

The plant-derived maytansinoids 4[1] and their microbial counterparts, the ansamitocins P-2 to P-4 5-7 (Figure 1) [2] exert strong in vitro and in vivo antitumor activity, blocking the assembly of tubulin into functional microtubules.[3] The biosynthesis of the ansamitocins involves the construction of the carbon framework on a type I modular polyketide synthase (PKS)[4] from 3-amino-5-hydroxybenzoic acid 1 (AHBA)[5] via chain extension by one “glycolate”, three propionate and three acetate units. The last PKS module holds the seco-proansamitocin 2a which is released and cyclized, presumably by an ansamycin amide synthase (gene asm9),[6] to yield the 19-membered macrocyclic lactam, proansamitocin 3 (Scheme 1).[7] The latter enzyme is particularly interesting because the corresponding chemical macrolactamizations are known to be challenging in part due to the lack of nucleophilicity of the aniline moiety.[7-9] The amide synthases are not part of the PKS.[10] but are separate enzymes with homology to arylamine acyl transferases.[4,11,12]
Scheme 1.
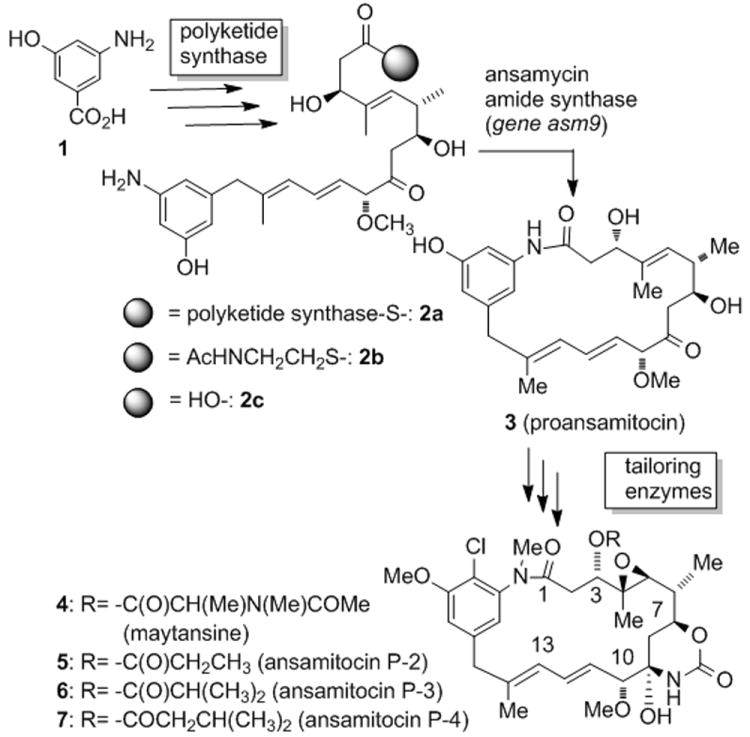
Biosynthesis of ansamitocins 5 - 7 via seco-proansamitocin 2a and proansamitocin 3 and comparison to maytansine 4.
We have studied the substrate specificity of the ansamitocin biosynthetic machinery[13,14] using a mutant strain (HGF073) of Actinosynnema pretiosum blocked in the biosynthesis of the PKS starter unit AHBA 1.[7] In mutasynthetic approaches we could show that the PKS, the amide synthase and the tailoring enzymes accept several substrates that are modified in the aromatic moiety of the biosynthetically advancing ansamitocins. As the amide synthase is supposed to utilize PKS-bound seco-proansamitocin 2a as substrate it is unclear whether simple thioester analogues or free caboxylic acids can also act as substrates in mutasbiosyntheses. Acceptance of very advanced biosynthetic intermediates and analogues would be highly desirable because it would allow to bypass the “conservative” PKS with modified substrates that the PKS machinery has difficulty to process. In addition, this approach would also shed light on mechanistic aspects of the amide synthase and on its substrate specificity. So far, the difficulty to access potential substrates of the amide synthase has been a major hurdle in this research. Although it is a time consuming task, total synthesis is a reliable strategy including access to analogues.
We have previously reported the total synthesis of N-acetylcysteamine-thioester (SNAC-ester) 2b.13 In the present work we describe the synthesis of seco-acid 2c and of the SNAC-ester of the new seco-proansamitocin derivative 16, as well as the utilization of all three advanced substrates in conversion experiments with A. pretiosum mutant HGF073.
