

Abstract
DNA-binding and modifying proteins show high specificity but also exhibit a certain level of promiscuity. Such latent promiscuous activities comprise the starting points for new protein functions, but this hypothesis presents a paradox: a new activity can only evolve if it already exists. How then, do novel activities evolve? DNA methyltransferases, for example, are highly divergent in their target sites, but how transitions toward novel sites occur remains unknown. We performed laboratory evolution of the DNA methyltransferase M.HaeIII. We found that new target sites emerged primarily through expansion of the original site, GGCC, and the subsequent shrinkage of evolved expanded sites. Variants evolved for sites that are promiscuously methylated by M.HaeIII [GG(A/T)CC and GGCGCC] carried mutations in ‘gate-keeper’ residues. They could thereby methylate novel target sites such as GCGC and GGATCC that were neither selected for nor present in M.HaeIII. These ‘generalist’ intermediates were further evolved to obtain variants with novel target specificities. Our results demonstrate the ease by which new DNA-binding and modifying specificities evolve and the mechanism by which they occur at both the protein and DNA levels.
INTRODUCTION
Much is known about the manner in which proteins interact with DNA to accomplish a wide variety of cellular functions in a highly specific manner. Our knowledge, however, of how these functions emerged in the course of evolution is limited. Several lines of evidence indicate that latent, weak, promiscuous functions comprise ample starting points for the evolution of new protein functions (1–3). Such activities can be dramatically improved via few mutations, and they ultimately become the main function (4). Indeed, the primary and promiscuous activities of evolutionary related families often overlap, such that the primary activity of one family is present as promiscuous, side activity in related families (3,5). Promiscuity fails to explain, however, how activities that do not pre-exist in a given proteome evolve. The acquisition of novel activities may proceed through ‘generalists’ intermediates that exhibit exceptionally wide ranges of promiscuous activities (6). It remains unknown, however, how common are such intermediates; under what circumstances and how do they emerge; and whether they can lead to novel specificities.
Here, we examined the evolution of M.HaeIII, a DNA methyltransferase that specifically targets GGCC sites, toward sites that are not recognized to any measurable degree by the wild-type enzyme. M.HaeIII belongs to the prokaryotic restriction–methylation system that includes hundreds of different enzyme families, each with different target DNA specificity. Their catalytic domains mediate the methyl transfer from the co-factor S-adenosylmethionine (SAM), and are relatively conserved (7,8). However, their target recognition domains (TRDs) diverged such that the evolutionary trajectories that led to the different methylation specificities we see today, cannot be inferred. For example, the TRDs of methyltransferases that target GC-contacting sites such as GGCC (M.HaeIII) and GCGC (M.HhaI), although showing structural resemblance (9), show only 26% sequence identity with more than 26 positional gaps due to insertions and/or deletions (Supplementary Figure S1). Further, as shown below, M.HaeIII shows no promiscuous methylation of GCGC sites, nor does M.HhaI with GGCC sites. The sequences and functions of these enzymes are therefore highly diverged, and the evolutionary trajectories that may connect these enzymes or, in fact, other DNA methyltransferase families, remain unknown.
Modifying the target specificities of DNA methyltransferases in the laboratory has also proven challenging. Enzymes with relaxed specificities were obtained by laboratory evolution (10). Increases in promiscuous ‘star’ activities (methylation of sites that differ from the original target site by one base) were also reported, but these resulted in enzymes that methylate non-palindromic sites (11–13). Here, we pursued a complete switch in target specificity: M.HaeIII was evolved to variants that methylate novel palindromic specificities such as GCGC and barely recognize M.HaeIII’s original target site. These non-overlapping specificities became accessible via ‘generalist’ intermediates that emerged under selection for M.HaeIII’s promiscuous methylation activities.
MATERIALS AND METHODS
Detailed experimental protocols are provided as Supplementary Data, available online.
Plasmids and strains
The M.HaeIII open reading frame (Supplementary Figure S3C) was cloned with a modified N-terminal His-tagged into pASK-IBA3+vector (IBA, Ampicillin resistance). The plasmid was modified to introduce the different methylation/restriction sites used for the selection of the different specificities (Supplementary Figure S3). Plasmids were transformed into Escherichia coli strain ER2267 (EcoK r- m- McrA- McrBC-Mrr-), or MC1061 [mcrA0 relA1mcrB1 hsdR2(r-m+), plus pGro7, Takara, chloramphenicol resistance], in which DNA methylation is not toxic (14). Transformants were selected by growth in the presence of ampicillin or both ampicillin and chloramphenicol accordingly.
Stabilization by consensus mutations
Orthologous sequences to M.HaeIII wild-type sequence were collected using Basic Local Alignment Search Tool for Proteins (BLASTP) within the rebase database (15). About 55 non-redundant family members (identity <95%), homologs to M.HaeIII were aligned using MUltiple Sequence Comparison by Log-Expectation (MUSCLE) (16). (Supplementary Figure S2). Eight positions in which M.HaeIII deviates from the most probable amino acid in a given position were identified and exchanges into the consensus amino acids were individually examined. Beneficial mutations were identified by higher methyltransferase activity in crude lysates (assayed with a DIG-biotin DNA substrate) (17) and higher soluble enzyme fraction indicated by SDS–PAGE. Four mutations with the largest stabilizing effects were introduced into wild-type M.HaeIII (C26A, I104K, M115L and F181L) to give the starting point for directed evolution (Supplementary Figure S2).
Library construction and selection
Random mutagenesis was performed by PCR using an error-prone polymerase (GeneMorph Mutazyme, Stratagene) and as template the stabilized M.HaeIII gene or the pool of M.HaeIII variants from the previous round and primers that flank the methylase’s open reading frame. The protocol was optimized to an average at 2.2 ± 1.6 mutations per gene. The mutated M.HaeIII genes were recloned into the modified pASK plasmid and transformed into E. coli MC1061 or ER2267. A strategy developed for cloning DNA methyltransferases (18) was adopted for selection (Figure 2A). Each round of evolution, or generation (noted as ‘G’), included three cycles of enrichment (transformation, growth, plasmid extraction and digestion with suitable restriction enzyme for the desired target site, see Supplementary Methods and Figure 2A for details). In each round, the methyltransferase expression levels were gradually reduced, starting from over-expression (0.2 µg/ml anhydrotetracycline inducer, with GroEL/ES over-expression, 0.05% arabinose) down to basal expression (no anhydrotetracycline). At the end of each round, 8–12 randomly chosen clones were isolated, sequenced and their in vivo activities were determined.
Figure 2.
