


Abstract
SR1 is a dual-function sRNA that acts as a base-pairing regulatory RNA on the ahrC mRNA and as a peptide-encoding mRNA on the gapA operon. The SR1-encoded peptide SR1P binds GapA thereby stabilizing gapA mRNA. Under glycolytic conditions, SR1 transcription is repressed by CcpN and CcpA. A computer-based search identified 23 SR1 homologues in Bacillus, Geobacillus, Anoxybacillus and Brevibacillus species. All homologues share a high structural identity with Bacillus subtilis SR1, and the encoded SR1P peptides are highly similar. In the Bacillus cereus group, the sr1p region is present in triplicate or duplicate resulting in longer SR1 species. In all cases, sr1 expression is under control of CcpN, and transcriptional lacZ fusions of nine examined SR1 homologues were sensitive to glucose. Two homologues showed an additional glucose-independent repression by CcpN and an unknown factor. A total of 10 out of 11 tested SR1P homologues complemented a B. subtilis Δsr1 strain in their ability to stabilize gapA mRNA, but only five of them bound GapA tightly. In vitro binding assays with six SR1/ahrC pairs suggest that—despite divergent primary sequences—the base-pairing function is also preserved. In summary, SR1 is an sRNA with two functions that have been conserved over ≈1 billion years.
INTRODUCTION
Small RNAs (sRNAs) are the largest class of post-transcriptional regulators in bacteria known to date (for reviews see (1, 2)). About 140 sRNAs are known in Escherichia coli and Salmonella. However, only about 25 of them have been assigned a biological function, indicating that defining their functions continues to be a challenging issue. In the last 3 years, systematic searches have been performed for several Gram-positive species (e.g. Bacillus subtilis (3, 4), Listeria monocytogenes (5), Staphylococcus aureus, (6, 7)) indicating that a plethora of sRNAs exists also in such genomes. sRNAs can be divided into two major groups: the first group regulate gene expression by a base-pairing mechanism with target mRNA, whereas the second group act by binding of small proteins. The major regulatory mechanisms applied by both cis- and trans-encoded base-pairing sRNAs include inhibition or activation of translation and promotion of RNA degradation or RNA stability. Additionally, inhibition of primer maturation, transcriptional attenuation and transcriptional interference have been found for some cis-encoded sRNAs (rev. in (8)). Interestingly, a few trans-encoded sRNAs have dual functions: they act both as base-pairing sRNAs and as peptide-encoding mRNAs. The first reported example was S. aureus RNAIII, which encodes δ-hemolysin (26 aa) and activates translation of hla mRNA (9) and, additionally, represses translation of spa, rot, sa1000/2353, coa (10). Later, the streptolysin SLS-ORF of Streptococcus Pel RNA (11) and the 43-codon-SgrT ORF on E. coli SgrS (12) were identified. SgrS and SgrT downregulate PtsG glucose transporter activity and have a physiologically redundant, but mechanistically distinct function in inhibition (12). In contrast, for the hyp7-ORF on Clostridium perfringens VR (13), the 37-codon PhrS-ORF of Pseudomonas aeruginosa (14), the 32-codon RivX-ORF (15) and the RSs0019-ORF of Rhodobacter sphaeroides (16) no functions have been elucidated so far.
The 205 nt RNA SR1 from the B. subtilis genome was found in our group by a combination of computer predictions and northern blotting (NB) (17). Previously, we have shown that SR1 acts by base pairing with its primary target, ahrC mRNA, the transcriptional activator of the rocABC and rocDEF arginine catabolic operons (18). SR1 inhibits translation initiation of ahrC mRNA by a novel mechanism: induction of structural changes downstream from the ribosome-binding site (19). SR1 is expressed under gluconeogenic and repressed under glycolytic conditions by CcpN, and, to a minor extent, CcpA (17). Thereby, CcpN requires ATP and a slightly acidic pH (20) to prevent promoter escape via direct contacts with the α-subunit of the B. subtilis RNA polymerase (21). Recently, we found that the 39-codon ORF on SR1 is translated into a peptide, designated SR1P. We demonstrated that SR1P binds GapA, thereby stabilizing the gapA operon mRNA by a hitherto unknown mechanism (22). SR1 is the first dual-function sRNA in B. subtilis.
Here, we provide a combination of a computer-based analysis of 23 SR1 and SR1P homologues from different Bacillales species and the in vivo and in vitro characterization of SR1 and SR1P homologues. We chose representatives of the two important Bacillus groups, the B. subtilis and the B. cereus group as well as from ungrouped Bacilli like B. megaterium, B. halodurans and Geobacillus kaustophilus for in vivo studies. All tested homologues were repressed by CcpN under glycolytic and expressed under gluconeogenic conditions. The two CcpN binding sites (BSs) were located at the same positions as in the B. subtilis sr1 gene. Whereas 10 of 11 SR1P homologues were functional in NB to stabilize B. subtilis gapA mRNA, only 5 SR1P homologues were able to bind B. subtilis GapA tightly indicating that their interaction with GapA is stronger than that of the other homologues. The computer-based analysis of the regions required for base pairing with the ahrC mRNA homologues predicted between 7 and 11 complementary regions. In vitro binding assays with six SR1/ahrC pairs revealed that the base-pairing function of SR1 is preserved. Eight SR1 homologues could be detected in their native hosts, but expression of B. pumilus SR1 seems to be repressed. In summary, we found that both SR1 functions—the base-pairing and the mRNA function—have been conserved during 0.9–1.3 billion years of evolution.
MATERIALS AND METHODS
Identification of SR1P homologues
The amino acid (aa) sequence of B. subtilis SR1P was used as a query in a BlastP search against the non-redundant protein sequence database at NCBI. Using the SR1P homologues found in the first search, the consensus sequence MGTIV CQXC(EN) XTIXH FEDEK (VTS)T(VT) LY G (KT) CXX XCXCX XXXXX XXX was derived. Using this consensus sequence as a query in an additional BlastP search against the same database resulted in more homologous proteins. Results from all searches were corrected with regard to redundancy, resulting in a list of 23 SR1P homologues. The aa sequences of the 23 SR1P homologues were aligned using ClustalW (Blosum protein weight matrix, 23).
Identification of SR1 homologues
The sr1 nucleotide sequence of B. subtilis was used as a query in a BlastN search against the Nucleotide collection database at NCBI. Furthermore, the sequences adjacent to all sr1p-encoding regions were analysed. Analyses of the loci of all obtained nucleotide sequences indicated the location of the sr1 homologues between the pdhD and the speA/cad genes. Investigation of the predicted sr1 locus in other Bacillus and Geobacillus species yielded a few more homologous sequences.
Phylogeny reconstruction
SR1 sequences from species of the Bacillus cereus group were split in two and three parts, respectively, each beginning at the characteristic RNA stem-loop structure and ending at the stop codon of the putatively encoded peptide. The resulting sequences and the SR1 sequence of Geob. kaustophilus, which was later used as a representative of the outgroup in the phylogeny, were aligned using MAFFT (24). A MODELTEST (25) was performed to determine the best-fitting model for the alignment. Using this model, a phylogeny was reconstructed using MrBayes (26), where 3 000 000 generations were generated, the burn in was set to 750 000 and a majority rule consensus tree including all compatible groups was constructed. Using the same methods, a second phylogeny containing the consensus sequence of the three copies of the Bacillus cereus group and SR1 units of other Bacillus species having only one copy of the dual-function RNA was determined.
Identification of base-pairing determinants in SR1 homologues
The aa sequence of B. subtilis ahrC was used as a query to identify ahrC homologues with BlastP in each species containing an sr1 homologue. IRNA, a local RNA–RNA interaction prediction tool (E. Barth, unpublished) was used to create interaction profiles between different regions of SR1 and ahrC mRNA based on the known interaction regions of B. subtilis SR1 and ahrC mRNA (18,19). IRNA determined optimal interaction profiles by optimizing a weighted base-pairing score as well as the total number of interactions.
Identification of CcpN and CcpA BSs in the promoter region of sr1 homologues
A simple string-matching algorithm was applied to identify BS of CcpN and CcpA in sr1 homologues. The consensus sequences used were 5′ WTGNAANCGNWNNCW for the cre site (CcpA BS) and 5′ TRTGHYATAYW (27) for the CcpN BS. The search was limited to the range between the SD sequence and 400 nt upstream of the sr1 transcriptional start site. Thereby, only hits with a maximum of two mismatches compared to the consensus sequences were allowed.