SNAC ester 2b (2.3 mg, 4.1 μmol) was added to a culture (50 mL) of A. pretiosum strain HGF073 (Scheme 2).[15] The culture was harvested after seven days and extracts were analyzed by UPLC-MS (ultra performance LC coupled ESI-MS). The spectra of the biosynthetic samples were compared with those of authentic AP-3 6 and found to be identical, with a parent ion at m/z 657 (M+Na)+ and collision-induced fragmentation giving daughter ion spectra with a base peak at m/z 569 (from m/z 657) due to loss of the ester function at C3. In earlier studies, we unequivocally could exclude background synthesis of AP-3 with blocked mutant HGF073.[17] The yield of AP-3 formed from SNAC ester 2b was 0.2 % based on quantitation by UV absorption at λ=248 nm after preparative HPLC purification.[16] That equals 2% of the incorporation seen with AHBA-supplementation (ca. 80 mg/L).
Scheme 2.
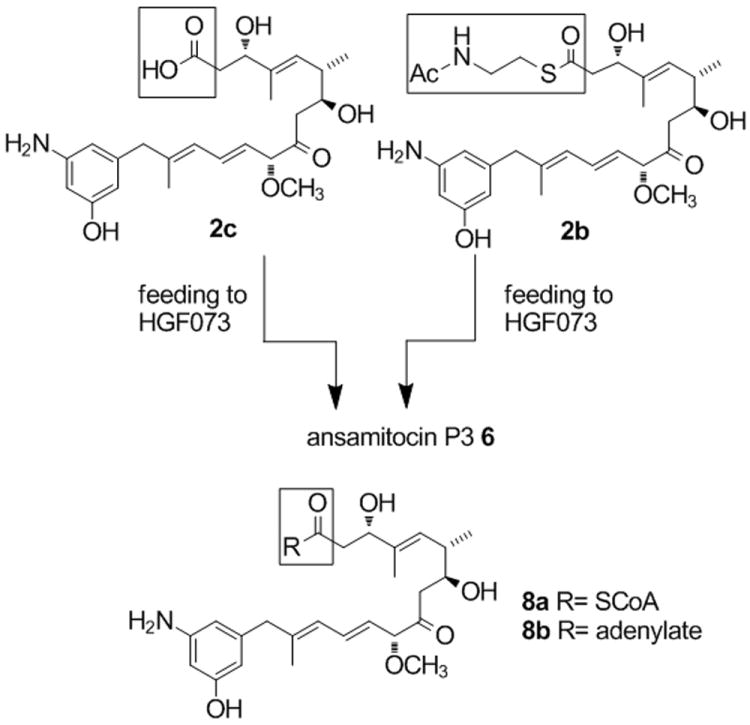
Feeding experiments with seco-proansamitocin 2c and SNAC-ester 2b yielding ansamitocin P3 6.
The SNAC ester 2b may have been accepted as substrate by the amide synthase directly or alternatively, 2b was first loaded onto the last PKS module by transesterification before macro-lactamization occurs. Surprisingly, seco-proansamitocin 2c (0.4 mg, 0.9 μmol), prepared from a published advanced synthetic precursor 9[13] via an established sequence (Scheme 3)[18], also gave AP-3 6 in 1.5 % yield compared to AHBA-supplementation (0.13 % absolute yield), when incubated with strain HGF073 of A. pretiosum (50 mL). Macrolactamization of 2c can only occur after activation of the carboxylate, e.g. to the CoA-ester 8a or the adenylate 8b. The amide synthase then promotes macrolactamization either directly or following transfer onto the final PKS module.
Scheme 3.
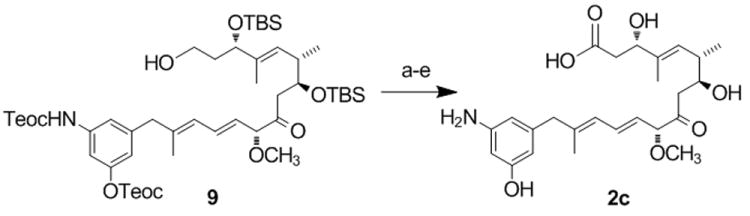
Synthesis of seco-proansamitocin 2c.[a]
[a] Reagents and conditions: (a) 9[13] DMSO, (COCl)2, -60 °C, 1 h then Et3N, -40°C (75%); (b) NaClO2, NaH2PO4 H2O, t-BuOH, 2-methyl-2-butene, 0 °C to rt, 30 min (86%); (c) HF-pyr, THF, rt, 6h, (48 %); (d) ZnCl2, MeNO2, ultrasound, rt, 1 h, (47%) (TBS= tert.-butyldimethylsilyl, Teoc= trimethylsilylethoxycarbonyl).