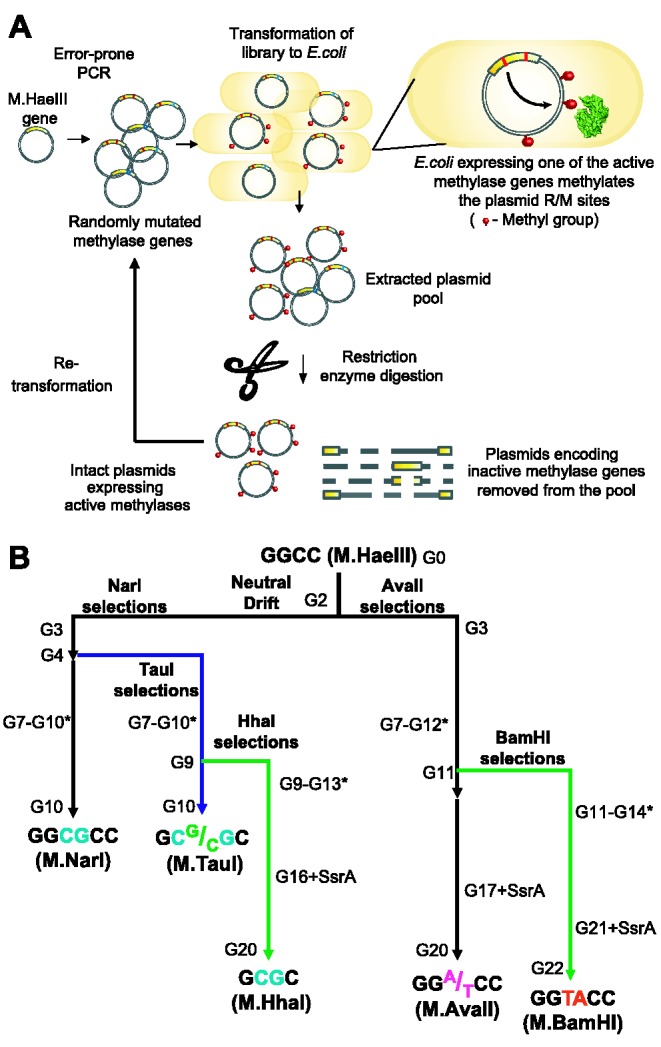
The selection methodology and trajectories for new and novel target specificities. (A) Selection by plasmid protection. M.HaeIII’s open reading frame was randomly mutated by error-prone PCR. The library is cloned and transformed to E. coli. Within each transformed cell, the expressed methyltransferase variant, if active, methylated its encoding plasmid and thereby protected it from digestion with the cognate restriction enzyme (18). Following digestion (e.g. with AvaII, for the GGA/TCC specificity), the surviving plasmids were retransformed, and subjected again to restriction for further enrichment of active methylase variants. After two cycles of enrichment (digestion and transformation), the plasmid DNA was extracted and the surviving M.HaeIII genes were amplified and randomly mutagenized (as a pool) for the next round. (B) Starting from DNA methyltransferase M.HaeIII, different trajectories for new (black lines) or novel target sites (blue and green lines) were followed. Noted as ‘G’ (or generation numbers) are the rounds of mutation and selection underlining these trajectories. The asterisk symbol denotes rounds in which negative selection against GGCC methylation was applied. SsrA denoted rounds in which the SsrA degradation tag was fused to the selected variants to increase the selection pressure for higher specific activity.
Negative selections
Enrichment for variants with reduced GGCC methylation activity was performed by digestion of the plasmid pool with NotI, located downstream the methyltransferase gene. The NotI site contains an HaeIII site (GCGGCCGC). Unmethylated NotI sites were digested thereby enabling ligation of a suitable linker (Supplementary Figure S4A). Variants that accommodated the linker were selectively amplified by PCR using a reverse primer that annealed to the ligated linker and a forward primer that annealed upstream to the methyltransferase gene. Negative selections were applied as specified in Figure 2B.
Methylation assays
Plasmid DNA was extracted (from library pools or individual variants), treated with the restriction enzymes (10–20 U, 2 h at 37°C) and analyzed by gel electrophoresis. The same procedure was applied with genomic DNA extracted with the Sigma kit. DNA substrates for in vitro assays were prepared by primer extension (26-nt forward templates carrying a single restriction/methylation site, 12-nt biotinylated reverse primers, exo- Klenow fragment polymerase, NEB, 1 h, 37°C). The double stranded DNA products were analyzed on 4% agarose type XI gels (Sigma). A list of all DNA substrates is available in Supplementary Methods. ‘Time-dependent methylation assays’ were performed as described previously (19), using H3-labeled SAM (∼0.3 µM) and different enzyme and DNA substrate concentrations (10–700 nM; Supplementary Figure S5B). Aliquots taken at different time points were quenched and transferred to streptavidin-coated ScintiPlate wells (PerkinElmer) and H3 levels were determined using the Wallac MicroBeta TriLux counter (PerkinElmer). KMDNA and vmax values were derived by fitting initial rates to the Michaelis–Menten model using GraphPad Prism. Error ranges relate to the standard errors observed in two or more independent experiments. ‘End-point assays’ aimed at detecting weak and promiscuous activities (Figures 1 and 3), were performed as above but at saturating, rather than initial rate conditions, namely, using high enzyme concentration (2–4 µM) and long incubation times during which favored DNA substrates were completely methylated (0.5–5 h).
Figure 1.
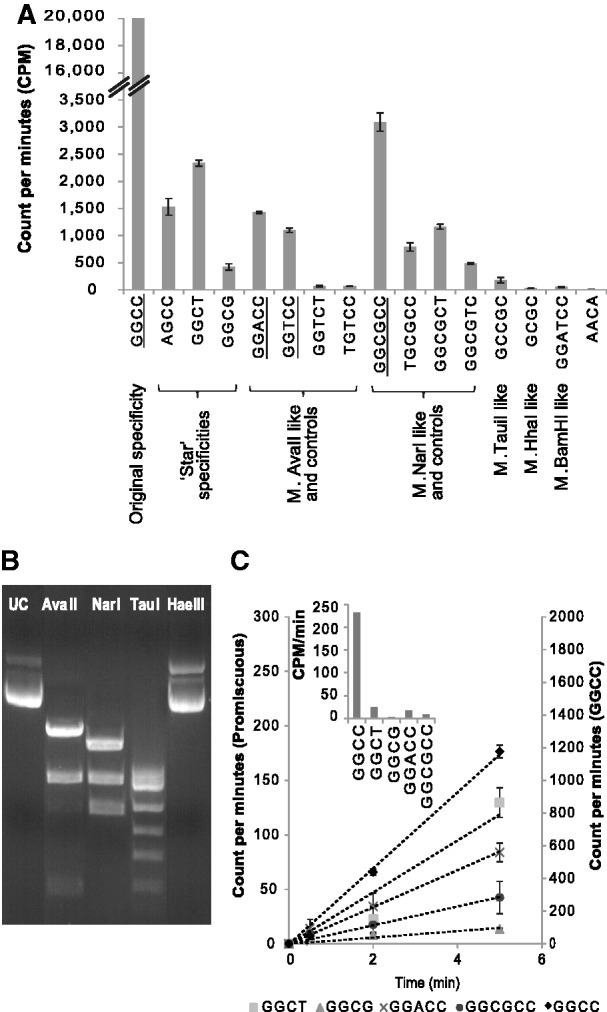
Methylation of different target sites by wild-type M.HaeIII for detection of promiscuous activities. (A) End-point methylation activity assay of wild-type M.HaeIII of the original GGCC sequence, of promiscuous non-palindromic ‘star sites’ (AGCC, GGCT and GGCG) (11), and of the newly identified expanded palindromic sites: GGA/TCC (M.AvaII-like) and GGCGCC (M.NarI-like). These palindromic sites show similar specificity as the original site as indicated by the lower methylation of related ‘star’ sites (controls sites) ([E]0 = 2 µM; [DNA substrate] = 0.67 μM; [3H−SAM] = 0.2 μM; 20% glycerol; 5-h incubation time at 37°C). (B) Plasmid protection assay: The encoding plasmid of wild-type M.HaeIII was transformed to E. coli and was expressed without induction. The plasmid was subsequently extracted and treated with different restriction enzymes as noted. (C) Rates of H3-methyl incorporation were measured with the original DNA target (GGCC, right Y-axis), and with different promiscuous target sites (left Y-axis). Insert: the derived initial velocities with the different target sequences. ([E]0 = 0.2 µM; [DNA substrate] = 0.5 μM; [3H−SAM] = 0.2 μM; 20% glycerol at 37°C). UC, uncut plasmid.
