA-CTD and also has a similar affinity to aDMA-CTD (Figure 3B). The presence of tryptophan in this position provides an additional dispersion interaction that is non-specific to the dimethylarginine motifs. Y566W mutant of the TDRD3 Tudor domain is therefore, able to bind both sDMA and aDMA peptides even though the
hydrogen bond between the aDMA amino group and the hydroxyl group of Y566. Y566F variant of TDRD3, which causes the loss of this hydrogen bond, abrogates the interaction between TDRD3 and aDMA. Interestingly, Y566W variant allows TDRD3 to form a complex with sDMA-CTD and also has a similar affinity to aDMA-CTD (Figure 3B). The presence of tryptophan in this position provides an additional dispersion interaction that is non-specific to the dimethylarginine motifs. Y566W mutant of the TDRD3 Tudor domain is therefore, able to bind both sDMA and aDMA peptides even though the  hydrogen bond is broken by this substitution. We conclude that the tyrosine residue at position 566 of the TDRD3 Tudor domain works as a selectivity filter for the aDMA-containing peptides.
hydrogen bond is broken by this substitution. We conclude that the tyrosine residue at position 566 of the TDRD3 Tudor domain works as a selectivity filter for the aDMA-containing peptides.
The results presented in this study extend our understanding of how the methylarginine binding Tudor domains coordinate their ligands. A comparison of the ligand-bound TDRD3 structure with other Tudor domains, supported by the mutational analysis and quantum chemical calculations provide a framework to understand dimethylarginine recognition at a molecular level.
ACCESSION NUMBERS
The atomic coordinates for the NMR ensemble of the TDRD3–aDMA-CTD complex have been deposited in the Protein Data Bank under accession code 2lto. The chemical shift assignments have been deposited in the Biological Magnetic Resonance Data Bank under accession code 18490.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1–7.
FUNDING
CEITEC-Central European Institute of Technology [CZ.1.05/1.1.00/02.0068] from European Regional Development Fund; Czech Science Foundation [P305/12/G034 and P305/10/1490]; Brno City Municipality Scholarship for Talented Ph.D. Students (to F.H.). Funding for open access charge: Czech Science Foundation.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Frank Löhr for the NMR pulse programs for the three-dimensional F1-13C/15N-filtered NOESY-[13C,1H]-HSQC experiments. T.S. did quantum chemical calculations, carried out NMR experiments and performed structure calculations. F.H. and E.K. carried out sample preparations, performed and analyzed the FA experiments. J.P. contributed to structure calculations. K.K. contributed to NMR experiments. R.S. conceived and designed the project and contributed to NMR experiments. All authors discussed the results and wrote the manuscript.
REFERENCES
- 1.Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol. Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siomi MC, Mannen T, Siomi H. How does the royal family of Tudor rule the PIWI-interacting RNA pathway? Genes Dev. 2010;24:636–646. doi: 10.1101/gad.1899210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, Lu Y, Espejo A, Wu J, Xu W, Liang S, Bedford MT. TDRD3 is an effector molecule for arginine-methylated histone marks. Mol. Cell. 2010;40:1016–1023. doi: 10.1016/j.molcel.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sims RJ, 3rd, Rojas LA, Beck D, Bonasio R, Schuller R, Drury WJ, 3rd, Eick D, Reinberg D. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science. 2011;332:99–103. doi: 10.1126/science.1202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20:2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 7.Corden JL. Transcription. Seven ups the code. Science. 2007;318:1735–1736. doi: 10.1126/science.1152624. [DOI] [PubMed] [Google Scholar]
- 8.Chapman RD, Heidemann M, Hintermair C, Eick D. Molecular evolution of the RNA polymerase II CTD. Trends Genet. 2008;24:289–296. doi: 10.1016/j.tig.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Buratowski S. The CTD code. Nat. Struct. Biol. 2003;10:679–680. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- 11.Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol. Cell. 2009;36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirose Y, Manley JL. RNA polymerase II and the integration of nuclear events. Genes Dev. 2000;14:1415–1429. [PubMed] [Google Scholar]
- 13.Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- 14.Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P. A structural perspective of CTD function. Genes Dev. 2005;19:1401–1415. doi: 10.1101/gad.1318105. [DOI] [PubMed] [Google Scholar]
- 15.Viladevall L, St Amour CV, Rosebrock A, Schneider S, Zhang C, Allen JJ, Shokat KM, Schwer B, Leatherwood JK, Fisher RP. TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol. Cell. 2009;33:738–751. doi: 10.1016/j.molcel.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh A, Shuman S, Lima CD. Structural insights to how mammalian capping enzyme reads the CTD code. Mol. Cell. 2011;43:299–310. doi: 10.1016/j.molcel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de la Mata M, Kornblihtt AR. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat. Struct. Mol. Biol. 2006;13:973–980. doi: 10.1038/nsmb1155. [DOI] [PubMed] [Google Scholar]