Strains and growth conditions
Bacillus subtilis strain DB104 (28), B. amyloliquefaciens FZB42, B. licheniformis ATCC14580, B. pumilus DSM27, B. thuringiensis DSM350, B. megaterium DSM319, B. halodurans DSM497 and Geob. kaustophilus DSM7263 were used. All strains are considered to be wild-type strains. They were grown at 37°C except Geob. kaustophilus that was grown at 54°C. TY medium was used as complex medium (18). For B. halodurans, TY was supplemented with 100 mM Na-sesquicarbonate pH 9.7.
Preparation of total RNA, RNA gel electrophoresis and NB
Preparation of total RNA, RNA gel electrophoresis on 6% denaturing polyacrylamide gels or 1.5% agarose gels and NB were carried out as described previously (18). For the detection of SR1 from B. amyloliquefaciens and B. licheniformis, the hybridization probe for B. subtilis SR1 was used. In contrast, separate probes had to be generated for the detection of the other SR1 homologues. The same holds true for the oligonucleotide probes against 5S rRNA used for reprobing (Supplementary Table S1).
Isolation of chromosomal DNA from different Bacillus species
Chromosomal DNA from all strains was isolated as described previously for B. subtilis (17).
Construction of plasmids for tet-inducible overexpression of sr1 homologues and mutants
A polymerase chain reaction (PCR) on chromosomal DNA of the different Bacillus species was performed with primers designed on the basis of the sr1 sequence retrieved from the NCBI database (primers are listed in Supplementary Table S1). The resulting fragments were subjected to a second PCR with primer SB1402 to add the Strep-tag sequence and the SR1 terminator from B. subtilis at the 3′ terminus, digested with HindIII and inserted into the pWSR1-HindIII vector. The sequence was confirmed. In the case of pWSR1/M60 (plasmids are listed in Supplementary Table S2) designed to analyse the effect of the native triplicate sr1 locus of B. thuringiensis in B. subtilis, only a single PCR reaction with the corresponding primer pair was performed, the resulting fragment digested with PstI and Acc65I cloned into the pWSR1 vector cleaved with the same enzymes. For the construction of pWSR1 derivatives comprising mutated B. subtilis sr1 genes, two single PCRs with mutant primer 1 and SB317 and mutant primer 2 and SB348 were performed, followed by a second PCR with primers SB317 and SB348. The final PCR fragment was digested with HindIII and inserted into the pWSR1-HindIII vector.
Construction of plasmids for transcriptional lacZ fusions and determination of β-galactosidase activity
The upstream regions of different sr1 species were amplified by PCR on chromosomal DNA of the corresponding Bacillus species as above using the primer pairs listed in Supplementary Table S1. The resulting fragments were digested with EcoRI and BamHI and cloned into pACC1 vector (22) cleaved with the same enzymes. In the wild-type cases, fragments were obtained that contained 87 bp upstream of the −35 box of psr1, the promoter and 10 nt downstream from the putative transcription start site. Additionally, plasmids with sequences from the sr1 homologues of B. subtilis, B. megaterium and B. halodurans were constructed that contained either only the promoter regions without regions upstream of the −35 boxes (pACS63, pACS65 and pACS67, respectively) or the promoter regions including only both CcpN BSs (pACS62, pACS64 and pACS66, respectively). The resulting plasmids were linearized with ScaI and inserted into the amyE locus of the B. subtilis DB104 chromosome by double crossing over. The resulting integrant strains were used for the determination of β-galactosidase activities as described previously (29).
Co-elution experiments with SR1P homologues and B. subtilis GapA
Co-elution experiments with Strep-tagged SR1P homologues and western blotting were performed as described recently (22).
Analysis of RNA–RNA complex formation
Both ahrC mRNA and SR1 were synthesized in vitro from PCR-generated template fragments with primer pairs indicated in Supplementary Table S1. SR1/ahrC complex formation studies were performed as described previously including tRNA as unspecific competitor (18).
Primer extension and 3′ RACE
Primer extension was performed as described previously (17). 3′ RACE was also performed as described (17), but with the following modifications: Total RNA from B. thuringiensis was ligated to intrinsic 16S rRNA instead to an artificial RNA adapter. Two subsequent PCR amplification steps were used, the first with outer primers SB1900/SB1894, and the second, nested PCR with inner primers SB1901/SB1897.
RESULTS AND DISCUSSION
Identification and gene synteny of 23 homologues of SR1
As we could show recently, the 39 aa ORF on B. subtilis SR1 is translated into a small peptide, SR1P, which was also found in some other Bacilli (22). To ascertain, whether SR1P is confined to Bacillus and Geobacillus species or is also present in other bacteria, a systematic search among all anotated bacterial genomes was undertaken as described in Materials and Methods section. This search identified 23 SR1 homologues, among them 18 Bacillus species, three Geobacillus species and, additionally, Anoxybacillus flavithermus WK1 and Brevibacillus brevis NBRC100599. A gene encoding SR1P was not found in B. selenitireducens and B. cellulosilyticus. No sr1 homologues were discovered in Gram-negative or in other Gram-positive bacteria. The gene synteny of 21 homologues is presented in Supplementary Figure S1. In all but one case, the sr1 genes are located between pdhD and speA/cad genes, but the number of genes between pdhD and sr1 on the one hand and sr1 and speA/cad on the other hand, differed. Whereas in all cases except for the B. cereus group and Brev. brevis, speA encoding arginine decarboxylase or cad encoding lysine decarboxylase was located immediately downstream from sr1, in the B. cereus group, tgl encoding transglutaminase was located between sr1 and speA/cad, and in B. mycoides, tgl and gene 35 850 were located between sr1 and speA. In Brev. brevis, five genes interrupted the sr1 and the speA gene. The region upstream of sr1 diverged much more with 6 and 10 genes located between pdhD and sr1 in B. pseudofirmus and B. halodurans, respectively, whereas in B. subtilis and B. pumilus, pdhD and sr1 were only separated by the slp gene. An alignment of all 23 SR1 sequences is presented in Supplementary Figure S2.
Based on the conserved location of the sr1 gene in the Bacillales, we searched upstream of the cad/speA genes in other Gram-positive bacteria for putative sr1 homologues, however, with no result.
Phylogeny
As summarized in Supplementary Table S3, most of the sr1 genes contain a bidirectional terminator shared with the downstream gene transcribed into the opposite direction. In contrast, the sr1 homologues of B. clausii, B. coagulans, Anoxybacillus, Brevibacillus and the three Geobacillus species carry unidirectional transcription terminators at their 3′ ends. Their downstream genes have their own terminators. All SR1 homologues, except those from the B. cereus group, were predicted to be about 200–230 nt long. The latter group has sr1 genes with triplicate or duplicate sr1p sequences, resulting in ≈600 nt or ≈400 nt long SR1 species comprising 3 subunits each coding for a peptide (SR1 I, SR1 II and SR1 III), respectively. The SR1 homologues from the B. subtilis group and B. pumilus are closely related and form a clade (Supplementary Figure S3). In this clade, the identity on nucleotide level for the sr1p coding region is between 83% and 92%. The B. clausii and B. pseudofirmus SR1 species are at the basal positions in the phylogeny and form a grade, but even their sr1p coding regions are between 81% and 84% identical with that of B. subtilis. Not surprisingly, the SR1 homologues from the three Geobacillus species cluster together and are more related to the B. cereus group than the SR1 species from the other Bacilli. B. megaterium, B. coahuilensis, B. coagulans and Anoxybacillus form a clade.
The presence of triplicate or duplicate sr1p genes prompted us to elucidate the order of duplications that gave rise to the additional sr1p copies. Therefore, we conducted an additional analysis. The resulting phylogeny reveals that there are three clades containing the copies I, II and III, respectively, distinguished (Figure 1). All three clades are comprised of copies from all species of the B. cereus group with the exception of clade III where B. cytotoxicus is missing as this species has only two SR1P copies. According to our phylogeny, copy III is the basal sr1 copy. This copy was duplicated in a common B. cereus group ancestor resulting in SR1 III and a SR1 I/II precursor. This precursor was duplicated once more resulting in SR1 I and SR1 II each of them encoding a peptide. The fact that the corresponding copies of the different species are more closely related to each other than the different copies of the same Bacillus species suggests that the most recent common ancestor of the B. cereus group Bacilli had already three SR1P copies. All three copies were retained during speciation events giving rise to the different B. cereus group members apart from B. cytotoxicus that lost the basal SR1P III copy.