Figure 3.

Methylation activities of the evolved variants from the 10th round (G10). Shown are the activities toward the new, evolved sites versus the original site, GGCC. The mutations in these variants are listed in Supplementary Table S2. (A) Plasmid protection was assayed under the same conditions as Figure 1B. All variants show a significant increase in protection of the evolved sites (full protection, in most variants) and partial, or even no protection against HaeIII digestion. Wild-type (WT) M.HaeIII shows only protection against HaeIII digestion. Arrows indicate the in vitro characterized variants. (B) In vitro methylation activity of purified Round 10 variants: N3 = NarI-selected variant, T2 = TauI-selected variant, A4 = AvaII-selected variant. ([E]0 = 2 µM; [DNA substrate] = 0.67 μM; [3H-SAM] = 0.25 μM; at 37°C). Aliquots of the reaction mixture were quenched at different times, and the level of methyl incorporation was determined. (C) End-point activity assay for the same variants. Noted in dark gray are the novel activities that were not selected for, and were undetectable in wild-type (A). C* represents a methylated base in hemimethylated substrates. ([E]0 = 4 µM for N3 and A4, 2 µM for T2; [DNA substrate] = 0.67 μM; [3H-SAM] = 0.2 μM; 5 h incubation time at 37°C).
Enzyme purification
Plasmids were transformed to MC1061::pGro7 cells. GroEL/ES and methyltransferase over-expression was induced with arabinose and anhydrotetracycline inducer after which the cell pellets were disrupted by addition of lysozyme and sonication. The methyltransferase variants were purified by Ni-NTA chromatography (Nickel-nitriloacetic acid column, QIAGEN) with the addition of 1 mM adenosine triphosphate (ATP) to dissociate the chaperons, concentrated and stored at −80°C with 10% glycerol.
RESULTS
Promiscuous targets of M.HaeIII
To identify its promiscuous activities, M.HaeIII was reacted with an array of different DNA target sites using a radiolabeled methyl donor 3H-SAM (19). We tested known ‘star’ sites of M.HaeIII (11), and palindromic sites based on the proposed mechanism of expansion or shrinkage of existing target sites (20). Two such sequences were clearly methylated: GGCGCC and GG(A/T)CC (Figure 1). These palindromic sequences contain the original GGCC sequence with an internal insertion (A/T or CG). The ‘star’ sequences of these targets (e.g. GGTCT versus GGTCC; Figure 1) showed much lower methylation, supporting the notion of recognition of an extended site (20). The newly identified extended sites comprised our first targets for directed evolution.
Increasing M.HaeIII’s evolvability
DNA methyltransferases, including M.HaeIII, have proven somewhat resistant to laboratory evolution despite the application of ultra-high-throughput selections (12,21). These difficulties may relate to the low stability of these enzymes. Indeed, when over-expressed in E. coli, M.HaeIII, an enzyme isolated from Haemophilus aegyptius, tends to aggregate. Low stability dramatically reduces the fraction of properly folded and functional mutants, and thereby limits the ability to acquire new functions (22). We therefore, applied new approaches aimed at circumventing the setbacks caused by the destabilizing effect of mutations, and by the stability-activity trade-offs that underline the evolution of new protein functions (22–24).
One approach to increase M.HaeIII’s stability and evolvability was by introducing ancestor/consensus mutations and, thereby increase its ability to accept a wider range of mutations (23). Accordingly, we identified a combination of four consensus mutations (C26A, I104K, M115L and F181L) that led to >6-fold increase in the concentration of soluble active enzyme (Supplementary Figure S2). Second, the libraries of mutated M.HaeIII genes were co-expressed with chaperones GroEL/ES to buffer the effects of destabilizing mutations (24,25) (Supplementary Figure S2D).
Third, M.HaeIII was neutrally drifted to accumulate a wide variety of mutations while maintaining its original GGCC specificity (26) (Figure 2A). To this end, M.HaeIII’s open reading frame was randomly mutated by error-prone PCR at an average of 2.2 ± 1.6 mutations per gene. The resulting library was cloned into an expression vector and transformed to E. coli. Within each transformed cell, the expressed methyltransferase variant, if active, methylated its encoding plasmid and thereby protected it from digestion with the cognate restriction enzyme (18) (Figure 2). The surviving plasmids were retransformed, and subjected again to HaeIII restriction for further enrichment of active methylase variants. After two cycles of enrichment (digestion and transformation), the plasmid DNA was extracted, and the surviving M.HaeIII genes were amplified and randomly mutagenized (as a pool) for the next round. Two such rounds of mutagenesis and selection by HaeIII digestion were performed to give an ensemble of polymorphic mutants that were all folded and functional with an average of 2.2 ± 1.5 mutations per gene. This neutrally drifted ensemble was used as the starting point for the selection of M.HaeIII variants that efficiently methylate target sites other than GGCC.