As a second complex substrate, we prepared the SNAC ester of 20-deoxy-seco-proansamitocin 16 which was chosen because we had observed that supplementing a culture of HGF073 with aminobenzoic acid 19 (260 mg, 1.5 mmol) unexpectedly yielded the corresponding ansamitocin derivative 18 in a very low yield as determined by UPLC-MS (Scheme 5).[19] The total synthesis approach relates to the successful preparation of SNAC ester 2b (Scheme 4).[13] Thus, starting from benzyl alcohol 10, intermediate propargyl aniline 12 was prepared via the N-Boc protected benzyl bromide which was subjected to a palladium-catalyzed cross coupling with alkinylindium derivative 11 (Scheme 3).[20] After desilylation, carboalumination,[21] during which the Boc-group was also removed, followed by Teoc protection yielded vinyl iodide 13 in good yield and with good stereocontrol.
Scheme 5.
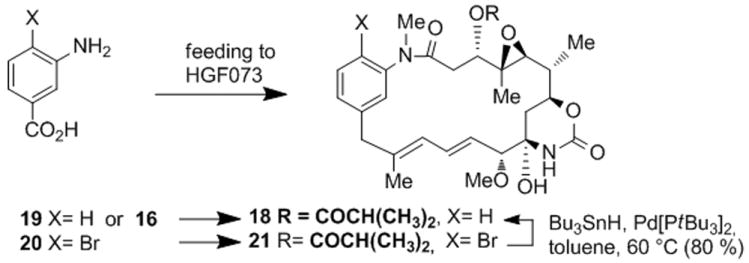
Preparation of deschloro-desmethoxy AP-3 18.
Scheme 4.
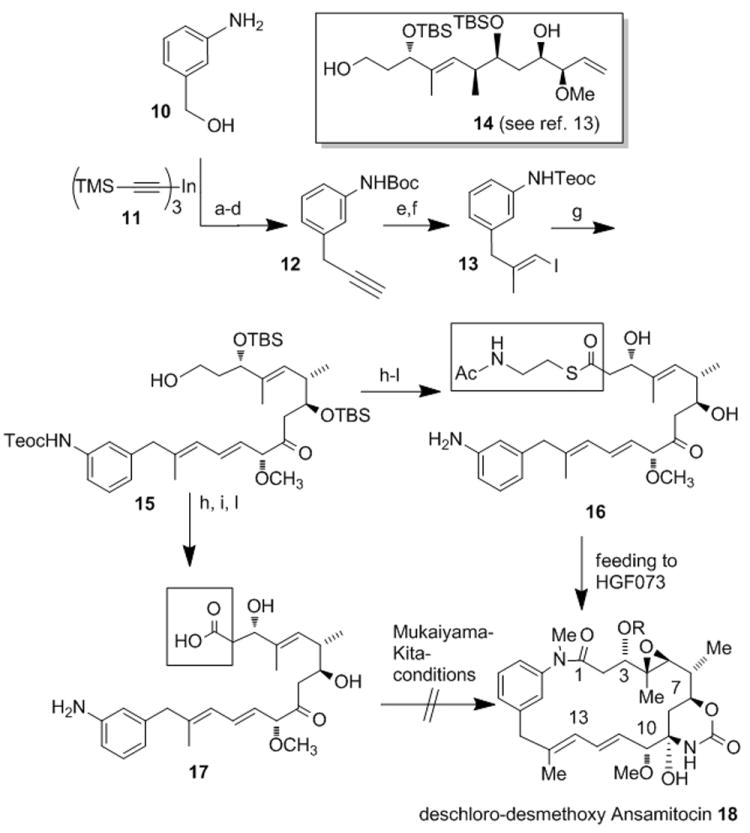
Total synthesis of SNAC ester 16 and seco acid 17[a] and feeding experiments with 16 towards deschloro-desmethoxy AP3 18.