Figure 1.

Bayesian phylogeny of the B. cereus group SR1 homologues. Our phylogeny suggests that the ancestral gene encoding SR1P was duplicated twice in the ancestor of the B. cereus group Bacilli. The first duplication generated copy III and an ancestral sequence for copies I and II. In the second duplication this ancestral sequence was duplicated once more to give rise to the copies I and II. All three copies were retained in subsequent speciation events through which the species of the B. cereus group were generated, except for the third copy in B. cytotoxicus, which was lost.
Since SR1 is regulated by CcpN, but the ccpN gene is not located in the vicinity of the sr1 gene, it was not possible to use the ccpN gene for a bioinformatics search for sr1 homologues as it was done by Horler and Vanderpool for SgrS/SgrR (30). However, we currently develop a programme that allows predicting small peptides in different bacteria. This programme will be applied to search for functional homologues (which might differ in primary nt and aa sequence) for SR1 and SR1P downstream from CcpN BSs.
Comparison of the corresponding sr1 promoter regions and the CcpN and CcpA BSs
The B. subtilis sr1 gene is transcribed from a perfect consensus promoter with the −35 box TTGACA and the −10 box TAATAT separated by a 17 bp spacer. A comparison of the promoters of all sr1 homologues revealed that all of them have −35 and −10 boxes separated by a 17 bp spacer region (Figure 2). The only exception is Brev. brevis psr1, which has an 18 bp spacer. All −10 boxes in the B. cereus group display the sequence TAAAAT, whereas in all other cases, either TAATAT (as in B. subtilis) or TATTAT is found, and, again, in Brev. Brevis deviantly TAAGAT. All −35 boxes have the consensus sequence TTGACD with D being A, G or T. The putative transcription start site is always an A, expect in B. pumilus, where it is G.
Figure 2.

BSs for CcpN in the promoter regions of sr1 homologues. The DNA sequences around the promoter regions of the sr1 homologues are shown. −35 and −10 boxes are in bold and underlined. Putative transcription start sites are shaded in light grey. The two putative BSs for CcpN are indicated in dark grey. The consensus sequence for the CcpN BS is TRTGHYATAYW (27), thereby R = purine, Y = pyrimidine, W = A or T, H = A, C or T.
Furthermore, in all instances, two BSs for CcpN that represses transcription of sr1 under glycolytic conditions (17) are present in nearly the same location as in B. subtilis. The two BSs for CcpN are located 6–9 bp upstream of the −35 box (3′ end of site I) and in the spacer of the −10 box of psr1. Only Brev. brevis BS I is located 19 bp upstream of the −35 box. Whereas in all cases, BS II is located in the 3′ half of the psr1 spacer and overlaps at the most the first T of the −10 box, B. coagulans BS II overlaps TATT of the −10 box. In the B. cereus group and B. coagulans, BSs I and II are arranged as inverted repeats, whereas in the B. subtilis group and all other species, they are arranged as two direct repeats. No clear definition in this respect is possible for B. clausii and B. halodurans BSs I and II.
Since CcpA has a minor influence on the glucose-dependent regulation of SR1 transcription, and at least one active CcpA BS (cre site) was found upstream of B. subtilis psr1 (17), we searched for putative cre sites in the vicinity of the sr1 promoters of all 23 homologues. As shown in Supplementary Figure S4, 21 sr1 homologues carry at least one hypothetical cre site. Thereby, cre sites upstream of the −35 box of psr1 were found in 12 cases, and downstream from or overlapping with the transcription start site were found in 19 cases. No putative CcpA BS was found at or in the vicinity of the Brev. brevis and at the Geob. kaustophilus sr1 promoters. Since in the majority of cases, cre sites are located either within the promoter region (e.g. bglPH, acu, amyE), where they inhibit transcription initiation, or downstream from the transcription start site (e.g. acsA, xyl, hut), where they might block elongation (31), we had shown previously that the cre site 275 nt upstream of the transcription start site of B. subtilis sr1 is active, whereas the cre site between nt +12 and +27 is not required (17).
Comparison of the secondary structures/coding properties of the SR1 homologues
The experimentally determined secondary structure of B. subtilis SR1 (19) was used to predict the secondary structures of all SR1 homologues. Eight of these predicted secondary structures are shown in Supplementary Figure S5. All SR1 homologues display a large stem-loop (SL1) at their 5′ end with a 3–12 nt apical loop and a considerable bulge (13–17 nt) in the 5′ half of the stem that contains the SR1P SD sequence. For B. subtilis SR1 SL1, we found that the double-stranded region at the basis of SL1 is required for RNA stability (M. Gimpel, unpublished). In the central part, between SL1 and the terminator stem-loop (TSL), a smaller stem-loop (SL2) is located with 4–7 paired nt and a 5–7 nt loop. The TSLs have a 3–9 nt loop and, except B. coagulans, a 12–15 bp stem region. An overview of the terminator sequences is given in Supplementary Table S3. In the B. cereus group, three copies of SL1 and SL2 are terminated by a single TSL. All start codons for the SR1P homologues are located in a partially double-stranded region on the 5′ half of SL1. All stop codons are located downstream from SL2 near the TSL, but in the Geobacilli, a longer sequence separates the stop codon from the TSL, resulting in a slightly longer (225–229 nt) SR1. In the B. cereus group, the stop codons of the first two sr1p sequences are located upstream of the next SL1, whereas the stop codon of the third copy is located upstream of the TSL. In the B. halodurans sr1 gene, the ≈1.4 kb long insertion element IS652 is inserted between the stop codon of sr1p and the transcription terminator.
Comparison of the SR1P homologues
All SR1P homologues comprise between 37 and 42 aa (Supplementary Figure S6). Interestingly, all members of the B. cereus group contain three sr1p copies, with the exception of B. cytotoxicus that contains two copies. All other species encode only one SR1P. All SR1P aa sequences comprise 3 highly conserved motifs: the N-terminal 7 aa MGTIVCQ, the central FEDEK region and the adjacent VTTLY motif. Furthermore, 3 cysteines at positions 9, 28 and 33 and the isoleucine at position 13 are highly conserved. A comparison of the three sr1p copies of the B. cereus group peptides shows that copy one is nearly identical in all cases, wheras the second and third copy vary. The first SR1P of all B. cereus group members shows 64% identity to the B. subtilis SR1P, whereas the last copy is more divergent with only 55–57% identity to B. subtilis SR1P. The highest variability can be found in the C-termini of all SR1 peptides. Furthermore, the peptides encoded by B. megaterium, B. halodurans, B. pseudofirmus, B. clausii, B. coahuilensis and B. coagulans reveal more deviations at positions 11–15 and starting from position 26 till the C-terminal end among each other and compared to all other peptides. In contrast, the Geobacillus species are similar to each other and more related to the B. cereus group peptide I reflecting their position in the phylogenetic tree (Supplementary Figure S3). With 64–71% identity they are more closely related to B. subtilis SR1P than the second and third peptides from the B. cereus group members (Supplementary Table S3).