Divergence toward M.HaeIII’s promiscuous activities
Selections to amplify the methylation of promiscuous target sites were performed as the neutral drift. However, instead of digesting with the cognate HaeIII, the plasmid pools of the various libraries were digested with restriction enzymes that recognize the target sites, such that only variants which methylated the new target sites survived [e.g. AvaII for the GG(A/T)CC sites; Figure 2B and Supplementary Figure S3]. To control the selection pressure, the methyltransferase gene libraries were cloned under the tet promoter (anhydrotetracycline induced expression). The selection plasmids also carried the desired methylation–restriction target sites. The GroEL/ES, the E. coli chaperonin, was over-expressed from a second plasmid (24) (Takara). Once the evolved libraries retained a satisfactory level of methylation activity (105 or more surviving clones), chaperonin co-expression was removed, and the methyltransferase expression level was reduced to the basal level (no inducer). The selection pressure was also augmented by recloning to plasmids that carried a larger number of target sites (Supplementary Figure S3).
Following this procedure, the neutrally drifted pool (denoted as G2) of M.HaeIII was initially evolved toward three different specificities (Figure 2B): the newly identified promiscuous sites, GGCGCC and GG(A/T)CC, comprised our first targets for evolution (digestion with NarI and AvaII, respectively). As we were primarily interested in trajectories that may have actually occurred in nature, we chose the GC(G/C)GC as the third target (digestion with TauI). Phylogenetically, methyltransferases with GC(G/C)GC specificity seem to be the closest paraologs of M.HaeIII, although the sequences of the TRDs of these two families are quite diverged (37% identity, more than 40 positional gaps; Supplementary Figure S1). M.HaeIII exhibited some methylation of GC(G/C)GC sites, although at barely detectable level (<10-fold, lower than with other promiscuous sites; Figure 1A).
As opposed to the first two target sites, no survivals were observed after TauI digestion, most likely due to the very weak promiscuous activity toward this site. However, after selection with NarI, we could obtain TauI-protected plasmids. Thus, the NarI G3 library was further mutated, split and selected with either NarI or TauI. Four additional rounds of random mutagenesis and selection were applied for these three different specificities (G4–G7; Figure 2B). By the seventh round, the pools of variants from all three trajectories showed marked increases in methylation of the target sites they were selected for, and some reduction in methylation of M.HaeIII’s original target, GGCC.
To increase the selectivity of the evolving variants, we co-selected for the survival of unmethylated GGCC plasmids (Supplementary Figure S4). Negative selection (denoted as star in Figure 2B) was applied starting from Round 7. However, we found that its main effect had been a parallel decrease in methylation of both the new and the original sites (as observed in plasmid protection assays in Supplementary Figure S4B). Thus, additional rounds of selection for the new target sites (positive selections) were necessary.
The emergence of novel activities
By the end of the 10th round (G10), several randomly picked variants from each trajectory were sequenced and their methylation activities were assayed (Figure 3 and Supplementary Table S1). Most tested variants showed complete protection from digestion by the restriction enzymes they were selected with, and decrease in the original GGCC methylation activity. Representative variants from each trajectory were also purified and characterized in vitro (Figure 3B). The evolved variants showed a significant increase in the rate of methylation of the evolved target sites but they were unstable and lost most of their activity during purification. The TauI-selected variant T2 showed the most dramatic improvement as methylation of GC(G/C)GC by wild-type M.HaeIII that could be barely detected (Figure 3B).
The G10 variants can be considered as bi-functional evolutionary intermediates—they methylated both the newly evolved target sites and the original one with only a mild preference toward the former (Figure 3B). However, a wider screen indicated that the G10 variants also methylated target sites that were neither selected for, nor methylated by wild-type M.HaeIII (Figure 3C). Thus, these variants behaved as ‘generalists’ (6).
Several novel target sites were detected (Figure 3C). Most notably, the TauI-selected variant [GC(G/C)GC] could also methylate GCGC, thus exhibiting HhaI-like methylation activity. Likewise, the AvaII selected variant [GG(A/T)CC] methylated GGATCC (BamHI) and GGTACC (KpnI) sites. In fact, the ‘generalist’ trend was already seen in the divergence of TauI methylation [GC(G/C)GC]. This activity could barely be detected in wild-type M.HaeIII, and could not be evolved directly. However, variants selected for NarI sites (GGCGCC) readily diverged to give TauI methylation [GC(G/C)GC].
Evolution toward novel target specificities
The ‘generalist’ G10 variants were used as the starting points for divergence toward novel target sites. Additional rounds of mutagenesis and selection were applied along two new trajectories (Figure 2B): The TauI-selected pool (G9) was now selected with HhaI for GCGC methylation; and, the AvaII selected pool (G10) was selected with BamHI for GGATCC methylation. The already pursued trajectory of GG(A/T)CC methylation (AvaII) was continued in parallel. Within two rounds, the selected pools showed a marked increase in plasmid protection against digestion with the cognate restriction enzymes. However, at this stage, the dynamic range of our selection system was exhausted (i.e. the enzyme was sufficiently active to protect its encoding plasmid, even at basal expression of the methyltransferase variants and without GroEL/ES over-expression). However, when selected variants were purified, their enzymatic methylation activity was found to be relatively low.
To increase the selection pressure and obtain higher catalytic efficiencies, we had to reduce the cellular enzyme doses, and thereby enforce the evolved variants to increase their specific enzymatic activity. Therefore, we fused an 11-amino acids SsrA degradation tag to the methyltransferase’s C-terminus. This tag targets the expressed enzyme variants for rapid degradation by the ClpXP protease (27). Indeed, as opposed to wild-type M.HaeIII, which fully protects its plasmid against HaeIII digestion at basal expression, upon fusion of the SsrA tag, protection could be observed only under over-expression (Supplementary Figure S4C). The pools selected for HhaI (G16), AvaII (G16) and BamHI (G20, Figure 2B) specificities were further mutated and recloned to a modified pASK vector that carried the SsrA tag (Supplementary Figure S3). The selections were initially performed with maximal level of expression (100 ng/ml of the anhydrotetracycline inducer), and the inducer’s levels were subsequently reduced (to 5 ng/ml in the HhaI and AvaII selections, and to 30 ng/ml in the BamHI selection). Under these conditions, the libraries showed measurable survival after digestion with the cognate restriction enzymes (>10%; Supplementary Figure S4D).
The evolved novel variants
Following additional rounds of selection with the SsrA tag (G20–G22, Figure 2B), several variants were randomly picked (Supplementary Table S1). To enable their production at a large scale and purification, the SsrA tag had to be removed. However, upon recloning without the SsrA tag, the evolved variants exhibited considerable toxicity, and viable trasnformants were found to carry additional mutations. Eventually, after sequencing many transformants, the tag was successfully removed from most variants. Their methylation activity could be tested in vivo (Figure 4), and subsequently with purified enzymes. Nonetheless, an inverse correlation was observed between the methylation activity of a variant and the growth rate of E. coli cells carrying it (Supplementary Figure S4E).
Figure 4.
