

Abstract
The spacing of peptides away from a hydrogel matrix dramatically impacts their availability and subsequent interactions with cells. Peptides were synthesized with monodisperse poly(ethylene glycol) spacers of different lengths that separate the peptide from the monomeric functionality which reacts during hydrogel polymerization. Specifically, bioactive RGD ligands were conjugated to PEG5, PEG11 or PEG27 spacers via solid phase techniques and then functionalized with an acryloyl end group. These acryloyl-PEGx-RGD conjugates were then copolymerized with PEGDA to form an inert hydrogel network decorated with RGD ligands for cell interactions. As the PEG spacer length increases, the RGD concentration required to support cell attachment and spreading decreases. The competitive detachment of hTCEpi cells in the presence of soluble linear RGD also shows non-linear dependence on the PEG spacer length, as more cells remained attached and spread on gels functionalized with longer PEG-RGD conjugates in comparison to the shorter PEG-RGD conjugates. The strategy and synthetic techniques developed here allow for reproducible control over peptide-hydrogel spacing and peptide concentration, and may be extended for incorporation of multiple peptides and to other hydrogel platforms.
Introduction
Well-defined cell-interactive hydrogels are controlled platforms that often attempt to mimic specific aspects of the native extracellular matrix. Most commonly, bioactive ligands are immobilized within an inert network that prevents non-specific protein adsorption. The synthetic inert platform described herein to investigate cell-material interactions is a poly(ethylene glycol) (PEG) hydrogel, but other common synthetic hydrogels include poly(2-hydroxyethyl methacrylate) and polyacrylamides. Such inert materials provide a “blank slate”,1 ensuring cell behavior such as migration, proliferation and differentiation can be interpreted with respect to the presentation of a specific ligand. Defined biomaterials have applications in regenerative medicine and tissue engineering by supporting the rational design of instructive cues in materials that replace diseased or damaged tissue. Analogously, these materials can be used in in vitro studies to decode the cooperative and synergistic interactions of multiple cues on cell behavior. A current limitation of these well-defined biomaterials is that it is difficult to systematically control ligand presentation within these networks.
In order to exploit the advantageous properties of well-defined synthetic hydrogels and gain insights into cell-material interactions, it is important to present cell-interactive peptides in a controllable and adaptable manner. Common strategies to immobilize peptides in PEG hydrogels include Michael-type addition,2 copolymerization with post-synthetically modified peptides via activated esters,3 and thiol-ene click reactions.4 These strategies have enabled investigators to probe the effects of an individual peptide’s diverse functional chemistry on cell behavior in a bio-inert context. The controlled environment of the well-defined cell-interactive hydrogels provide a platform where often several factors can be regulated, including type of ligand, bioactivity of ligand, ligand concentration, and temporal and spatial ligand presentation.5,6 In existing synthetic approaches, however, it is inherently difficult to control incorporation efficiency, retain high purity of post-modified moieties and systematically vary peptide presentation, particularly the peptide-hydrogel spacing. Peptide presentation refers to the arrangement of RGD in the network and specifically the accessibility of RGD for cell interactions, including effects of steric interference from background material and the ligands proximity to the background network. Ligand accessibility has been investigated with self-assembled monolayer systems,7 on poly-acrylonitrile beads,8 and supramolecular nanofibers,9 while in hydrogel systems, ligands within 5 to 10 nanometres of the hydrogel surface are typically assumed to be accessible to the cells.3,10,11 Emerging applications of synthetic hydrogels that mimic the extracellular matrix require simple, adaptable chemistries that allow for the presentation of distinct peptide ligands with controlled density and spacing.
To explore the effect of peptide-hydrogel spacing on cell behavior, we selected a model system of PEG and the bioactive Arg-Gly-Asp (RGD)12 ligand. PEG hydrogels derivitized with RGD cell adhesion ligands have been used extensively throughout the biomaterial literature to explore cell-material interactions and shown to be biologically relevant for corneal epithelial cells.13 The spacing between the peptide and the hydrogel matrix, however, has not been systematically explored to date and may be a factor overlooked by many studies in which the availability of peptide for cell interactions is assumed. In the seminal study of RGD-hydrogel spacing, Hern et al. demonstrated that a PEG77 (PEG3400) spacer between the RGD ligand and hydrogel counterpart encouraged specific receptor-ligand interactions while an un-spaced RGD only resulted in non-specific interactions.3 To our knowledge there has not been a systematic investigation of peptide-hydrogel spacing that spans a range of PEG spacer lengths. We hypothesize that the peptide-hydrogel spacing in well-defined synthetic hydrogels is a critical variable that controls the availability and presentation of bioactive cell adhesion ligands such as RGD.