Comparison of the complementary regions between the SR1 homologues and the corresponding ahrC/arg homologues
An alignment of the coding regions for all ahrC/arg homologues is shown in Supplementary Figure S7, whereas Supplementary Figure S8 shows all complementary regions of SR1 and ahrC RNA. As highlighted in Supplementary Figure S5, all SR1 homologues experimentally analysed in this study contain between 7 and 11 regions that are complementary to their respective ahrC RNA-homologues. Supplementary Table S3 provides an overview of the length of the complementary regions. The location of these regions is more or less conserved, but the primary sequences are highly divergent. An overview of the complementary regions of all 23 SR1 homologues and their target mRNAs that might be involved in base pairing is presented in Supplementary Figure S8. In all cases, the first two complementary regions are found 5′ (red) and 3′ (green) and overlapping with the base of SL1. The other regions are distributed along the single-stranded regions and the central stem-loop SL2 as in B. subtilis. The last complementary region (yellow) is always found at the TSL, either in the 5′ part of the stem as in B. subtilis, B. licheniformis, B. pumilus, Geob. kaustophilus or at the loop of the TSL as in B. amyloliquefaciens, B. anthracis, B. megaterium and Geob. kaustophilus. In B. subtilis, this region (designated G) is most important for the initial interaction with the 5′ complementary region (G′) of the ahrC target RNA ≈90 bp downstream of the ahrC ribosome BS (19). In B. thuringiensis, two ahrC homologues exist, arg2 highly similar to B. subtilis ahrC, and the more divergent arg1. As shown in Supplementary Figure S5, B. thuringiensis SR1 is predicted to interact with both arg mRNAs using different complementary regions. The interaction pattern for SR1/arg2 is similar to the interaction patterns of the other SR1 homologues with their respective ahrC/arg genes. In contrast, the interaction pattern for SR1/arg1 involves the central instead of the base regions of SL1. B. anthracis encodes only arg2, and the arg2/SR1 interaction pattern resembles that of the B. thuringiensis arg2/SR1 pattern.
Binding assays of SR1/ahrC mRNA pairs
To analyse the predicted interactions of SR1 homologues with their cognate ahrC mRNAs experimentally, in vitro binding studies were performed for the SR1/ahrC RNA pairs of B. subtilis, B. amyloliquefaciens, B. licheniformis, B. megaterium, Geob. kaustophilus and B. thuringiensis. B. halodurans and B. pumilus were not included, because the sr1 gene in B. halodurans is interrupted by IS652 and did not yield a distinct SR1 species in northern blots, and sr1 is apparently not expressed in B. pumilus (see below and Figure 4). For B. thuringiensis, in vitro transcribed first, second and third copies of SR1 were analysed separately, since RNA/RNA complexes formed with species larger than 400 nt do not run into native gels. SR1 species were generated in vitro using T7 RNA polymerase, 5′ end-labelled, gel-purified and used for binding assays with unlabelled ahrC-RNA species of about 350–380 nt comprising all predicted regions for base pairing (Supplementary Figure S8). B. thuringiensis SR1 I and SR1 II did not form complexes with arg2 RNA (not shown). In all other cases, SR1/ahrC RNA complexes were formed (Figure 3A), albeit with varying efficiencies. The most efficient interactions were observed for B. subtilis, B. licheniformis and Geob. kaustophilus, where at the highest ahrC RNA concentration between 30% and 42% of labelled SR1 was bound, whereas in the other cases, only ≈10% of SR1 were bound. These differences might be due to differences in number and/or accessibility of the complementary regions or the requirement of additional helper proteins for complex formation in vivo. Previously, we have shown in B. subtilis that the initial contact between SR1 and ahrC RNA requires complementary region G (19). This region is located in the 5′ half of the SR1 TSL downstream of the coding region for SR1P. The complementary region G′ is found about 90 nt downstream of the ahrC RBS (ribosome binding site). In B. subtilis, SR1 binds to this region and induces structural changes ≈40–60 nt downstream of the RBS that prevent initiation of ahrC translation (19, see model in Supplementary Figure S11). Therefore, we investigated if ahrC RNA species of the other Bacilli that comprise only region G′ could be bound by the cognate SR1 species. Complexes with this short ahrC RNA (arg2 in B. thuringiensis) were observed for B. subtilis, B. amyloliquefaciens, B. licheniformis, B. megaterium and Geob. kaustophilus but not for B. thuringiensis SR1 III (Figure 3B). One explanation for the failure of SR1 III to bind to arg2-G′ might be that in vivo the interaction partner of arg2 is full-length (587 nt) B. thuringiensis SR1, whose structure differs from the short species used in these experiments. However, due to technical constraints, we could not investigate such a long sRNA in the binding assays (see above). Predicted interactions with B. thuringiensis SR1 and arg1 mRNA that does not exist in B. subtilis, could not be confirmed in binding assays with SR1/arg1 species comprising all complementary regions. Therefore, it is not clear if this predicted interaction is relevant.
Figure 4.
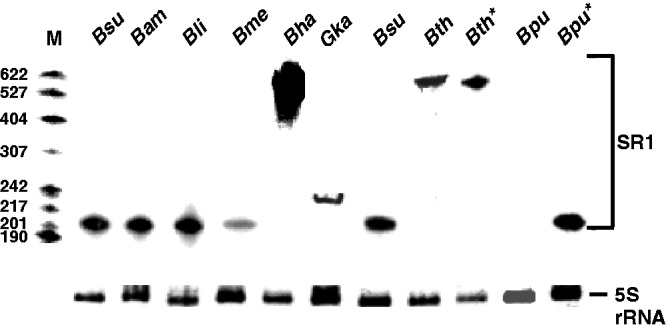
Expression of SR1 in eight Bacillus/Geobacillus species. All strains were grown as described in Materials and Methods section. At the onset of stationary growth phase, samples were withdrawn, total RNA prepared and subjected to NB. For hybridization, [α-32P] dATP-labelled DNA probes for the respective SR1 homologues were used. To compare the amounts of RNA loaded onto the gel, signals for 5S rRNA obtained by ethidium-bromide staining are shown. In the case of B. thuringiensis and B. pumilus, the sr1 gene was expressed in parallel from a high-copy vector (pWSR1/M60 and pWSR1/M56; Supplementary Table S2) in B. subtilis (lanes labelled with asterisk) and loaded next to the total RNA isolated from the original host.
Figure 3.

Binding assays of SR1/ahrC RNA pairs from different Bacillus species. Binding experiments were performed as described in Materials and Methods section. Autoradiograms of gel-shift assays are shown. The concentration of unlabelled ahrC species is indicated in nM. (A) Binding assays with homologous SR1/ahrC pairs from B. subtilis, B. amyloliquefaciens, B. licheniformis, B. megaterium, Geob. kaustophilus and B. thuringiensis III. SR1 species from these Bacilli were 5′ end-labelled with [γ32P]-ATP and used at a final concentration of 4 nM (at least 10-fold lower concentration than ahrC RNA) in all experiments. ahrC-RNA species of the indicated length comprising the central part of ahrC with all complementary regions (Supplementary Figures S7 and S8) were used. (B) Binding assays with homologous SR1/ahrC pairs as above. ahrC RNAs containing only region G′ were paired with the SR1 homologues comprising all complementary regions. (C) Regions that can base pair in the experiment shown in (B). Below, the distance between the 5′ end of region G′ and the 3′ end of the corresponding ahrC start codons is shown for each ahrC RNA/SR1 pair.
In the binding assays, the region located next to the ahrC SD sequence was used as region G′ (Figure 3C). At primary sequence level, there is no conservation in regions G and G′ among the SR1 and ahrC homologues (see above and Supplementary Figures S2 and S8). Even the distance between the ahrC start codon and G′ and the GC-content of G/G′ vary (Supplementary Figures S8). In the cases analysed experimentally, the distance between G′ and ahrC start codon was between 25 nt and 83 nt. The G/G′ double-strand comprised between 6 and 10 contiguous bp that were in one case interrupted by one bulged-out nucleotide. In the majority of the SR1 species, G is located either at the 5′ arm of the terminator stem as in B. subtilis, B. licheniformis and B. pumilus (Supplementary Figures S2, S5), and would need in vivo an enzyme to open up the double-stranded region as proposed previously (19). In other cases, G is located at the terminator loop (B. amyloliquefaciens, B. megaterium, Geob. kaustophilus and B. thuringiensis) and is in B. thuringiensis and Geob. kaustophilus preceded by a second complementary region in the 5′ half of the stem.
The results of the in vitro binding assays suggest that the repression of ahrC translation by SR1 is conserved, and that—as previously found in B. subtilis—the G/G′ base pairing is—despite all variability—sufficient to initiate binding.