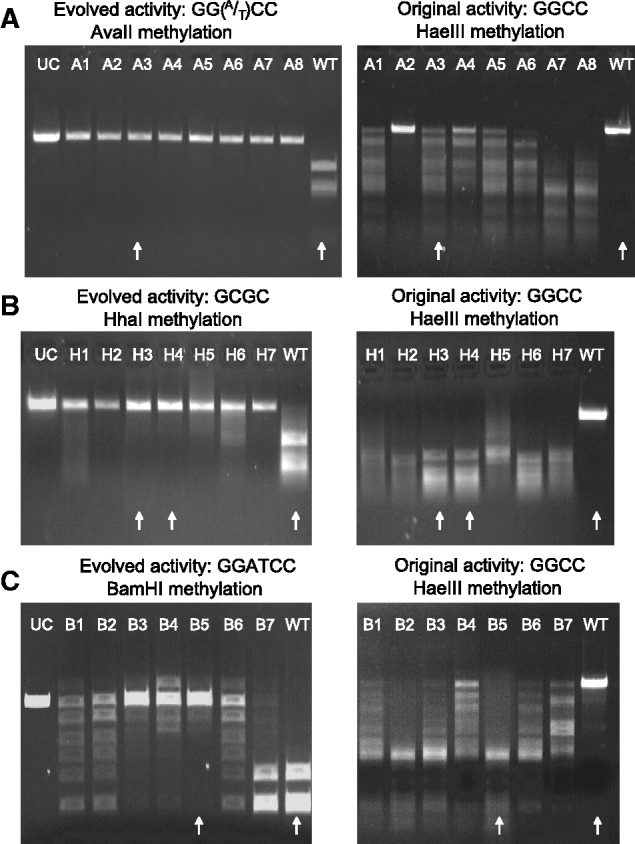
Methylation activities of variants evolved toward new and novel target specificities. Shown is a plasmid protection assayed under the same conditions as Figure 1B (digested in addition to the different restriction enzymes as noted, in parallel with NcoI digestion for plasmid linearization). Arrows indicate the in vitro characterized variants. (A) Randomly picked variants from the last round (G20) of selection for methylation of AvaII sites [GG(A/T)CC]. All eight variants confer complete protection of the AvaII sites selected for (i.e. complete methylation, left panel) and variable degrees of protection of the original sites (e.g. complete protection by A2 versus no protection with A7). UC = plasmid treated only with NcoI. M.HaeIII grown and digested under the same conditions. (B) Seven randomly picked HhaI-selected variants (GCGC) from G20. These variants completely lost the original activity as indicated by no protection against HaeIII digestion. (C) Seven randomly picked BamHI-selected variants (GGATCC) from G21 showed a marked increase in the protection of the evolved target site, as well as a reduction in protection against HaeIII. WT, wild-type.
The tested variants showed high methylation activities as detected by the plasmid protection assay of the evolved target sites (Figure 4). Unlike the intermediate variants from G10, methylation activity of the original GGCC target was marginal in some variants and undetectable in others. That the G20–G22 variants methylation rates are comparable with wild-type was also indicated by their ability to fully protect the genomes of their host E. coli cells, even at basal expression levels (more than 30 000 HhaI, GCGC sites,e.g. Supplementary Figure S5A).
Kinetic constants were determined by measuring the initial rates of methylation with purified enzymes at different concentrations of target DNA substrates (Table 1 and Supplementary Figure S5B). The variant displaying the highest catalytic efficiency (kcat/KM) was the M.AvaII-like A3 that was selected toward GG(A/T)CC sites. Its kcat/KM for the evolved GG(A/T)CC target site was 7.6 × 106 min−1M−1 (51-fold increase relative to wild-type; and comparable with the wild-type’s rate with GGCC). A3’s rate toward the original GGCC target was 5.3 × 105 (13-fold lower than wild-type). Overall, it exhibited a shift of ∼660-fold in specificity.
Table 1.
Kinetic parameters for methylation of different DNA target sites by wild-type M.HaeIII and its evolved variants.
| AvaII selected | BamHI selected | HhaI selected |
|||
|---|---|---|---|---|---|
| Wild-type | A3 | B5 | H3 | H4 | |
| Original target site GGCC | |||||
| kcat (min−1) | 0.4 (±0.02) | 0.09 (±0.01) | 0.11 (±0.02) | 0.02 (±0.0) | 0.02 (±0.0) |
| KM (nM) | 58 (±14) | 165 (±43) | 728 (±211) | 94 (±23) | 121 (±40) |
| kcat/KM (M−1·min−1) | 6.9 × 106 | 5.3 × 105 | 1.5 × 105 | 1.7 × 105 | 1.4 × 105 |
| Fold changea | 0.08 | 0.02 | 0.02 | 0.02 | |
| Evolved target site GGACC | |||||
| kcat (min−1) | 0.03 (±0.0) | 0.53 (±0.01) | |||
| KM (nM) | 189 (±32) | 70 (±6) | |||
| kcat/KM (M−1·min−1) | 1.5 × 105 | 7.6 × 106 | |||
| Fold changea | 50.7 | ||||
| Evolved target site GGTACC | |||||
| kcat (min−1) | ND | 0.14 (±0.01) | |||
| KM (nM) | ND | 193 (±38) | |||
| kcat/KM (M−1·min−1) | <30c | 7.3 × 105 | |||
| Fold change | >2.4 × 104 | ||||
| Evolved target site GGATCC | |||||
| kcat (min−1) | ND | 0.07 (±0.0) | |||
| KM (nM) | ND | 116 (±26) | |||
| kcat/KM (M−1·min−1) | <30c | 5.6 × 105 | |||
| Fold changea | >1.9 × 104 | ||||
| Evolved target site GCGC | |||||
| kcat (min−1) | ND | 0.1 (±0.0) | 0.12 (±0.01) | ||
| KM (nM) | ND | 42 (±6) | 53 (±10) | ||
| kcat/KM (M−1·min−1) | <30c | 2.4 × 106 | 2.2 × 106 | ||
| Fold changea | >8 × 104 | >7.3 × 104 | |||
| Specificity shiftb | >6.6 × 102 | >1.1 × 106 / >8.6 × 105 | >3.2 × 106 | 3.2 × 106 | |
aThe ratio of kcat/KM values of the evolved variant versus wild-type M.HaeIII. Marked in bold.
bThe overall change in ratio of kcat/KM values for the new or novel evolved target site versus the original GGCC site. Marked in bold.
cThe minimal detectable activity, given the background reads, enzyme concentrations and incubation times applied, corresponds to a kcat/KM value of ≤30 M−1·min−1.
ND, not detected.
Even larger changes in specificity were exhibited by HhaI-selected variants H3 and H4, and by the BamHI-selected variant B5. Wild-type M.HaeIII shows no methylation of these sites, GCGC and GGATCC, even at the highest enzyme concentrations (Figure 1). In these cases, >104-fold increases in methylation rates were observed because wild-type M.HaeIII exhibits no detectable methylation of these sites (given the background reads, maximal enzyme concentrations and incubation times applied, the minimal detectable activity corresponds to a kcat/KM value of <30 min−1M−1). Additionally, the HhaI selected variants H3 and H4 wild-type-like catalytic efficiency toward the evolved GCGC sequence (kcat/KM of ∼2.3 × 106 min−1M−1), and much lower methylation rate of the original GGCC site (∼45-fold decrease relative to wild-type M.HaeIII).