To address this hypothesis, we synthesized monodisperse, functional pegylated cell adhesion molecules to be incorporated into a hydrogel platform. Different lengths of hydrophilic PEG spacers were used to separate an acryloyl functional group from the RGD cell adhesion ligand and effectively control the distance between RGD and an inert polymer network (Fig. 1a). The monoacrylated PEGx-RGD ligands were copolymerized with PEG diacrylate (PEGDA) macromers, where the PEG spacer functions as a flexible tether that impacts the presentation and accessibility of the ligand. Cell attachment and spreading assays were used to probe the availability of the covalently bound ligands for cell interactions and competitive binding assays studied the specificity and relative affinity of the receptor-ligand interactions. We show peptide-hydrogel spacing within the range of 0 to 27 PEG units significantly influences the cell-material interactions, where longer spacers (27 PEG units) promote cell attachment and spreading at lower concentrations than short spacers. These results highlight the role of spacing and presentation of bioactive ligands in an inert network. The use of acryloyl-PEGx-RGD conjugates is likely to be broadly applicable to any hydrogel system that is radically polymerized (i.e. PHEMA, polyacrylamide), and control over spacing can be used as a general tool to regulate accessibility and presentation of the ligand.
Fig. 1.

a) The PEGx-peptide conjugates are copolymerized with PEGDA macromers via photopolymerization to form a hydrogel network. b) RGD peptides were covalently conjugated to monodisperse PEG spacers using solid phase methods. The range of monodisperse PEG spacers include x = 5, 11 and 27. c) Table of peptide purity measured by integrating under the peaks of the HPLC chromatogram shows the final purity of the PEGx-RGD conjugates. d) The final amount of peptide incorporated into a hydrogel was compared to the initial amount of peptide in hydrogels functionalized with 1.1mM PEGx-RGD ligands as measured using a colorimetric microBCA assay. **0.001 ≤ p < 0.01 compared to the no spacer condition.
Experimental
Poly(ethylene glycol) acrylation
3.4 kDa PEGDA was synthesized according to previously published methods.14 Briefly, PEG (Hampton Research) with a molecular weight of 3350 g mol−1 was dried overnight under vacuum at 140C and stored under nitrogen. Triethylamine (Sigma-Aldrich) was dried by passing through an alumina column. PEGDA was prepared by combining 0.02mmol ml−1 PEG in anhydrous toluene (Acros Organics) with 0.08mmol ml−1 acryloyl chloride (Sigma-Aldrich) and 0.06 mmol ml−1 triethyl-amine. The reaction proceeded for 4 h in a nitrogen environment at 70C. Triethlyamine salts were filtered. The PEG was precipitated in cold diethyl ether, collected and dried for 48 h. The product was dialyzed using cellulose ester dialysis tubing with a 500–1000 MWCO (Spectrum Laboratories) for 4 h with three changes of Milli-Q H2O (Millipore). The lyophilized product was verified with 1H NMR with a Bruker AC-300 instrument and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry with a Bruker Reflex II instrument. The percent acrylation for all batches of PEGDA, as calculated from the 1H NMR data, was greater than 80%.
Peptide synthesis
Acryloyl-peptide (x = 0) synthesis
The peptide GGGRGDSP was synthesized on solid rink amide resin using fluorenylmethoxycarbonyl (Fmoc) chemistry on a CS Bio 36S automated peptide synthesizer. After Fmoc deprotection, the terminal amine group was modified with an acryloyl functional group by reacting with excess acryloyl chloride and triethylamine for 5 min in dry dichloromethane (DCM). The peptide was released from the resin and the protecting groups were removed by incubating the peptide for four hours in 95% trifluoroacetic acid (TFA)/2.5% Milli-Q H2O/2.5% triisopropylsilane (TIS). Lyophilized acryloyl-RGD conjugates were analyzed with MALDI-TOF and the purity of synthesized peptides was evaluated via reverse phase high performance liquid chromatography (HPLC) using a C18 analytical column (Shimadzu) with a gradient of 5–95% H2O with 0.1% TFA/acetonitrile and a flow rate of 0.9 mL min−1. The peptides were purified using reverse phase HPLC with a C18 semi-prep column (Grace). The scrambled equivalent was synthesized via the same procedure, but with the GGGRDGSP peptide.
Acryloyl-PEGx-peptide (x = 5,11,27) synthesis
The peptide GGGRGDSP was synthesized on solid sieber amide resin using Fmoc chemistry on a CS Bio 36S automated peptide synthesizer. A monodisperse heterobifunctional Fmoc-NH-PEGx-COOH spacer (x = 5, 11, 27) (EMD Chemicals Inc.) was reacted to the terminal glycine for 4–16 h. After Fmoc deprotection, the terminal amine group was modified with an acryloyl functional group by reacting with excess acryloyl chloride and triethylamine for 5 min in dry DCM. The peptide was released from the resin (2% TFA/5% TIS/93% DCM), residual solvent was removed, and then the protecting groups were removed (95% TFA/2.5% Milli-Q H2O/2.5% TIS). Lyophilized acryloyl-PEGx-RGD conjugates were analyzed and purified with the same techniques as mentioned above for the acryloyl-peptide (x = 0). The scrambled equivalent was synthesized via the same procedure, but with the GGGRDGSP peptide.