[a] Reagents and conditions: (a) Boc2O, Et3N, dioxane/ H2O (2.5:1), rt, 12 h, (82 %); (b) PPh3, CBr4, CH2Cl2, rt, 1 h (83 %); (c) Pd(dppf)Cl2, THF, 65 °C, 4 h (98%); (d) TBAF*3H2O, THF, -20 °C, 1 h (88%); (e) i. AlMe3, Cp2ZrCl2, (CH2Cl)2, rt, 1 h, then addition of 11[13] at 0°C rt, 72 h, ii. I2, THF, -30 °C to 0 °C, 1.5 h (53%); (f) TeocCl, NaHCO3, CH2Cl2, rt, 10 min (91%); (g) 14, Pd(OAc)2, CsCO3, Bu4NBr, NEt3, DMF, rt, 2 h (68 %); (h) DMSO, (COCl)2, -60 °C, 1 h then Et3N, -40°C (90%); (i) NaClO2, NaH2PO4 H2O, t-BuOH, 2-methyl-2-butene, 0 °C to rt, 30 min (85%); (j) AcHN(CH2)2SH, DIC, DMAP, CH2Cl2, rt, 2 h (56%); (k) HF*pyr (70% HF) / THF, rt, 24 h (62 %); (l) ZnCl2, MeNO2, ultrasound, rt, 80 min (63 %; towards 17: 11%). (TMS= trimethylsilyl Boc= tert-butyloxycarbonyl; dppf= bis(diphenylphosphino) ferrocene, TBAF= tetra-n-butylammonium fluoride; DIC= diisopropyl carbodiimide, DMAP= 4-dimethylamino pyridine
In the following, the Heck reaction allowed to merge vinyl iodide 13 and complex alkene 14[13] under Jeffery conditions[22] furnishing the coupling product 15 in 68% yield. Simultaneous oxidation of both hydroxy groups and further oxidation of the aldehyde moiety to the carboxylic acid was followed by SNAC-ester formation. Finally, the silyl ethers were removed by treatment with the HF*pyr complex and deprotection of the Teoc-group was achieved with ZnCl2 under ultrasound conditions to yield the desired SNAC ester 16. Standard functional group manipulations led to seco acid derivative 17 using alcohol 15 as branching point. Interestingly, we were unable to chemically cyclize seco-proansamitocin 17 under various conditions which for example included the Mukaiyama (N-methyl-2-chloropyridinium iodide, Et3N)[23] and Kita (ethoxyacetylene, cat. [RuCl2(p-cymene)]2[24] protocols. The difficulty of smoothly achieving macrolactamization in ansamycin synthesis has been encountered before for which the reduced nucleophilicity of the arylamino group is likely responsible.[8]
In contrast, when SNAC-ester 16 (1.2 mg, 2.2 μmol) was incubated with HGF073, the new deschloro-desmethoxy AP-3 18 (14 μg, 1.1 %) was formed. Again, UPLC-MS served to identify the metabolite revealing the parent ion at m/z 571 (M+H)+ and collision-induced fragmentation giving daughter ion spectra with a base peak at m/z 483 (from m/z 571). In order to confirm the structure of 18, we conducted another mutational biosynthesis with Actinosynnema pretiosum HGF073 using bromoaminobenzoic acid 20 as mutasynthon, which yielded 19-bromo-20-demethoxy AP-3 21[15] in good yield (up to 15 mg/L). Pd-catalyzed debromination finalized the alternative synthesis of 19-deschloro-20-demethoxy AP-3 18. The product was identical in all respects with the fermentation product collected from feeding SNAC ester 16. The semisynthetic material also served for quantifying the feeding experiment with SNAC-ester 16. This was achieved with an authentic standard utilizing diagnostic UV absorption at λ = 248 nm.[16] The degree of incorporation for all three seco-acid derivatives is rather low and in magnitude is comparable with incorporation yields of tri- or tetraketide PKS intermediates.[17] This does not necessarily mean that their further processing is inefficient. In fact, we encountered severe stability problems with all three open chain substrates 2b, 2c and 16 during the last steps of total synthesis. These included dehydration (at C2-C3), retro aldol reaction (at C3) and double bond isomerization (C10 to C14), just to name those we could clearly identify. Obviously, only when cyclization has occurred undesired degradations are suppressed.[25] Therefore, it cannot be excluded that substantial amounts of the free seco-acid derivatives decomposed during the seven days in the fermentation broth.
The new AP-3 derivative 18[26] was tested for its inhibitory effects on the proliferation of different cancer cell lines in comparison to AP-3 6. It showed very strong activity in the pg/ml range against all cancer cell lines tested. In the case of cervix and prostate carcinoma 18 had an even higher activity than AP-3 6.