Overall, the activity changes observed in all the evolved variants were manifested primarily in higher kcat values, and only minor changes in KM were observed. This trend is in agreement with the tendency of DNA methyltransferases to bind cognate and non-cognate target sites with comparable affinities, but methylate them with very different kcat values (28). The one exception was the BamHI-selected variant B5. Although selected toward GGATCC methylation, the methylation rate of GGTACC was similar (7.3 × 105 and 5.6 × 105 min−1M−1, respectively). Curiously, the 46-fold reduction in its catalytic efficiency with the original GGCC site was achieved mainly through an increase in KM.
To assess the specificity of the newly evolved variants, the methylation of a wide range of DNA target sites was tested (Supplementary Figure S5C). This was done by an end-point assay, similar to the one used for detecting the promiscuous activities of M.HaeIII and the intermediate variants (Figure 1). In all cases, and despite the end-point format, a clear preference was observed toward the evolved target sites relative to the original GGCC site. Further, no cross-reactivity was detected between the HhaI and the AvaII/BamHI trajectories—i.e. the HhaI-selected variants showed no methylation of GGA/TCC or GGATCC sites, and vice-versa (also tested in vivo, Supplementary Figure S5D). However, in the same manner that the selection for promiscuous sites opened the door for novel methylation sites that were not selected for, the newly evolved variants also recognized other target sequences. In conjunction with the trend we observed with wild-type and the intermediate variants, promiscuous methylation of extended palindromic versions of the target sites selected for could be observed [e.g. GC(A/T)GC and GC(AT/TA)GC in HhaI-selected variants]. However, the methylation of non-palindromic ‘star’ sites was mostly apparent. In fact, the HhaI-selected variants (GCGC) were found to methylate GCGN or NCGC sites, where N corresponds to any base. Nonetheless, sites in which any one of the two inner bases was modified (GTGC, GCTC), or both external bases were modified (e.g. TCGT), showed essentially no methylation. It was clear, therefore, that the methylation selectivity of these variants was primarily obtained through recognition of the three first bases, either on the ‘plus’ or on the ‘minus’ strand.
DISCUSSION
Despite natural methyltransferases being highly divergent, and the ease of selection (active DNA methyltransferases protect their own encoding genes from restriction), the laboratory divergence of new methylation target sites has thus far proven challenging. Three major elements made it possible. The first one is boosting evolvability by the incorporation of stabilizing, compensatory mutations (23), by chaperonin buffering (24) and by neutral drift (26). Their roles in enabling the trajectories described here support the notion that the destabilizing effects of mutations comprise a major limiting factor in protein evolution (22,23,29–32).
The second element regards the expansion-shrinkage mode by which new methylation target sites emerge. Earlier attempts made use of ‘star’ activities and only yielded mild changes in specificity (10), or methylation of non-palindromic sites (12,13). An alternative to the ‘star’ mode is the expanded site mode. As indicated (20), M.EcoRV, a GATATC methyltransferase, recognizes its substrate in a similar manner as EcoDam (GATC) whereby, DNA bending enables the accommodation of the expanded site. This proposed evolutionary mode (20) is strongly supported by our results: we found that wild-type M.HaeIII promiscuously methylates expanded sites such as GG(A/T)CC, and could be readily evolved to methylate them with high efficiency and specificity. Via divergence to further expanded target sites, non-overlapping specificities such as GGATCC emerged. Finally, by shrinkage of the evolved GC(G/C)GC target site, variants that methylate the novel target site GCGC (M.HhaI-like) emerged. Indeed, the natural methyltransferases for our evolving target specificities (like M.HhaI for GCGC) are only remotely related to the starting point M.HaeIII, and the sequence and structural homology is largely restricted to their catalytic domains. However, as demonstrated here, recognition of an expanded, or a shrunken target site, might have been the mechanism for changing DNA recognition specificity in ancestral methyltransferases. Further selection for specificity and drift, blurred nearly all remnants of common ancestry.
The third divergence-driving element is ‘generalist’ intermediates. These comprised the missing link between the non-overlapping specificities of GGCC (M.HaeIII) and GCGC (M.HhaI). Upon selection to improve latent, promiscuous activities, novel activities that were neither present in the starting point, nor selected for, emerged. What might be the molecular basis for emergence of generalists? The early mutations occurred in residues that directly contact the GGCC target site (Figure 5). For instance, in wild-type M.HaeIII, Arg225 interacts with the first guanine in M.HaeIII’s target site [GGCC, (9)], and mutations into Ser or Gly were the first to be fixed in the NarI trajectory (methylation of GGCGCC, and thereby might facilitated recognition of NarI partial sequence CGCC). In a similar manner, the first mutations in the AvaII trajectory [methylation of GG(A/T)CC] occurred in Ser224 [to Gly and in Arg227 (to His) that interact with the internal bases of GGCC and facilitate the ability to methylate the expanded target site (9)]. Thus, as indicated by the broad activity patterns (Figure 1), selection for methylation of new sites led to mutations in ‘gate-keeper’ residues that loosened contacts with the original target site, rather than create new contacts with the evolving target sites.
Figure 5.

The location of mutations that underlined the laboratory divergence of new and novel target specificities. Ribbon diagrams are based on the wild-type M.HaeIII’s crystal structure (Protein data bank (PDB) code: 1dct). The cognate DNA substrate (GGCC) is denoted in lines, with the corresponding bases numbered (G1, G2, C3, C4). M.HaeIII residues in which mutations occurred are denoted as sticks. (A) First shell mutations fixed along the selections for AvaII methylation and the subsequent BamHI (GGTACC) selection. (B) Like-wise for the NarI and TauI selections and, the subsequent HhaI trajectory. In all these trajectories, the mutations fixed in the first rounds were in residues directly contacting the exchanged DNA bases of the GGCC methylation site. (C) All type of positions at which mutations were fixed throughout the trajectory leading to the M.HhaI-like methyltransferase (shown are positions with mutation rates >25%). In red, first shell mutations in direct contact with the DNA target site. In green, remote mutations in regions previously implicated with conformational changes that relate to DNA binding and methylation (based on structural alignment with M.HhaI) (33,34). In blue, stabilizing mutations, including the initially introduced consensus mutations and stabilizing mutations that accumulated later in the trajectory. The mutations observed in the AvaII and BamHI selection trajectories are illustrated in Supplementary Figure S5F.