Both regulation of ahrC translation and gapA mRNA stability occur in stationary growth phase. Therefore, the question arises if sr1p translation and SR1/ahrC base-pairing interfere. On the one hand, in all SR1 homologues, between one and four complementary regions are located downstream from the translated region (Supplementary Figures S2, S5 and S8), and in five Bacillus species SR1/ahrC RNA-complexes could form when only one of these complementary regions was present (Figure 3). However, it cannot be excluded that complementary regions B–F, which were previously shown to have only a marginal effect on the SR1/ahrC interaction in B. subtilis (19), have a higher impact on ahrC RNA binding in the other SR1 homologues. As these regions overlap with the sr1p ORF, binding of B to F might hinder sr1p translation. On the other hand, the sr1p ORF is poorly translated, as a translational sr1p-lacZ fusion did not yield measurable β-galactosidase activities in B. subtilis (17), and only the addition of a 3xFLAG tag allowed to visualize SR1P in western blots (22). However, taken together, the mutual interference of the two SR1 functions cannot be entirely excluded.
Since the primary sequences of the regions in ahrC or arg, which are required for the initial interaction with homologous SR1 species are highly different, it cannot be assumed that heterologous SR1 species are able to complement a Δsr1 B. subtilis strain for the effect on AhrC, and, thus, the rocABC and rocDEF operons.
Identification of SR1 homologues in eight different species in northern blots
For a comparative experimental analysis of SR1 homologues, nine species were chosen: B. subtilis, B. amyloliquefaciens, B. licheniformis, B. pumilus (the first three belonging to the B. subtilis group, the latter one closely related), B. thuringiensis and B. anthracis (B. cereus group) and the three ungrouped B. halodurans, B. megaterium and Geob. kaustophilus. All strains except B. anthracis were grown in TY complex medium under the appropriate growth conditions (see Materials and Methods section), RNA isolated and subjected to NB with the respective probes. SR1 homologues from B. subtilis, B. amyloliquefaciens and B. licheniformis were detected with a probe against B. subtilis SR1 (Figure 4). For the other SR1 species, the cognate probes were used. As predicted, SR1 from Geob. kaustophilus is with 229 nt larger in size than the B. subtilis, B. amyloliquefaciens, B. licheniformis and B. megaterium SR1 homologues. Interestingly, in B. halodurans, a mixture of SR1 species of different lengths between 400 nt and 650 nt was observed, which apparently results from unspecific termination of sr1 transcription within insertion element IS652.
In B. thuringiensis, a ≈580 nt long species encoding the three SR1P peptides was expressed. To exclude that a processing into smaller SR1 species escaped our attention, we performed primer extension and 3′ RACE experiments using primers complementary to different SR1 regions. Primer extension with primers located within each of the three sr1p regions resulted in a single clear signal corresponding to the 5′ end of a 587 nt RNA (Supplementary Figure S9). 3′ RACE yielded the predicted 3′ end of a 587 nt SR1 as well as signals representing various degradation products, which were evenly distributed and did not indicate specific processing sites. Therefore, the triplicate SR1 of B. thuringiensis is not processed into three shorter species.
Although high amounts of total RNA were isolated from B. pumilus, the corresponding SR1 homologue could not be detected in northern blots with the cognate SR1 probe. The expression of B. megaterium sr1 was weaker than that of the other sRNAs. One reason could be transcriptional down-regulation of this RNAs in its native host. This result prompted us to analyse the strength of the nine different SR1 promoters.
Experimental analysis of the promoter strength and the role of CcpN for nine selected SR1 species
To this end, transcriptional lacZ-fusions were constructed comprising 87 bp upstream of the −35 box and 10 nt downstream from the putative transcription start sites containing both CcpN BSs and about 40 nt upstream of them and integrated into the amyE locus of the chromosome of B. subtilis strains DB104 and DB104 (ΔccpN::phleo). The latter strain was used to investigate the effect of CcpN, i.e. transcription repression in the presence and absence of CcpN under glycolytic conditions (17). Cultures were grown in SP medium with and without 2% glucose and β-galactosidase activities determined. In the absence of glucose, activities were high (between 1567 MU and 4169 MU) for all sr1 promoters except those of B. halodurans and B. megaterium (Table 1). In all cases, in the presence of 2% glucose, ≈7–20-fold lower β-galactosidase activities were determined in DB104. This result confirms that the two CcpN BSs found in nearly the same locations as in B. subtilis psr1 are active. In the absence of CcpN, the β-galactosidase activities were—except for B. halodurans and B. megaterium—in the same range as in the absence of glucose, verifying that CcpN is responsible for this effect. In B. halodurans and B. megaterium, the promoter strength was ≈10-fold lower in the absence of glucose compared to the other species, but increased about 3.6–4.6-fold when CcpN and glucose were not present. This result suggests in these two species an additional glucose-independent repression by CcpN, which has not been reported so far. Surprisingly, a small (1.5–1.9-fold) but distinctive glucose repression was still observed in the absence of CcpN in B. subtilis, B. licheniformis, B. halodurans and B. megaterium. Since none of the constructed plasmids contained a cre site (Supplementary Figure S4), this residual glucose repression cannot be attributed to CcpA, but must be due to another, still unknown glucose-dependent factor.
Table 1.
β-galactosidase activities of wild type and ccpN knockout strains
| lacZ fusion | β-galactosidase activity (Miller units) in |
||||||
|---|---|---|---|---|---|---|---|
| DB104 (−Glc) | DB104 (+Glc) | GRF | ΔccpN (−Glc) | ΔccpN (+Glc) | GRF | F | |
| pACT87 Bsu | 2282 ± 171 | 205 ± 39 | 11.1 | 2181 ± 146 | 1453 ± 156 | 1.5 | 1.0 |
| pACS54 Bli | 1743 ± 34 | 129 ± 15 13 | 13.6 | 1687 ± 180 | 961 ± 95 | 1.8 | 1.0 |
| pACS55 Bam | 1567 ± 65 | 77 ± 19 | 20.4 | 1584 ± 111 | 983 ± 105 | 1.0 | 1.0 |
| pACS56 Bth | 4169 ± 88 | 428 ± 23 | 9.7 | 4224 ± 32 | 3736 ± 71 | 1.0 | 1.0 |
| pACS57 Bpu | 1785 ± 40 | 158 ± 23 | 11.3 | 2510 ± 139 | 2020 ± 195 | 1.2 | 1.4 |
| pACS58 Bha | 216 ± 19 | 22 ± 3 | 9.7 | 998 ± 127 | 526 ± 54 | 1.9 | 4.6 |
| pACS59 Bme | 223 ± 21,5 | 30 ± 14 | 7.4 | 809 ± 80 | 512 ± 76 | 1.6 | 3.6 |
| pACS60 Ban | 2341 ± 117 | 146 ± 22 | 16.3 | 2440 ± 126 | 2315 ± 38 | 1.1 | 1.0 |
| pACS61 Gka | 1714 ± 113 | 160 ± 36 | 10.7 | 1681 ± 106 | 1350 ± 21 | 1.2 | 1.0 |
| pACS62 Bsu C | 2300 ± 57 | 206 ± 11 | 11.2 | 2135 ± 18 | 1687 ± 76 | 1.3 | 0.9 |
| pACS63 Bsu P | 2196 ± 74 | 1590 ± 71 | 1.4 | 2042 ± 79 | 1496 ± 127 | 1.4 | 0.9 |
| pACS64 Bha C | 270 ± 14 | 58 ± 18 | 4.7 | 1100 ± 44 | 666 ± 10 | 1.7 | 4.0 |
| pACS65 Bha P | 1934 ± 1 | 1182 ± 166 | 1.7 | 1895 ± 47 | 1491 ± 26 | 1.3 | 1.3 |
| pACS66 Bme C | 322 ± 2.1 | 49 ± 34 | 6.7 | 1052 ± 62 | 522 ± 36 | 2.0 | 3.3 |
| pACS67 Bme P | 1736 ± 80 | 1582 ± 7 | 2.0 | 1571 ± 77 | 1611 ± 43 | 1.0 | 0.9 |
Bsu, B. subtilis; Bli, B. licheniformis; Bam, B. amyloliquefaciens; Bth, B. thuringiensis; Bpu, B. pumilus; Bha, B. halodurans; Bme, B. megaterium; Ban, B. anthracis; Gka, Geob. kaustophilus. All pAC derivatives contain the promoter, both CcpN sites and about 67 more upstream bp. Cultures were grown in SP medium with 2% or without glucose. All values represent averages of at least three independent determinations with five transformants grown in parallel; GRF, glucose repression factor, the ratio between the values obtained without and with glucose in wild-type DB104 and DB104 (ΔccpN::phleo), respectively. F, ratio of the values obtained for ΔccpN and DB104 without glucose. A value >1 suggests a glucose-independent repression by CcpN. Lower part: C, pAC derivative comprises promoter and both CcpN BS, but no further upstream sequences; P, pAC derivative contains only the promoter and CcpN BS II, but lacks BS I (Figure 2).