Acryloyl-PEG77-peptide synthesis
Peptides were modified with an acyrloyl-PEG77-succinimidyl valerate (Laysan Bio Inc.) as described elsewhere.3 Briefly, lyophilized GGGRGDSP peptides (synthesized on rink resin with CS Bio 36s automated peptide synthesizer, HPLC purity = 72%) were reacted with acryloyl-PEG77-succinimidly valerate at a 1.1 : 1 ratio in 50mM sodium bicarbonate buffer for 4.5 h. The lyophilized PEG77-RGD was characterized with MALDI-TOF and HPLC. Acryloyl-PEG77-RGD conjugates were purified via dialysis using cellulose ester dialysis tubing with a 500–1000 MWCO (Spectrum Laboratories) for 4 h with three changes of Milli-Q H2O (Millipore). The scrambled equivalent was synthesized via the same procedure, but with the GGGRDGSP peptide (HPLC purity = 77%).
Photoinitiator synthesis
Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was synthesized as previously described.15 In a nitrogen environment, 5 mmoles of 2,4,6-trimethylbenzoyl chloride (Sigma-Aldrich) were added dropwise with an addition funnel to an equimolar amount of dimethylphosphonite. After 24 h, four fold excess lithium bromide dissolved in 2-butanone was added. The mixture was heated in a 55C oil bath for 10 min and then cooled to room temperature. Next, the product was washed with 2-butanone and filtered 2 times. The product was dissolved in methanol and precipitated in diethyl ether. The product was filtered and allowed to dry. The structure was confirmed with 1H NMR.
PEGDA polymerization/hydrogel formation
PEGDA (10% w/w) was dissolved in 1 × phosphate buffer saline (1 × PBS, pH 7.4) (Invitrogen) and mixed with LAP (2.2mM). Acryloyl-PEGx-RGD conjugates were weighed and dissolved in 1xPBS to form a 20mM stock solution. Based on the molecular weight and incorporation efficiency (see next sub-section), peptide conjugates were aliquoted into PEGDA prepolymer solutions to target a final peptide concentration in the hydrogel between 0.2 and 1.6mM. The solutions were polymerized between Sigmacoted (Sigmacote; Sigma-Aldrich) glass plates separated with a 0.5mm thick Teflon® PTFE skived sheet (McMaster-Carr) spacer by exposure to a UV light source (365λ, 8.7mW cm−2) for 1000 s. Hydrogels were sterilized prior to cell culture in an ethanol solution (70 v/v) and placed under a UV light source (265λ) for 15 min. Prior to plating, surfaces were stored in media overnight.
Incorporation efficiency of peptide conjugates
PEGDA hydrogels with the respective acryloyl-PEGx-RGD conjugates (0.8mM and 1.6mM) were prepared as described above. For each condition, six 3mm diameter circles were cut with disposable biopsy punches (Integra Miltex) and placed in 96 well plates. From the six surfaces, three surfaces were hydrated in 150μl 1 × PBS. The 1 × PBS was exchanged every thirty minutes for a minimum of three exchanges. The other three surfaces were controls, and only a minimal amount of 20μl of 1 × PBS was added for swelling and never exchanged with fresh 1xPBS. Next, all hydrogel surfaces were degraded using 60 μl of 0.5M sodium hydroxide and then neutralized with 0.5M hydrochloric acid. The final volume in each well was equivalent. The microBCA Protein Assay Kit (Thermo Scientific) microplate procedure was used to colorimetrically quantify peptide content. In addition to the BSA standard curve, an RGD standard curve was also generated using known amounts of peptide GGGRGDSP.
Equilibrium swelling ratio
Functionalized gels, prepared as previously described, were cut into 3mm diameter circles, and then incubated in deionized water for 72 h. Gels were weighed (wet weight, ws), lyophilized, and weighed again (dry weight, wd). Six replicates of hydrogels functionalized with1.6mM acryloyl-PEGx-RGD, for each spacer length were tested (0.8mM for PEG77-RGD). The equilibrium swelling ratio (Qm) was determined, where Qm=ws/wd.