The relaxation in specificity enabled generalist intermediates to methylate not only the original and the newly evolved target sites but also other sites not selected for. Thus, a complete release of the burden of selection and transient non-functionalization, were not found to be a prerequisite for the acquisition of a new specificity, at least not in our experiment. Which mutations, then, mediated the switch in favor of the novel target sites and the loss of the original GGCC preference? (Figure 5) One example might be Arg243Gln that appeared at the early stages of the HhaI selection (G10). This mutation resulted in significantly enhanced methylation of the evolved GCGC site and a decline in methylation of the original GGCC site (Supplementary Figure S5E).
Most mutations occurred, however, in residues not directly interacting with the target DNA (Figure 5C). Several of the remote mutations locate to regions that undergo large conformational changes upon DNA binding (e.g. Leu80Hys/Cys in the HhaI and in the BamHI selections, respectively) (33,34). These mutations may have shifted the conformational equilibrium in favor of the conformations that target the new sites (35–37). Other remote mutations act as stability compensators, or global suppressors that compensate for the loss of stability associated with the mutations that mediate new activities (31). A clear example is Asn262Tyr that appeared in all the evolved variants regardless of their specificity and also comprises the consensus in the M.HaeIII family (Supplementary Table S2). Additionally, some of the consensus mutations that failed to stabilize wild-type M.HaeIII (Supplementary Figure S2A) appeared in evolved variants with new specificities (e.g. Gly77Ala and Val283Leu in the BamHI and HhaI evolved variants, respectively; Supplementary Table S1). This indicates the particular utility of consensus mutations (and ancestral ones) in promoting the acquisition of new functions (23).
Although our evolved variants methylate their target sites with catalytic efficiencies that match those of natural methyltransferases, they do not match the latter’s specificity. The HhaI selected variants, H3 and H4, exhibited kcat/KM values of ∼2.3 × 106 min−1M−1. However, they only show ∼10-fold preference toward the evolved GCGC target versus the original GGCC target (in ratio of kcat/KM values; Table 1 and Supplementary Figure S3B). The ‘star’ activities of these variants are also very high (Supplementary Figure S5C). The methylation of sites that relate neither to the original target sequence (GGCC) nor to any of the evolved ones, and were not tested in our assays, is not likely to occur (Supplementary Figure S5D), but cannot be completely ruled out at this stage.
Specificity is a hallmark of the restriction–methylation system and the key to its biological function in protection against foreign DNA in general, and particularly in maintaining cross-species barriers (38). There are, however, few examples of natural methyltransferases with similar relaxed specificity (39). For example, the CviJI methyltransferase is less specific toward the last base of its target sequence RGC(Y/G), than the endonuclease (RGCY) (40). Relaxed specificity could be typical to evolutionary intermediates whereby the methylase diverges beyond the coverage of its cognate restriction enzyme. However, contrary to the current dogma of absolute specificity, broad specificity of the restriction enzyme may provide more efficient protection, as recently shown for phage defense by KpnI nuclease (41). The specificity of action of the restriction–methylation system has implications beyond foreign DNA. Broad methylation specificity might also be deleterious, possibly due to methylated cytosines being prone to deamination (42,43). Indeed, our most active variants became toxic once the SsrA tag that suppressed expression was removed (Supplementary Figure S4E). It may also be that, by itself, selection toward a new target site does not induce absolute specificity, and negative selection against alternative sites might be necessary. However, the selection we applied against the original GGCC specificity resulted in reduced rates with both the newly evolved and the original sites (Supplementary Figure S4B). Indeed, trade-offs between fidelity and processivity are observed throughout, be it ribosomes (44) or DNA methyltransferases (previously reported M.HaeIII variant with broadened ‘star’ activity exhibits >10-fold higher kcat/KM values with the original GGCC site) (12).
To conclude, our results suggest that ‘generalist’ intermediates—i.e. enzyme variants that exhibit activities toward novel target sites that were neither selected for, nor present in the starting point, comprise a missing link that bridges remotely related methyltransferases with non-overlapping specificities such as GGCC and GCGC (certainly with respect to their TRDs). Generalists may also comprise the progenitors of other highly diverged enzyme families (1,3,6). The acquisition and loss of promiscuous activities is therefore a dynamic process. Drift, i.e. accumulation of neutral mutations with respect to the native activity, can reduce or enhance existing promiscuous activities (45,46). However, as shown here, mutations that drive the acquisition of new functions also give rise to other activities, primarily by alleviating specificity constraints. The trajectories leading from one activity to another therefore open new side roads that may in turn lead to other new activities.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Tables 1 and 2, Supplementary Figures 1–5, Supplementary Methods and Supplementary Reference [47].
FUNDING
Funding for open access charge: Israel Science Foundation.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
D.S.T. and L.R.-S. designed the experiments, analyzed data and wrote the manuscript. L.R-S. performed the experiments. D.S.T. is the Nella and Leon Benoziyo Professor of Biochemistry.
REFERENCES
- 1.Jensen RA. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 2.O'Brien PJ, Herschlag D. Catalytic promiscuity and the evolution of new enzymatic activities. Chem. Biol. 1999;6:R91–R105. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 3.Khersonsky O, Tawfik DS. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 4.Aharoni A, Gaidukov L, Khersonsky O, McQ Gould S, Roodveldt C, Tawfik DS. The ‘evolvability’ of promiscuous protein functions. Nat. Genet. 2005;37:73–76. doi: 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- 5.Glasner ME, Gerlt JA, Babbitt PC. Evolution of enzyme superfamilies. Curr. Opin. Chem. Biol. 2006;10:492–497. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura I, Ellington AD. In vitro evolution of beta-glucuronidase into a beta-galactosidase proceeds through non-specific intermediates. J. Mol. Biol. 2001;305:331–339. doi: 10.1006/jmbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 7.Posfai J, Bhagwat AS, Posfai G, Roberts RJ. Predictive motifs derived from cytosine methyltransferases. Nucleic Acids Res. 1989;17:2421–2435. doi: 10.1093/nar/17.7.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyl-transferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995;253:618–632. doi: 10.1006/jmbi.1995.0577. [DOI] [PubMed] [Google Scholar]