These results demonstrate for the first time that open chain seco acid precursors can in principal be cyclized by A. pretiosum when a mutant blocked in AHBA-biosynthesis is utilized. We also showed that not only activated thioesters are cyclized but also the free carboxylic acid can be accepted and is transformed into the corresponding macrolactam. Overexpression and isolation of the amide synthase should pave the way to study its mechanism in detail and will allow synthetic chemists to explore the use of the enzyme for preparative macrolactamizations using other long chain amino acids as substrates. Such investigations are currently pursued in our laboratories.
Table 1.
Antiproliferative activity, IC50 [ng/mL], of 18 compared to 6 with different cancer cell lines.
| Cell line | ||||||
|---|---|---|---|---|---|---|
| KB-3-1 Cervix carc. | U-937 Lymp-homa | PC-3 Prostate carc. | SK-OV-3 Ovarian carc. | A-431 Epiderm carc. | A-549 Lung carc. | |
| 18 | 0.022 | 0.015 | 0.020 | 0.040 | 0.080 | 0.095 |
| 6[a] | 0.11 | 0.0035 | 0.035 | 0.030 | 0.050 | 0.095 |
The IC50 with primary human fibroblasts from foreskin was 3.0 ng/ml.
Acknowledgments
We thank the US National Institutes of Health (grant CA 76461), the Deutsche Forschungs= gemeinschaft (grant Ki 397 / 13-1) and the Fonds der Chemischen Industrie for financial support. We are grateful to T. Knobloch for helpful discussions. We thank M. Quitschalle and Dr. T. Frenzel for expert synthetic support.
Footnotes
Supporting Information Available: Synthesis and spectra of new compounds, protocols of feeding experiments and their analysis.
Supporting information for this article is available on the WWW under http://www.chembiochem org or from the author.
References
- 1.a) Kupchan SM, Komoda Y, Court WA, Thomas GJ, Smith RM, Karim A, Gilmore CJ, Haltiwanger RC, Bryan RF. J Am Chem Soc. 1972;94:1354–1356. doi: 10.1021/ja00759a054. [DOI] [PubMed] [Google Scholar]; b) Kupchan SM, Komoda Y, Branfman AR, Sneden AT, Court WA, Thomas GJ, Hintz HPJ, Smith RM, Karim A, Howie GA, Verma AK, Nagao Y, Dailey RG, Jr, Zimmerly VA, Sumner WC., Jr J Org Chem. 1977;42:2349–2357. doi: 10.1021/jo00434a001. [DOI] [PubMed] [Google Scholar]; c) Bryan RF, Gilmore CJ, Haltiwanger RC. J Chem Soc Perkin II. 1973:897–901. [Google Scholar]
- 2.Higashide E, Asai M, Ootsu K, Tanida S, Kozai Y, Hasegawa T, Kishi T, Sugino Y, Yoneda M. Nature. 1977;270:721–722. doi: 10.1038/270721a0. [DOI] [PubMed] [Google Scholar]
- 3.Reviews: Kirschning A, Harmrolfs K, Knobloch T. C R Chimie. 2008:1523–1543.; Cassady JM, Chan KK, Floss HG, Leistner E. Chem Pharm Bull. 2004;52:1–26. doi: 10.1248/cpb.52.1.
- 4.Yu T-W, Bai L, Clade D, Hoffmann D, Toelzer S, Trinh KQ, Xu J, Moss SJ, Leistner E, Floss HG. Proc Nat Acad Sci U S A. 2002;99:7968–7973. doi: 10.1073/pnas.092697199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hatano K, Akiyama S-L, Asai M, Rickards RW. J Antibiot. 1982;35:1415–1417. doi: 10.7164/antibiotics.35.1415. [DOI] [PubMed] [Google Scholar]
- 6.Spiteller P, Bai L, Shang G, Carroll BJ, Yu T-W, Floss HG. J Am Chem Soc. 2003;125:14236–14237. doi: 10.1021/ja038166y. [DOI] [PubMed] [Google Scholar]
- 7.Meyer A, Brünjes M, Taft F, Frenzel T, Sasse F, Kirschning A. Org Lett. 2007;9:1489–1492. doi: 10.1021/ol0702270. [DOI] [PubMed] [Google Scholar]
- 8.Kashin D, Meyer A, Wittenberg R, Schöning K-U, Kamlage S, Kirschning A. Synthesis. 2007:304–319. [Google Scholar]