Cell culture
Telomerase-immortalized human corneal epithelial cells (hTCEpi), graciously donated by James Jester from University of California, Irvine,16 were cultured in epithelial medium containing a 3 : 2 ratio of Ham’s F-12: Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen) supplemented with 2.5% fetal bovine serum, 0.4μg ml−1 hydrocortisone, 8.4ng ml−1 cholera toxin, 5μg ml−1 insulin, 24μg ml−1 adenine, 10 ng ml−1 epidermal growth factor, 100 units penicillin, and 100μg ml−1 streptomycin.17,18 hTCEpi cells in epithelial medium were plated on 100mm tissue culture plates containing a mitomycin-c treated Swiss 3T3 fibroblast layer. Cells were incubated at 37C and 5% CO2 until they reached approximately 70% confluence. Cells were used between passages 45 to 55. For all experiments, 6 replicates of each substrate were used and cells were plated at a density of 15 000 cells cm−2. Cells were incubated on surfaces for 24 h after plating to allow for attachment and spreading. To investigate projected cell area and cell number, we stained for actin-filaments and nuclei. Surfaces were fixed with 1% para-formaldehyde in 1x PBS (Electron Microscopy Sciences) at room temperature for 20 min. The cells were then permeablized with 0.1% (v/v) Triton X-100 (Sigma Aldrich) in 1x PBS for 5 min. The cells were then incubated in 1% (w/w) bovine serum albumin (Sigma Aldrich) in 1x PBS for 20 min to block non-specific binding. Next, cells were incubated in 5μg mL−1 of TRITC-phalloidin (Sigma Aldrich) containing 0.1μg mL−1 4′,6-dia-midino-2-phenylindole (DAPI) (Invitrogen) in 1x PBS for 45 min. Eight 20X images of each surface were taken using a Zeiss Axiovert 100M microscope.
Soluble RGD assay
hTCEpi cells were seeded on PEGDA hydrogels functionalized with the respective acryloyl-PEGx-RGD conjugates (1.6mM), prepared as described above. Phase images of the substrates were taken 4 h after cell seeding. Either soluble linear GGRGDSP (0.5mM) or cyclic RGDfV (0.25mM) (BaChem Americas, Inc.) was added to the medium. After 24 h, six 10X phase images were taken of each surface. This procedure was repeated on TCPS, PEGDA, and with soluble cyclic RADfV. For this experiment, 4 replicates of each condition were used.
Statistical analysis
Values reported are the means ± the SD. Experiments were analyzed using analysis of variance (ANOVA). When variability was determined to be significant (P < 0.05), the Bonferroni multiple comparison test was used to determine significance (P < 0.05) between groups. Significance was further divided into “statistically significant” (0.01 ≤ P < 0.05), “very significant” (0.001 ≤ P < 0.01), and “extremely significant” (P < 0.001).
Results and discussion
Using solid phase techniques, peptides can be conjugated to pure monodisperse heterobifunctional PEG reagents (Fmoc-NH-(PEG)x-COOH) where x is 5, 11 or 27 (Fig. 1b). The resulting PEG-peptide conjugates can be functionalized with an acryloyl group while on the solid phase resin. By synthesizing and modifying the acryloyl-PEGx-RGD conjugates on a solid phase support the products are easily separated from soluble reagents and solvents resulting in high yield, and excess reagents are used to drive reactions to completion.19 Acryloyl-PEGx-RGD conjugates were exactly characterized by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry and purified using high performance liquid chromatography (HPLC) for a final purity of greater than 86% for all conjugates (Fig. 1c). It is important to note that acryloyl-PEG77-peptide conjugates used in this study were synthesized via a polydisperse monoacrylated-PEG77-N-hydroxy succinimide ester, as this type of spacer has been a standard spacer used in previous studies to present peptide ligands to cells.3,20–22
Two advantages of using a monodisperse spacer over the popular polydisperse spacer (i.e. PEG77) are that: 1) the final products are clearly identified with standard peptide characterization tools (MALDI-TOF, HPLC) to verify monomer functionality and the addition of the PEG spacer and 2) the purification via HPLC is straightforward, where product peaks are distinct, sharp and symmetric. In addition to being pure, the monodisperse bioactive conjugates are functionalized with acryloyl groups to exploit an immobilization scheme that potentially limits changes in physical properties of the hydrogel as peptide epitope concentrations increase.23 Together, the purity and functionality of the monodisperse conjugates afford the freedom required to be used to present multiple epitopes or high epitope density schemes. For example, it is critical to use exactly characterized and purified conjugates for applications that require reproducible and controlled incorporation of multiple epitopes. Likewise, for high epitope density presentation schemes designed to maximize the number of ligand-receptor binding events, constant physical properties would be desirable to avoid confounding variables.
Acryloyl-PEGx-RGD conjugates were efficiently incorporated into the bulk PEGDA hydrogel network via photoinitiated radical chain growth copolymerization. Exposure of 10% (w/w) PEG 3400 diacrylate and acryloyl-PEGx-RGD (0 to 1.6mM) mixtures to UV light (365nm, 8.7 mW cm−2) in the presence of the photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphos-phinate resulted in photo-copolymerization. Copolymerization, as a means to incorporate bioactive ligands, exploits the high reactivity and efficient conversions characteristic of acrylate-radical photoinitiated polymerization.24 The efficiency of acryloyl-PEGx-RGD incorporation into PEGDA hydrogels was measured using a microBCA protein assay, where the final peptide concentration after removal of non-covalently bound molecules from the hydrogel was compared to the peptide concentration in the initial reaction mixture. The measured incorporation efficiency of 1.1 mM acryloyl-PEGx-RGD conjugates into PEGDA hydrogels was 70 ± 4% for all acryloyl-PEGx-RGD ligands (Fig. 1d). Similar incorporation efficiency was also demonstrated with other peptide concentrations (data not shown). The incorporation efficiency is likely a function of total acrylate conversion during polymerization and the physical characteristics of the peptides (i.e. hydrophobicity). All concentrations reported hereafter are the initial concentrations multiplied by the individual incorporation efficiency values to reflect the final peptide concentration in the hydrogel for each respective spacer length. The incorporation data suggests copolymerization of PEGDA macromers and acryloyl-PEGx-RGD conjugates is a highly reproducible method to incorporate known concentrations of pure peptides into a hydrogel network.