- 9.Reinisch KM, Chen L, Verdine GL, Lipscomb WN. The crystal structure of HaeIII methyltransferase convalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Cell. 1995;82:143–153. doi: 10.1016/0092-8674(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 10.Timar E, Groma G, Kiss A, Venetianer P. Changing the recognition specificity of a DNA-methyltransferase by in vitro evolution. Nucleic Acids Res. 2004;32:3898–3903. doi: 10.1093/nar/gkh724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen HM, Tawfik DS, Griffiths AD. Promiscuous methylation of non-canonical DNA sites by HaeIII methyltransferase. Nucleic Acids Res. 2002;30:3880–3885. doi: 10.1093/nar/gkf507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen HM, Tawfik DS, Griffiths AD. Altering the sequence specificity of HaeIII methyltransferase by directed evolution using in vitro compartmentalization. Protein Eng. Des. Sel. 2004;17:3–11. doi: 10.1093/protein/gzh001. [DOI] [PubMed] [Google Scholar]
- 13.Chahar S, Elsawy H, Ragozin S, Jeltsch A. Changing the DNA recognition specificity of the ecoDam DNA-(adenine-N6)-methyltransferase by directed evolution. J. Mol. Biol. 2009;395:79–88. doi: 10.1016/j.jmb.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 14.Raleigh EA, Wilson G. Escherichia coli K-12 restricts DNA containing 5-methylcytosine. Proc. Natl Acad. Sci. USA. 1986;83:9070–9074. doi: 10.1073/pnas.83.23.9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 18.Szomolanyi E, Kiss A, Venetianer P. Cloning the modification methylase gene of Bacillus sphaericus R in Escherichia coli. Gene. 1980;10:219–225. doi: 10.1016/0378-1119(80)90051-7. [DOI] [PubMed] [Google Scholar]
- 19.Roth M, Jeltsch A. Biotin-avidin microplate assay for the quantitative analysis of enzymatic methylation of DNA by DNA methyltransferases. Biol. Chem. 2000;381:269–272. doi: 10.1515/BC.2000.035. [DOI] [PubMed] [Google Scholar]
- 20.Jurkowski TP, Anspach N, Kulishova L, Nellen W, Jeltsch A. The M.EcoRV DNA-(adenine N6)-methyltransferase uses DNA bending for recognition of an expanded EcoDam recognition site. J. Biol. Chem. 2007;282:36942–36952. doi: 10.1074/jbc.M706933200. [DOI] [PubMed] [Google Scholar]
- 21.Lee YF, Tawfik DS, Griffiths AD. Investigating the target recognition of DNA cytosine-5 methyltransferase HhaI by library selection using in vitro compartmentalisation. Nucleic Acids Res. 2002;30:4937–4944. doi: 10.1093/nar/gkf617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc. Natl Acad. Sci. USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bershtein S, Goldin K, Tawfik DS. Intense neutral drifts yield robust and evolvable consensus proteins. J. Mol. Biol. 2008;379:1029–1044. doi: 10.1016/j.jmb.2008.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Tokuriki N, Tawfik DS. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009;459:668–673. doi: 10.1038/nature08009. [DOI] [PubMed] [Google Scholar]
- 25.Fares MA, Ruiz-Gonzalez MX, Moya A, Elena SF, Barrio E. Endosymbiotic bacteria: groEL buffers against deleterious mutations. Nature. 2002;417:398. doi: 10.1038/417398a. [DOI] [PubMed] [Google Scholar]
- 26.Gupta RD, Tawfik DS. Directed enzyme evolution via small and effective neutral drift libraries. Nat. Methods. 2008;5:939–942. doi: 10.1038/nmeth.1262. [DOI] [PubMed] [Google Scholar]
- 27.Neuenschwander M, Butz M, Heintz C, Kast P, Hilvert D. A simple selection strategy for evolving highly efficient enzymes. Nat. Biotechnol. 2007;25:1145–1147. doi: 10.1038/nbt1341. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, MacMillan AM, Verdine GL. Mutational separation of DNA binding from catalysis in a DNA cytosine methyltransferase. J. Am. Chem. Soc. 1993;115:5318–5319. [Google Scholar]
- 29.DePristo MA, Weinreich DM, Hartl DL. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat. Rev. Genet. 2005;6:678–687. doi: 10.1038/nrg1672. [DOI] [PubMed] [Google Scholar]
- 30.Soskine M, Tawfik DS. Mutational effects and the evolution of new protein functions. Nat. Rev. Genet. 2010;11:572–582. doi: 10.1038/nrg2808. [DOI] [PubMed] [Google Scholar]
- 31.Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Zeldovich KB, Chen P, Shakhnovich EI. Protein stability imposes limits on organism complexity and speed of molecular evolution. Proc. Natl Acad. Sci. USA. 2007;104:16152–16157. doi: 10.1073/pnas.0705366104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Youngblood B, Buller F, Reich NO. Determinants of sequence-specific DNA methylation: target recognition and catalysis are coupled in M.HhaI. Biochemistry. 2006;45:15563–15572. doi: 10.1021/bi061414t. [DOI] [PubMed] [Google Scholar]
- 34.Zhou H, Purdy MM, Dahlquist FW, Reich NO. The recognition pathway for the DNA cytosine methyltransferase M.HhaI. Biochemistry. 2009;48:7807–7816. doi: 10.1021/bi900502g. [DOI] [PubMed] [Google Scholar]
- 35.Kalodimos CG, Biris N, Bonvin AM, Levandoski MM, Guennuegues M, Boelens R, Kaptein R. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004;305:386–389. doi: 10.1126/science.1097064. [DOI] [PubMed] [Google Scholar]
- 36.Tzeng SR, Kalodimos CG. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- 37.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–207. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 38.Jeltsch A. Maintenance of species identity and controlling speciation of bacteria: a new function for restriction/modification systems? Gene. 2003;317:13–16. doi: 10.1016/s0378-1119(03)00652-8. [DOI] [PubMed] [Google Scholar]
- 39.Wilson GG, Murray NE. Restriction and modification systems. Annu. Rev. Genet. 1991;25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 40.Shields SL, Burbank DE, Grabherr R, van Etten JL. Cloning and sequencing the cytosine methyltransferase gene M. CviJI from Chlorella virus IL-3A. Virology. 1990;176:16–24. doi: 10.1016/0042-6822(90)90225-g. [DOI] [PubMed] [Google Scholar]
- 41.Vasu K, Nagamalleswari E, Nagaraja V. Promiscuous restriction is a cellular defense strategy that confers fitness advantage to bacteria. Proc. Natl Acad. Sci. USA. 109:E1287–1293. doi: 10.1073/pnas.1119226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehrlich M, Norris KF, Wang RY, Kuo KC, Gehrke CW. DNA cytosine methylation and heat-induced deamination. Biosci Rep. 1986;6:387–393. doi: 10.1007/BF01116426. [DOI] [PubMed] [Google Scholar]
- 43.Lieb M. Spontaneous mutation at a 5-methylcytosine hotspot is prevented by very short patch (VSP) mismatch repair. Genetics. 1991;128:23–27. doi: 10.1093/genetics/128.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johansson M, Zhang J, Ehrenberg M. Genetic code translation displays a linear trade-off between efficiency and accuracy of tRNA selection. Proc. Natl Acad. Sci. USA. 109:131–136. doi: 10.1073/pnas.1116480109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amitai G, Devi-Gupta R, Tawfik DS. Latent evolutionary potentials under the neutral mutational drift of an enzyme. HFSP. 2007;1:67–78. doi: 10.2976/1.2739115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bloom JD, Romero PA, Lu Z, Arnold FH. Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biol. Direct. 2007;2:17. doi: 10.1186/1745-6150-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.