- 18.Munoz MJ, de la Mata M, Kornblihtt AR. The carboxy terminal domain of RNA polymerase II and alternative splicing. Trends Biochem. Sci. 2010;35:497–504. doi: 10.1016/j.tibs.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 19.David CJ, Boyne AR, Millhouse SR, Manley JL. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65-Prp19 complex. Genes Dev. 2011;25:972–983. doi: 10.1101/gad.2038011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S, Andrade J, Levezinho H, Gut M, Eick D, Gut I, et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat. Struct. Mol. Biol. 2011;18:977–983. doi: 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- 21.Ahn SH, Kim M, Buratowski S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol. Cell. 2004;13:67–76. doi: 10.1016/s1097-2765(03)00492-1. [DOI] [PubMed] [Google Scholar]
- 22.Egloff S, O'Reilly D, Chapman RD, Taylor A, Tanzhaus K, Pitts L, Eick D, Murphy S. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science. 2007;318:1777–1779. doi: 10.1126/science.1145989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson SA, Kim H, Erickson B, Bentley DL. The export factor Yra1 modulates mRNA 3′ end processing. Nat. Struct. Mol. Biol. 2011;18:1164–1171. doi: 10.1038/nsmb.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacKellar AL, Greenleaf AL. Cotranscriptional association of mRNA export factor Yra1 with C-terminal domain of RNA polymerase II. J. Biol. Chem. 2011;286:36385–36395. doi: 10.1074/jbc.M111.268144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu K, Guo Y, Liu H, Bian C, Lam R, Liu Y, Mackenzie F, Rojas LA, Reinberg D, Bedford MT, et al. Crystal structure of TDRD3 and methyl-arginine binding characterization of TDRD3, SMN and SPF30. PLoS One. 2012;7:e30375. doi: 10.1371/journal.pone.0030375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tripsianes K, Madl T, Machyna M, Fessas D, Englbrecht C, Fischer U, Neugebauer KM, Sattler M. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat. Struct. Mol. Biol. 2011;18:1414–1420. doi: 10.1038/nsmb.2185. [DOI] [PubMed] [Google Scholar]
- 27.Liu K, Chen C, Guo Y, Lam R, Bian C, Xu C, Zhao DY, Jin J, MacKenzie F, Pawson T, et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain. Proc. Natl Acad. Sci. USA. 2010;107:18398–18403. doi: 10.1073/pnas.1013106107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu H, Wang JY, Huang Y, Li Z, Gong W, Lehmann R, Xu RM. Structural basis for methylarginine-dependent recognition of Aubergine by Tudor. Genes Dev. 2010;24:1876–1881. doi: 10.1101/gad.1956010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cote J, Richard S. Tudor domains bind symmetrical dimethylated arginines. J. Biol. Chem. 2005;280:28476–28483. doi: 10.1074/jbc.M414328200. [DOI] [PubMed] [Google Scholar]
- 30.Chen C, Jin J, James DA, Adams-Cioaba MA, Park JG, Guo Y, Tenaglia E, Xu C, Gish G, Min J, et al. Mouse Piwi interactome identifies binding mechanism of Tdrkh Tudor domain to arginine methylated Miwi. Proc. Natl Acad. Sci. USA. 2009;106:20336–20341. doi: 10.1073/pnas.0911640106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 32.Bax A, Grzesiek S. Methodological advances in protein NMR. Accounts Chem. Res. 1993;26:131–138. [Google Scholar]
- 33.Peterson RD, Theimer CA, Wu H, Feigon J. New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA-protein complexes. J. Biomol. NMR. 2004;28:59–67. doi: 10.1023/B:JNMR.0000012861.95939.05. [DOI] [PubMed] [Google Scholar]
- 34.Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage lambda N-peptide/boxB RNA complex. J. Am. Chem. Soc. 1997;119:6711–6721. [Google Scholar]
- 35.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 36.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 37.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, et al. AMBER 10. San Francisco: University of California; 2008. [Google Scholar]
- 38.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 40.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges - the Resp Model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 41.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 42.Vriend G. What If - a molecular modeling and drug design program. J. Mol. Graphics. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 43.Heyduk T, Lee JC. Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl Acad. Sci. USA. 1990;87:1744–1748. doi: 10.1073/pnas.87.5.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 45.Tao J, Perdew JP, Staroverov VN, Scuseria GE. Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003;91:146401. doi: 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- 46.Turney JM, Simmonett AC, Parrish RM, Hohenstein EG, Evangelista FA, Fermann JT, Mintz BJ, Burns LA, Wilke JJ, Abrams ML, et al. Psi4: an open-source ab initio electronic structure program. WIREs: Comput. Mol. Sci. 2012;2:556–565. [Google Scholar]
- 47.Luisi B, Orozco M, Sponer J, Luque FJ, Shakked Z. On the potential role of the amino nitrogen atom as a hydrogen bond acceptor in macromolecules. J. Mol. Biol. 1998;279:1123–1136. doi: 10.1006/jmbi.1998.1833. [DOI] [PubMed] [Google Scholar]
- 48.Jeziorski B, Moszynski R, Szalewicz K. Perturbation-theory approach to intermolecular potential-energy surfaces of Van-Der-Waals complexes. Chem. Rev. 1994;94: 1887–1930. [Google Scholar]
- 49.Cubero E, Luque FJ, Orozco M. Is polarization important in cation-pi interactions? Proc. Natl Acad. Sci. USA. 1998;95:5976–5980. doi: 10.1073/pnas.95.11.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D, Hu S, Tarakeshwar P, Kim KS, Lisy JM. Cation-pi interactions: A theoretical investigation of the interaction of metallic and organic cations with alkenes, arenes, and heteroarenes. J. Phys. Chem. A. 2003;107:1228–1238. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.