The experimental platform used in this study was specifically designed in such a way as to minimize changes in the physical properties of the hydrogel networks. Evidence that matrix properties were independent of concentration and spacer length of the acryloyl-PEGx-RGD conjugates was derived from swelling experiments. The equilibrium swelling ratio (Qm) of hydrogels containing 1.6 mM PEGx-RGD, for x = 0,5,11,27 and 0.8mM for PEG77-RGD gels are shown in Fig. 2. For all of the hydrogels, Qm was essentially constant with a value of 11.5 ± 1.4, and only the hydrogel functionalized with no spacer (x = 0) exhibits a Qm that is statistically different than the other gels. In addition, the statistical difference of swelling ratios was only observed for this concentration; for all other concentrations tested (0.8mM and 0.4mM for x = 0,5,11,27 and 0.4mM and 0.2mM for x = 77) there was no significant difference between the equilibrium swelling ratios for the different spacers (data not shown). To place these results in context, in studies in which the physical properties of hydrogels are purposefully varied to induce observable changes in cell behavior, the differences in the equilibrium swelling ratios of the hydrogels were of order five or more (a percent change of 36% or greater).25
Fig. 2.
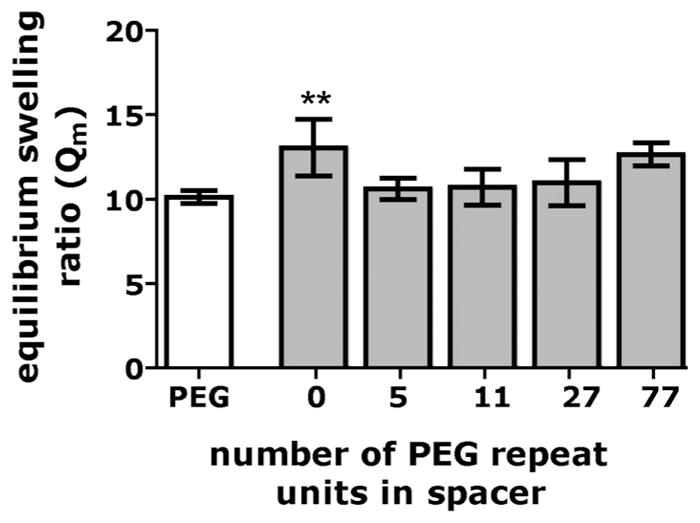
Equilibrium swelling ratio of the 1.6mM PEGx- functionalized hydrogels RGD (0.8mM for PEG77-RGD)for the highest concentrations of peptide tested in this study. **0.001 ≤ p < 0.01 compared to PEG5-RGD.
It is also known that the physical properties of the hydrogels may also be a function of addition of mono-acrylated monomers, in this case the acryloyl-PEGx-RGD conjugates. The concentrations of conjugates used in our experiment, however, are low, and the equilibrium swelling ratios of PEGx-RGD functionalized hydrogels were similar to hydrogels synthesized with just PEGDA (Qm = 10.1 ± 0.3). These results are consistent with those of Beamish et al. Using a similar system of mono- and di-acrylated moieties, they showed that for 10wt% PEGDA hydrogels polymerized with a range of concentrations of mono-acrylated PEG (Mn = 5000), there was only a slight decrease in the equilibrium swelling ratio as the concentration of the mono-acrylated component increased from 0mM to 10mM.23
Telomerase-immortalized human corneal epithelial (hTCEpi) cell attachment was modulated by the peptide concentration and the length of the PEG spacer (Fig. 3a, 3c, 3d). hTCEpi cells were targeted for cell-culture experiments as previous studies suggest that RGD functionalized surfaces support corneal epithelial cell attachment, spreading, migration and wound healing.13 We are also interested in hydrogel materials that can be molded with topographic features and functionalized with peptides for the development of synthetic basement membrane constructs for corneal prosthetic devices.26 The following results using hTCEpi cells, however, are not necessarily exclusive for this specific cell type and are most likely generalizable to other cell types with the capacity for integrin-RGD mediated interactions. There was a two-phase trend in 24 h hTCEpi cell attachment. The first phase was characterized by a small but gradual increase in cell number from the 0mM negative control, followed by the onset of cell attachment where there was a noticeable transition region. The transition was followed by the second phase where the cell attachment increased rapidly with increasing peptide concentrations to approach an upper limit of cell attachment defined by the tissue culture polystyrene (TCPS) positive control. Between phase 1 and phase 2, we likely approach a critical RGD density required for stable integrin adhesions that support cell attachment and spreading. Hydrogel surfaces functionalized with RGD (no spacer) or PEG5-RGD did not support significant hTCEpi cell attachment, remaining in phase 1 across the range of concentrations tested. In contrast, hydrogels fabricated with longer linkers, PEG27-RGD or PEG77-RGD exhibited both phase 1 and phase 2 behavior. Interestingly, as the PEG spacer length increased, the ligand concentration needed to a) support cell attachment and b) approach the upper limit of cell attachment decreased. Specifically, a two-fold increase in the PEG27-RGD concentration (1.6mM) was necessary to attain the analogous high hTCEpi cell count on surfaces functionalized with PEG77-RGD (0.8mM).