To corroborate that indeed CcpN was responsible for the unanticipated results obtained with B. halodurans and B. megaterium, six additional pAC derivatives were constructed. Three of them contain only the psr1 region with CcpN BS II, but lack BS I upstream of the −35 box in B. subtilis (pACS63), B. halodurans (pACS65) and B. megaterium (pACS67). The other three contain the sr1 promoters with only the two CcpN BSs, but without further upstream sequences (pACS62, pACS64 and pACS66 for B. subtilis, B. halodurans and B. megaterium, respectively). The measured β-galactosidase activities (Table 1) indicate that the three promoters without CcpN BS I have almost the same strength (1500–2000 MU) as one would expect from the nearly identical sequences of their −35 and −10 boxes. However, as soon as two CcpN BSs were present, the promoter activity decreased to the values obtained with the longer constructs (Table 1) under non-glycolytic conditions. Although a certain degree of CcpN-dependent, glucose-independent repression was found in vivo at the gapB and pckA promoters in B. subtilis (32), and at all three promoters in vitro (20), we did not observe such a repression in vivo at psr1. Since the location of the CcpN BS is identical at all sr1 promoters in contrast to their location at the gapB and pckA promoters, where BS I overlaps the −10 box and BS II the region around +20, this cannot be the reason for the observed differences. A comparison of the nt sequence of CcpN BS I for all sr1 promoters analysed in this report reveals a difference in two apparently critical nt: HY of the consensus sequence TRTGHYATAYW is in all cases analysed experimentally TT, TC (in B. pumilus) or AT (in Geob. kaustophilus), whereas it is AC in B. megaterium and B. halodurans. Therefore, we hypothesize that the glucose-independent repression at these two promoters is due to a more efficient binding of CcpN at BS I. However, the 1.5-fold higher values in the ΔccpN strain (-glucose) in the absence of BS I compared to the presence of BS I suggest the additional binding of a yet unknown glucose-independent regulator in this region. The 1.7- and 2.0-fold glucose-dependent transcriptional repression also observed with these shorter fusions in the absence of both CcpN and any cre sites points to a supplementary small effect of an unknown glucose-dependent regulator (see above). Future research will be aimed at the identification of this factor.
Ability of 11 SR1P homologues to stabilize B. subtilis gapA RNA and to interact with B. subtilis GapA protein
As shown previously by NB, gapA operon mRNA is barely detectable in an SR1 knockout strain after growth in complex medium till onset of stationary phase (22). Inducible overexpression of wild-type SR1 encoding SR1P from a multicopy vector can complement this defect, leading to stabilization and, as a consequence, visualization of gapA mRNA. SR1P interacts with GapA protein, and this interaction stabilizes gapA mRNA by a hitherto unknown mechanism (22). To analyse the ability of SR1P homologues to stabilize B. subtilis gapA mRNA, sr1 homologues from B. amyloliquefaciens, B. licheniformis, B. pumilus, B. thuringiensis, B. anthracis, B. halodurans, B. megaterium and Geob. kaustophilus were inducibly overexpressed in B. subtilis (Δsr1::cat) under control of the tet operator. All SR1P homologues were tagged at the C-terminus with a Strep tag followed by the B. subtilis SR1 transcription terminator. In the case of B. thuringiensis, three plasmids were constructed, each of them encoding one of the three different peptides SR1/P1, P2 and P3 (Figure 5 and Supplementary Figure S6). Cells were grown in TY medium until onset of stationary phase, induced with anhydro-tetracycline for 15 min, harvested, and RNA was isolated and subjected to NB. In spite of their differences in the aa sequences, 10 of 11 SR1P homologues were able to complement the absence of B. subtilis SR1P (Figure 5A and D) i.e. to stabilize gapA mRNA. Only B. megaterium SR1P was not functional. The expression and gapA stabilizing function of the entire B. thuringiensis sr1 gene encoding the three peptides was analysed, too. It resulted in a ≈580 nt long sRNA in northern blots, which is the size of an unprocessed SR1 RNA (Figure 5A) and was as functional in complementation of the sr1 knockout strain as each single sr1p encoding region.
Figure 5.

Comparative analysis of SR1 homologues in northern blots and co-elution experiments. (A) Northern blots. Bacillus subtilis strains were grown under the appropriate conditions in TY medium (see Results section) until the onset of stationary phase, samples taken, total RNA prepared, treated with glyoxal, separated on 1.5% agarose gels, blotted onto nylon membrane and hybridized with a [α-32P] dATP-labelled DNA probe for B. subtilis gapA (three species of 1.2 kb, 2 kb and 6 kb) and reprobed with specific [α-32P] dATP-labelled DNA probes for the respective SR1 homologues. For the correction of loading errors, filters were reprobed with a [γ-32P]-ATP-labelled oligonucleotide specific for 5S rRNA (Supplementary Table S1). Wild type, B. subtilis strain DB104. All the other strains are DB104 (Δsr1::cat) containing either an empty vector or the vector with the corresponding sr1 gene. In the case of B. thuringiensis sr1, four vectors were analysed expressing the first, the second or the third copy of sr1p (I, II or III) or the entire sr1 gene with all three sr1p copies (I-III) (Supplementary Figures S5 and S6). The numbers below the gel indicate the relative amounts of gapA mRNA calculated by using the loading control (signal for 5S rRNA). (B) Co-elution experiments. Nine SR1P homologues were expressed with a C-terminal Strep tag (tag not shown in the sequence) from a tet-inducible promoter on the corresponding multicopy vector pWSR1/MX (Supplementary Table S2) in B. subtilis strain DB104 (Δsr1::cat). The ability of each SR1P homologue to tightly bind and co-elute B. subtilis GapA was investigated in a co-elution assay with a streptactin column as described previously (22). Plasmid pWSR1 expresses sr1 without Strep tag. Bsu mut expresses a mutated sr1 which is not functional in stabilization of gapA mRNA in northern blots. An aliquot of each elution fraction E3 was separated on a 17.5% SDS-Tris-glycine PAA gel along with purified GapA and a size marker (M) in kD and stained with Coomassie blue. (C)Western blot analysis of B. halodurans and B. megaterium SR1P flow-through, washing and elution fractions as an example for a very weak interaction with the heterologous B. subtilis GapA. Here, GapA is already visible in the washing fractions. (D) Overview of the complementation of B. subtilis Δsr1p by 10 different SR1P homologues. On the one hand, the ability of each SR1 homologue to complement a B. subtilis sr1 knockout strain by stabilizing the gapA mRNA was analysed by NB (see (A)). On the other hand, the ability of the SR1P homologues to interact with B. subtilis GapA was investigated by a co-elution assay (see (B)). +, complementation in northern blot/co-elution of B. subtilis GapA, −, no complementation/co-elution. Ba (B. anthracis) P1 is identical to Bth P1. (E) Overview of the complementation of B. subtilis Δsr1p by five B. subtilis SR1P mutants with substituted or lacking cysteine residues.