- 9.Meyer A, Kirschning A. Synlett. 2007:1264–1268. [Google Scholar]
- 10.Pompeo F, Mushtaq A, Sim E. Protein Expr Purific. 2002;24:138–151. doi: 10.1006/prep.2001.1550. [DOI] [PubMed] [Google Scholar]
- 11.a) August PR, Tang L, Yoon YJ, Ning S, Müller R, Yu TW, Taylor M, Hoffmann D, Kim CG, Zhang X, Hutchinson CR, Floss HG. Chem Biol. 1998;5:69–79. doi: 10.1016/s1074-5521(98)90141-7. [DOI] [PubMed] [Google Scholar]; b) Rascher A, Hu Z, Viswanathan N, Schirmer A, Reid R, Nierman WC, Lewis M, Hutchinson CR. FEMS Microbiol Lett. 2003;218:223–230. doi: 10.1016/S0378-1097(02)01148-5. [DOI] [PubMed] [Google Scholar]
- 12.Chemically amide synthases perform analogous transformations to the thioesterases responsible for macrolactonization and –lactamization of polyketides and nonribosomal peptides (NRP): Kopp F, Marahiel MA. Nat Prod Rep. 2007;24:735–749. doi: 10.1039/b613652b.; Trauger JW, Kohli RM, Mootz HD, Marahiel MA, Walsh CT. Nature. 2000;407:215–218. doi: 10.1038/35025116.; Trauger JW, Kohli RM, Walsh CT. Biochemistry. 2001;40:7092–7098. doi: 10.1021/bi010035r.; Kohli RM, Trauger JW, Schwarzer D, Marahiel MA, Walsh CT. Biochemistry. 2000;40:7099–7108. doi: 10.1021/bi010036j.; Kohli RM, Takagi J, Walsh CT. Proc Natl Acad Sci U S A. 2002;99:1247–1252. doi: 10.1073/pnas.251668398.; Kohli R, Burke MD, Tao J, Walsh CT. J Am Chem Soc. 2003;125:7160–7161. doi: 10.1021/ja0352202.; Kohli RM, Walsh CT. Chem Commun. 2003:297–307. doi: 10.1039/b208333g.
- 13.Frenzel T, Brünjes M, Quitschalle M, Kirschning A. Org Lett. 2006;8:135–138. doi: 10.1021/ol052588q. [DOI] [PubMed] [Google Scholar]
- 14.Taft F, Brünjes M, Floss HG, Czempinski N, Grond S, Sasse F, Kirschning A. ChemBioChem. 2008;7:1057–1060. doi: 10.1002/cbic.200700742. [DOI] [PubMed] [Google Scholar]
- 15.Parallel fermentations were carried out with the wild-type strain and with mutant HGF073 supplemented with AHBA and without supplementation.
- 16.Details on quantifying the amount of fermentation products produced are given in the supporting information.
- 17.Taft F, Brünjes M, Knobloch T, Floss HG, Kirschning A. J Am Chem Soc. 2009;131:3812–3813. doi: 10.1021/ja8088923. [DOI] [PubMed] [Google Scholar]
- 18.For the details on the synthesis of 2c from 9 refer to the supporting information.
- 19.Formation of 18 was determined to be 0.5% in comparison to supplementation with AHBA (see also ref. [16]).
- 20.Pérez I, Pérez Sestelo J, Sarandeses LA. J Am Chem Soc. 2001;123:4155–4160. doi: 10.1021/ja004195m. [DOI] [PubMed] [Google Scholar]
- 21.a) Negishi E-i, Van Horn DE, Yoshida T. J Am Chem Soc. 1985;107:6639–6647. [Google Scholar]; b) Negishi E-i, Kondakov SY, Choueiry D, Kasai K, Takahashi T. J Am Chem Soc. 1996;118:9577–9588. [Google Scholar]
- 22.Jeffery T. Tetrahedron. 1996;52:10113–10130. [Google Scholar]
- 23.a) Saigo K, Usui M, Kikuchi K, Shimada E, Mukaiyama T. Bull Chem Soc Jpn. 1977;50:1863–1866. [Google Scholar]; b) Roush WR, Coffey DS, Madar DJ. J Am Chem Soc. 1997;119:11331–11332. [Google Scholar]
- 24.Kita Y, Maeda H, Omori K, Okuno T, Tamura Y. Synlett. 1993:273–274. [Google Scholar]
- 25.Indeed, one may argue that macrocyclization is not only a tool of Nature to create macrocycles that are able to adopt different defined conformations to suit the receptor’s requirements but also allows to chemically stabilize open chain polyketide metabolites.
- 26.Antiproliferative activities of 21 are described in reference [14].