Fig. 3.

a) Number of hTCEpi cells attached after 24 h to hydrogel surfaces functionalized with 0.2 to 1.6mM PEGx-RGD ligands (where x ranges from 0 to 77). Cell count refers to the total number of cells counted in 6 random images per surface, with 6 replicates of each condition. b) Projected hTCEpi cell area after 24 h on hydrogel surfaces functionalized with 0.2 to 1.6 mM PEGx-RGD ligands. The outline of actin stained filaments was used to measure the projected cell area, which was averaged per cell on 6 random images per surface, with 6 replicates per condition. *0.01 ≤ p < 0.05, ***p < 0.001 compared to the PEGDA control (0mM). The dashed line corresponds to the cell count or cell area on TCPS. Fluorescent images of hTCEpi cells on c) no spacer-RGD and d) PEG27-RGD hydrogels at a concentration range of 0.2 mM to 1.6 mM.
Similar to the hTCEpi cell attachment data, hTCEpi cell spreading was also controlled by the ligand concentration and length of the PEG spacer (Fig. 3b). The same trends observed with the hTCEpi cell attachment data were observed for hTCEpi cell spreading. The onset of spreading, when rounded hTCEpi cells transitioned into spread morphology, was followed by a rapid increase in spreading, until the projected hTCEpi cell area approached that of the positive TCPS control. Significantly, the onset of hTCEpi cell spreading occurred at the same peptide concentrations as the onset of cell attachment for each PEGx-RGD conjugate. The PEG spacer length also influenced the onset of cell spreading and maximum projected hTCEpi cell area. On PEG27-RGD surfaces, significant hTCEpi cell spreading (1000 μm2) is achieved at a concentration of 1.6 mM, while comparable spreading on PEG77-RGD surfaces is achieved at a concentration of 0.8 mM. Both the hTCEpi cell attachment and spreading data suggest peptide concentration in combination with PEG spacer length dictates the mechanism or manner in which the cells can interact with the covalently bound ligands.
Our work supports previous studies that have shown that interactions between cell surface integrins and RGD ligands can result in biospecific attachment of cells to substrates, where the RGD ligand density regulates the number of cells attached.3,27,28 In addition to RGD density, other studies have revealed additional factors that impact cell-substrate interactions, including ligand presentation, specifically nanoscale clustering.29,30 In this work, we show that the peptide-hydrogel spacing dictates the accessibility of the RGD ligand within a hydrogel network. Although hydrogels were functionalized with similar RGD concentrations, RGD ligands attached via shorter spacers were not accessible in the hydrogel, yet RGD ligands attached via longer spacers were accessible. Furthermore, the 0.8 mM to 1.6 mM RGD concentration range that supports cell attachment and spreading in this study is similar to that previously reported for other synthetic hydrogel systems. For example, Hern et al. reported that concentrations of 1mM RGD supported fibroblast spreading,3 and Burdick et al. showed that concentrations of 4 mM RGD supported osteoblast spreading.31 These RGD concentrations reported for hydrogel systems, by us and others, are significantly larger than the concentration required for fibroblast spreading on modified alkylsiloxane surfaces (10 fmol cm−2)27 even if one assumes ligands within 10 nanometres of the hydrogel surface are available for cell interactions. This discrepancy highlights the importance of ligand accessibility within a heterogeneous inert network for cell-substrate interactions.
The specific interaction between bound RGD ligands and the integrin receptors of hTCEpi cells was confirmed via two methods. In the first method, scrambled acryloyl-PEGx-RDG conjugates were incorporated into PEGDA hydrogels. For all of the PEGx-RDG conjugates tested (x = 0,5,27,77) at concentrations of 1.6 mM and 0.8 mM, there was no significant hTCEpi cell attachment (Supplementary Figure 1†). In the second method, soluble RGD competed against the covalently immobilized PEGx-RGD conjugates for interactions with integrins. Four hours after cells were plated on 1.6 mM PEGx-RGD functionalized hydrogel surfaces, either 0.5 mM soluble linear RGD or 0.25 mM soluble cyclic RGDfV (Fig. 4a) was added to the media. The percent of cells that remained attached to the surface after 24 h, as well as the percent of attached cells that had spread, was compared to the nominal condition where no soluble RGD was added to the media.