To investigate whether the SR1P homologues were able to interact with B. subtilis GapA, the strains were cultivated as above, induced, cells harvested, protein extracts prepared and loaded onto a streptactin column as described (22). Elution fractions were analysed on SDS-Tris-glycin-PAA gels for co-elution of B. subtilis GapA (Figure 5B). Interestingly, only in the presence of four SR1P homologues—B. subtilis, B. amyloliquefaciens and Geob. kaustophilus SR1P as well as B. thuringiensis SR1P/P1 (which is identical to P1 from B. anthracis)—B. subtilis GapA was co-eluted (Figure 5B). In the other cases, GapA was detected in the washing fractions by western blotting (as shown for B. halodurans and B. megaterium SR1P (Figure 5C) indicating that its interaction with the heterologous SR1P was weaker than with B. subtilis SR1P. Apparently, with the exception of B. megaterium SR1P, even the weak interactions were sufficient for a stabilizing effect on B. subtilis gapA mRNA in vivo as detected in the northern blots. The overview of the co-elution data in conjunction with the corresponding SR1 peptide sequences (Figure 5D) reveals that two regions of SR1P differ markedly in those peptides that were impaired in GapA binding: the EAIHY region (aa 11–15) and the VTTLY (aa 21–25) region, which are separated by the highly conserved FEDEK motif. As we have shown previously, the highly heterogenous C-terminus is not important for SR1P function, as the C-terminal 9 aa of B. subtilis SR1P can even be deleted without functional consequences (22). The highly divergent regions might contain candidate aa for the interaction surface with GapA. An alignment of the aa sequences of the corresponding 23 GapA proteins is shown in Supplementary Figure S10. All GapA proteins display between 85% and 97% identity on aa level with B. subtilis GapA, except for the B. cereus group members that show 80% identity.
Role of Cysteine residues in SR1P
To analyse the importance of the six cysteine residues for the ability of SR1P to stabilize gapA mRNA, mutants M2 (C6T), M3 (D8L, C9T), M7 (C28S, C29S), M32 (C6S, C9S) and M54 (C28S, C29S and Δ31-39) were analysed in NB. The results with mutants M7 and M54 (Figure 5E) show that the four C-terminal cysteine residues C28, C29, C32 and C34 are not necessary for the function of SR1P in gapA stabilization. However, whereas a single substitution of C6 nor C9 could be tolerated (M2, M3), a double substitution (M32) abolished the functionality of SR1P. This indicates that at least one of the N-terminal cysteine residues—C6 or C9—is required. Additionally, to rule out that the six cysteine residues in wild-type SR1P form a Zn finger motif that coordinates Zn2+, we analysed the functionality of SR1P in the presence of EDTA, a chelator for divalent cations. To this end, three co-elution experiments were performed in parallel with crude protein extracts from DB104 (Δsr1:cat, pWSR1/M25) in the presence of either 50 mM EDTA, 0 mM EDTA or 1 mM ZnCl2. The chelator and ZnCl2 were added after sonication and before loading the extracts onto the streptactin columns. No differences between the amounts of co-eluted GapA were observed (not shown), indicating that SR1P is not a Zn finger protein.
CONCLUSIONS
So far, only a few studies have been published that deal with the computational search for sRNA homologues and their experimental investigation. These concern sRNAs restricted to Gram-negative bacteria, mostly enterobacteriaceae: SgrS (30, 33), GlmY/GlmZ (34) and GcvB (35).
Here, we present the first analysis for an sRNA from Gram-positive bacteria. We identified 23 homologues of the dual-function sRNA SR1 in Bacillus, Geobacillus, Anoxybacillus and Brevibacillus species. No homologues were found in other bacteria, neither Gram-positive nor Gram-negative ones. Hence, SR1 originated between 0.9 and 1.3 billion years ago as this is the timescale in which the species encoding SR1 diverged (36). The expression of all sr1 homologous genes is under control of transcriptional repressor CcpN, which binds at nearly the same positions at all sr1 promoters. Although the promoters of the tested nine homologues were active and all but those of B. halodurans and B. megaterium were of similar strength in B. subtilis, SR1 could not be detected in total RNA isolates from B. pumilus. This suggests that a still unknown, species-specific regulator might control sr1 expression in B. pumilus. Furthermore, a so far unprecedented glucose-independent 3–4-fold repression of sr1 transcription by CcpN was observed in B. halodurans and B. megaterium. Interestingly, different contributions of σ54 and σ70 and a two-component system (TCS) to the transcription of GlmY/Z homologues from different species were found (31). Thus, other regulatory principles might be also used for sr1 homologues, which were not included in this experimental study.
All homologues encode both highly similar SR1P peptide homologues of 37–42 aa and contain 7–11 short regions complementary to the cognate coding regions of ahrC/arg. On primary sequence level, the peptide-encoding regions are more conserved than the base-pairing regions. However, on structural level, the location and length of the complementary regions as well as their ability to base pair with ahrC mRNA, is also conserved (Figure 3 and Supplementary Figures S2, S5 and S8). As previously found in B. subtilis, the G/G′ base pairing is sufficient in most cases to initiate binding between SR1 and ahrC mRNA (for a model, see Supplementary Figure S11). This suggests that the inhibition of ahrC/arg translation is also conserved. An interference of SR1P synthesis and SR1/ahrC mRNA base pairing cannot be entirely excluded, although the translational efficiency of sr1p is very low, and the regions decisive for the initial interaction are located downstream of the sr1p stop codon.
Our results are in contrast to those of Horler and Vanderpool (30) who found that the base-pairing function of SgrS is more conserved than the peptide-encoding function. The species that encode SgrS/SgrR diverged between 0.4 and 0.7 billion years ago (36). Recently, a functional homologue of SgrS, TarA, which regulates ptsG was discovered in Vibrio cholerae. In contrast to SgrS, TarA is smaller (100 nt versus 227 nt), does not contain the 43 aa SgrT ORF and is not downregulated by SgrR but upregulated by transcription activator ToxT (37). TarA escaped the computational approach by Horler and Vanderpool, because they used as search criteria the neighbourhood of sgrS and sgrR encoding its E. coli regulator, which is conserved in a variety of Gram-negative bacteria. Presently, we cannot exclude that in non-related bacteria SR1 functional homologues exist that are either controlled by other transcription factors or whose SR1P sequences differ significantly from those analysed here. Since CcpN is encoded in a number of firmicutes (32), among them Staphylococcus aureus, Enterococcus faecalis or L. monocytogenes, we used S. aureus as an example to search for peptides comprising 20–50 aa in the vicinity of whose encoding genes/promoters is at least one CcpN BS with no more than one deviation from the B. subtilis consensus. However, none of the peptides found by this search showed any similarity on aa level to SR1P. The same was true for all other peptides of this size range predicted in S. aureus with our programme. This is not too surprising, because the GapA proteins of B. subtilis and S. aureus share only 53% aa identity compared to at least 80% identity between the GapA proteins of the 23 analysed Bacilli. Given that all these peptide homologues must bind their homologous GapA proteins, it is difficult to imagine a high similarity on aa level between peptides of less-related species. However, the discovery of 23 homologous SR1P will contribute to the elucidation of interacting regions/residues between SR1P and GapA. Once this region is identified, new bio-computational tools can be developed to search for functional SR1P homologues in distantly related bacteria.
Strikingly, all B. cereus group members, which contain more than one sr1p copy, are pathogens. Why do all these pathogens contain two or three sr1p copies? Recently, it has been proposed by Papenfort et al. that conserved sRNAs with seemingly unrelated functions constitute a reservoir of regulators that act to tame foreign genes and to integrate them into existing regulatory networks (38). Alternatively, the pool of already existing regulators might be expanded in order to regulate virulence genes. It is tempting to speculate that the two duplications of sr1p in the B. cereus group are such an enlargement that might allow these pathogens to adapt to the adverse environment of their hosts.
In summary, there are many commonalities between the 23 SR1 homologues showing that the two functions have been remarkably conserved during 0.9–1.3 billion years of evolution. This timescale is much longer than the time of conservation for SgrS for which only one of the two functions, the base-pairing function, was maintained during the whole evolution of this sRNA. This demonstrates the importance of SR1. Nevertheless, there are some interesting differences between the members of the SR1 family in transcriptional regulation as well as the gene duplications in the pathogenic B. cereus group, which are subjects for further research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3 and Supplementary Figures 1–11.
FUNDING
Deutsche Forschungsgemeinschaft [Br1552/6-3 to S.B.]. Funding for open access charge: Deutsche Forschungsgemeinschaft DFG.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank S. Pohl (Newcastle), who performed the two PCR reactions from the B. anthracis sr1 upstream and coding regions and sent us the gel-purified fragments. We are grateful to R. Borriss (Berlin), G. Steinborn (Gatersleben), R. Biedendieck (Braunschweig), M. Miethke (Marburg), and S. Graf (Mainz) for sending us the B. amyloliquefaciens FZB42, B. licheniformis ATCC14580, B. megaterium DSM319, B. halodurans DSM497 and Geob. kaustophilus DSM7263 strains, respectively.