Fig. 4.

a) The percent of cells on 1.6 mM functionalized surfaces (0.8mM for PEG77-RGD) at t = 24 after the addition of 0.5 mM soluble linear RGD or 0.25 mM soluble cyclic RGDfV shows that there is a slight decrease in the number of cells attached (compared to the control where no soluble RGD was added) after the addition of soluble linear RGD and in contrast, adding cyclic RGDfV results in a large reduction in cell number on the surface after 24 h. b) hTCEpi cells plated on surfaces modified with 1.6mM PEG11-RGD conjugates were spread after 24 h on both no soluble RGD and 0.5mM linear RGD, but rounded on 0.25mM soluble cyclic RGDfV.
The competitive detachment of hTCEpi cells in the presence of soluble linear RGD was dependent on the PEG spacer length. In the presence of 0.5mM soluble linear RGD (Fig. 4a), we observed that there were fewer cells on PEG5-RGD surfaces than on PEG11-RGD, PEG27-RGD and PEG77-RGD surfaces. In addition, for all PEGx-RGD functionalized surfaces, there was at least a 15% decrease in the percent of cells attached when soluble linear RGD was present as compared to the control condition with no soluble RGD (Fig. 4a). A large majority of the attached cells, however, had spread by 24 h (Fig. 4c, 5a), a trend that was consistent across surfaces. Cell spreading in the presence of soluble linear RGD suggests that: 1) the integrin affinity for RGD tethered to the surface is greater than the integrin affinity for soluble RGD, which could be evidence of cell-mediated RGD rearrangement or a concentration that mimics clustered RGD on the surface, or 2) there is a cellular mechanotransduction response generated from the tethered ligand-cell interaction.32 In the case of the former hypothesis, the data suggests that on surfaces functionalized with RGD conjugated to spacers greater than or equal to 11 PEG units, epithelial cells had a higher ligand-receptor binding affinity than to surfaces functionalized with shorter PEG-RGD conjugates.
Fig. 5.

a) hTCEpi cells plated on surfaces modified with 1.6mM PEGx-RGD conjugates were spread after 24 h after presence of 0.5mM linear RGD (similar to control spreading), but rounded in presence of 0.25mM soluble cyclic RGD. ***p < 0.001 compared to the corresponding “no soluble RGD” data. b) The percent of cells on a 1.6mM PEG27-RGD surface at t = 24 after the addition of 0.5mM soluble scrambled linear RDG or 0.25mM soluble scrambled cyclic RADfV shows that a large number of cells remain attached after the addition of scrambled soluble peptides. There was no statistical difference between the scrambled and control condition (no soluble RGD). There were few attached cells on the control PEGDA surface (not treated with soluble RGD). c) hTCEpi cells plated on TCPS show no change in cell spreading from the control substrate that was not treated with soluble RGD, even when treated with soluble linear RGD or cyclic RGD (scale bar = 50 μm).
In contrast to the soluble linear RGD data, the addition of soluble cyclic RGDfV resulted in significant detachment of cells and prevented cells from spreading, independent of the PEG spacer length (Fig. 4a, 4d). The comparison of the cellular response to cyclic and linear forms of soluble RGD provides a rudimentary measurement for affinity of the hTCEpi cell to the covalently bound PEGx-RGD ligands, as the integrin receptor has a higher affinity for cyclic RGDfV than for linear RGD.33 The 90% decrease in hTCEpi cell attachment and the inhibition of spreading for all surfaces after the addition of soluble cyclic RGDfV suggests that this soluble molecule competes with a specific interaction between integrin receptors and available PEGx-RGD ligands on the hydrogel. There was no significant change in hTCEpi cell spreading on TCPS control surfaces upon the addition of soluble cyclic RGDfV or linear RGD (Fig. 5c), as expected. Also, scrambled cyclic RADfV did not alter the percent of cells attached on any of the surfaces (Fig. 5b). These results mutually indicate that hTCEpi cells attach bio-specifically and with high affinity to the PEGx-RGD modified surfaces.