REFERENCES
- 1.Gottesman S, Storz G. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a003798. pii: a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brantl S. Bacterial chromosome-encoded small regulatory RNAs. Future Microbiol. 2009;4:85–103. doi: 10.2217/17460913.4.1.85. [DOI] [PubMed] [Google Scholar]
- 3.Irnov K, Sharma CM, Vogel J, Winkler WC. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res. 2010;38:6637–6651. doi: 10.1093/nar/gkq454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen S, Nielsen HB, Jarmer H. Transcriptionally active regions in the genome of Bacillus subtilis. Mol. Microbiol. 2009;73:1043–1057. doi: 10.1111/j.1365-2958.2009.06830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, et al. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 6.Geissmann T, Chevalier C, Cros M-J, Boisset S, Fechter P, Noirot C, Schrenzel J, François P, Vandenesch F, Gaspin C, et al. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res. 2009;37:7239–7257. doi: 10.1093/nar/gkp668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohn C, Rigoulay C, Chabelskaya S, Sharma CM, Marchais A, Skorski P, Borezée-Durant E, Barbet R, Jacquet E, Jacq A, et al. Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res. 2010;38:6620–6636. doi: 10.1093/nar/gkq462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brantl S. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr. Op. Microbiol. 2007;10:102–109. doi: 10.1016/j.mib.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Morfeldt E, Taylor D, von Gabain A, Arvidson S. Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J. 1995;14:4569–4577. doi: 10.1002/j.1460-2075.1995.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 2007;21:1353–1366. doi: 10.1101/gad.423507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mangold M, Siller M, Roppenser B, Vlamincks BJ, Penfound TA, Klein R, Novak R, Novick RP, Charpentier E. Synthesis of group A streptococcal virulence factors is controlled by a regulatory RNA molecule. Mol. Microbiol. 2004;53:1515–1527. doi: 10.1111/j.1365-2958.2004.04222.x. [DOI] [PubMed] [Google Scholar]
- 12.Wadler CS, Vanderpool CK. A dual function for a bacterial small RNA: SgrS performs base-pairing dependent regulation and encodes a functional polypeptide. Proc. Natl Acad. Sci. USA. 2007;104:20454–20459. doi: 10.1073/pnas.0708102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu T, Yaguchi H, Ohtani K, Banu S, Hayasi H. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol. Microbiol. 2002;43:257–265. doi: 10.1046/j.1365-2958.2002.02743.x. [DOI] [PubMed] [Google Scholar]
- 14.Sonnleitner E, Gonzalez N, Sorger-Domenigg T, Heeb S, Richter AS, Backofen R, Williams P, Hüttenhofer A, Haas D, Bläsi U. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 2011;80:868–885. doi: 10.1111/j.1365-2958.2011.07620.x. [DOI] [PubMed] [Google Scholar]
- 15.Roberts SA, Scott JR. RivR and the small RNA RivX: the missing links between the CovR regulatory cascade and the Mga regulon. Mol. Microbiol. 2007;66:1506–1522. doi: 10.1111/j.1365-2958.2007.06015.x. [DOI] [PubMed] [Google Scholar]
- 16.Berghoff B, Glaeser J, Sharma CM, Vogel J, Klug G. Photooxidative stress-induced and abundant small RNAs in Rhodobacter sphaeroides. Mol. Microbiol. 2009;74:1497–1512. doi: 10.1111/j.1365-2958.2009.06949.x. [DOI] [PubMed] [Google Scholar]
- 17.Licht A, Preis S, Brantl S. Implication of CcpN in the regulation of a novel untranslated RNA (SR1) in B. subtilis. Mol. Microbiol. 2005;58:189–206. doi: 10.1111/j.1365-2958.2005.04810.x. [DOI] [PubMed] [Google Scholar]
- 18.Heidrich N, Chinali A, Gerth U, Brantl S. The small untranslated RNA SR1 from the B. subtilis genome is involved in the regulation of arginine catabolism. Mol. Microbiol. 2006;62:520–536. doi: 10.1111/j.1365-2958.2006.05384.x. [DOI] [PubMed] [Google Scholar]
- 19.Heidrich N, Moll I, Brantl S. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res. 2007;35:4331–4346. doi: 10.1093/nar/gkm439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Licht A, Golbik R, Brantl S. Identification of ligands affecting the activity of the transcriptional repressor CcpN from Bacillus subtilis. J. Mol. Biol. 2008;380:17–30. doi: 10.1016/j.jmb.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Licht A, Brantl S. The transcriptional repressor CcpN from Bacillus subtilis uses different repression mechanism at different promoters. J. Biol. Chem. 2009;284:30032–30038. doi: 10.1074/jbc.M109.033076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gimpel M, Heidrich N, Mäder U, Krügel H, Brantl S. A dual-function sRNA from B. subtilis: SR1 acts as a peptide encoding mRNA on the gapA operon. Mol. Microbiol. 2010;76:990–1009. doi: 10.1111/j.1365-2958.2010.07158.x. [DOI] [PubMed] [Google Scholar]
- 23.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2984. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 24.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- 26.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 27.Eckart RA, Brantl S, Licht A. Search for additional targets of the transcriptional regulator CcpN from Bacillus subtilis. FEMS Microbiol. Lett. 2009;299:223–231. doi: 10.1111/j.1574-6968.2009.01754.x. [DOI] [PubMed] [Google Scholar]
- 28.Kawamura F, Doi RH. Construction of a Bacillus subtilis double mutant deficient in extracellular alkaline and neutral proteases. J. Bacteriol. 1984;160:442–444. doi: 10.1128/jb.160.1.442-444.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brantl S. The copR gene product of plasmid pIP501 acts as a transcriptional repressor at the essential repR promoter. Mol. Microbiol. 1994;14:473–483. doi: 10.1111/j.1365-2958.1994.tb02182.x. [DOI] [PubMed] [Google Scholar]
- 30.Horler RSP, Vanderpool CK. Homologs of the small RNA SgrS are broadly distributed in enteric bacteria but have diverged in size and sequence. Nucleic Acids Res. 2009;37:5465–5476. doi: 10.1093/nar/gkp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stülke J, Hillen W. Regulation of carbon catabolism in Bacillus species. Annu. Rev. Microbiol. 2000;54:849–880. doi: 10.1146/annurev.micro.54.1.849. [DOI] [PubMed] [Google Scholar]
- 32.Servant P, Le Coq D, Aymerich S. CcpN (YqzB), a novel regulator for CcpA-independent catabolite repression of Bacillus subtilis gluconeogenic genes. Mol. Microbiol. 2005;55:1435–1451. doi: 10.1111/j.1365-2958.2005.04473.x. [DOI] [PubMed] [Google Scholar]
- 33.Wadler CS, Vanderpool CK. Characterization of homologs of the small RNA SgrS reveals diversity in function. Nucleic Acids Res. 2009;37:5477–5485. doi: 10.1093/nar/gkp591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Göpel Y, Lüttmann D, Heroven AK, Reichenbach G, Dersch P, Görke B. Common and divergent features in transcriptional control of the homologous small RNAs GlmY and GlmZ in Enterobacteriaceae. Nucleic Acids Res. 2011;39:1294–1309. doi: 10.1093/nar/gkq986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma C, Darfeuille F, Plantinga T, Vogel J. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007;21:2804–2817. doi: 10.1101/gad.447207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Battistuzzi FU, Feijao A, Hedges SB. A genomic timescale of prokaryote evolution: insight into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evol. Biol. 2004;4:44. doi: 10.1186/1471-2148-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richard AL, Withey JH, Beyhan S, Yildiz F, DiRita VJ. The Vibrio cholerae virulence regulatory cascade controls glucose uptake through activation of TarA, a small regulatory RNA. Mol. Microbiol. 2010;78:1171–1181. doi: 10.1111/j.1365-2958.2010.07397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papenfort K, Podkaminski D, Hinton JC, Vogel J. The ancestral SgrS RNA discriminates horizontally acquired Salmonella mRNAs through a single G-U wobble pair. Proc. Natl Acad. Sci. USA. 2012;109:E757–E764. doi: 10.1073/pnas.1119414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.