Collectively, the attachment, spreading and soluble RGD results highlight the significance of a PEG spacer on the accessibility of an RGD ligand and the affinity of the RGD-integrin interaction within the context of hydrogels. The spacer between an adhesive peptide and the bulk hydrogel significantly reduces the minimum nominal peptide concentration that supports hTCEpi cell attachment and spreading. The function of the PEG spacer can be interpreted several ways, including the following hypotheses: 1) it increases the number of ligands available to cell by extending the tether length, 2) it decreases the steric hindrance that occurs from surrounding molecules7,34 and thus increases the accessibility of the ligand, 3) it enables the local rearrangement of the biological motifs to allow for more effective receptor clustering,35 or 4) it increases ligand flexibility for a higher ligand-receptor binding affinity. Estimates of the maximum increase in ligand number only due to extension (hypothesis 1 above), however, do not adequately explain the results. If one assumes that ligands within 10 nm of hydrogel surface are available for cell-interactions, that the PEG spacer assumes helical conformation (average monomer extension of 2 Å)36 and that all peptides extend and orient towards the surface, one can estimate that there is a two-fold increase in ligand number available to cells due to PEG spacer extension from PEG11 to PEG77. However, the experimental data suggest that similar cell attachment and spreading on PEG11 and PEG77 functionalized surfaces occurs when there is a four-fold increase of the PEG11-RGD concentration over the PEG77-RGD concentration. Interestingly, there is also a non-linear relationship between PEG spacer length and both cell attachment and cell spreading (Supplementary Figure 2†), which suggests that there is not a simple, proportional increase in cell-ligand interactions as the spacer length is increased. We expect that there are likely multiple mechanisms simultaneously at play which provide the advantages of the PEG spacer noted in this study, potentially including a combination of the hypotheses previously mentioned. Recent advances in investigating local cell-mediated arrangement of bioactive ligands using FRET measurements37 may provide possible approaches to further identify the role of a spacer between peptides and hydrogels.
Conclusion
The described method for fabrication of pure, monodisperse acryloyl-PEGx-RGD conjugates provides an adaptable and controlled approach to investigating peptide-hydrogel spacing. Here we synthesized pure functionalized PEGx-RGD conjugates to demonstrate the importance of peptide-hydrogel spacing for cell-material interactions. We showed that photoinitiated copolymerization is a robust method that can be used to efficiently incorporate bioactive acryloyl-PEGx-RGD ligands into a hydrogel network. The peptide concentration in combination with PEG spacer length dictated the amount of RGD available for epithelial cell interactions. Specifically, the minimum peptide concentrations that supported cell attachment and spreading decreased as the distance between the RGD and hydrogel increased. This approach to present peptides is likely generalizable to most hydrogel chemistries and is highlighted by the ability to decrease the minimum peptide concentration needed for cell attachment and spreading. While the incorporation of one bioactive ligand, RGD, is shown, the approach can potentially be used for controllable incorporation of multiple cues that better mimic the native extracellular matrix. The results presented in this paper demonstrate that the hydrogel-peptide spacing critically impacts the presentation and availability of bioactive ligands and is a necessary consideration in the development of well-defined biomaterials.
Supplementary Material
Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant No. NSF CHE-9208463 (NMR) and NSF CHE-9520868 (MALDI-TOF) and the National Institutes of Health under Grant No. NEI-1RO1EY016134-O1A2 and NEI-1RO1EY017367-O1A.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c1sm06589k
Contributor Information
William L. Murphy, Email: wlmurphy@wisc.edu.
Paul F. Nealey, Email: nealey@engr.wisc.edu.
References
- 1.Harris JM. Introduction to biotechnical and biomedical applications of poly(ethylene glycol) In: Harris JM, editor. Poly(ethylene glycol) chemistry: biotechnical and biomedical applications. Plenum Press; New York: 1992. [Google Scholar]
- 2.Elbert DL, Hubbell JA. Biomacromolecules. 2001;2:430–441. doi: 10.1021/bm0056299. [DOI] [PubMed] [Google Scholar]
- 3.Hern DL, Hubbell JA. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 4.Salinas CN, Cole BB, Kasko AM, Anseth KS. Tissue Eng. 2007;13:1025–1034. doi: 10.1089/ten.2006.0126. [DOI] [PubMed] [Google Scholar]
- 5.DeForest CA, Polizzotti BD, Anseth KS. Nat Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houseman BT, Mrksich M. Biomaterials. 2001;22:943–955. doi: 10.1016/s0142-9612(00)00259-3. [DOI] [PubMed] [Google Scholar]
- 8.Beer JH, Springer KT, Coller BS. Blood. 1992;79:117–128. [PubMed] [Google Scholar]
- 9.Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, Stupp SI. Biomaterials. 2007;28:4608–4618. doi: 10.1016/j.biomaterials.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 10.Rowley JA, Madlambayan G, Mooney DJ. Biomaterials. 1999;20:45–53. doi: 10.1016/s0142-9612(98)00107-0. [DOI] [PubMed] [Google Scholar]
- 11.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ. Nat Mater. 2010;9:518–526. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruoslahti E, Pierschbacher MD. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 13.Fong E, Tzlil S, Tirrell DA. Proc Natl Acad Sci U S A. 2010;107:19302–19307. doi: 10.1073/pnas.1008291107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin CC, Metters AT. J Biomed Mater Res, Part A. 2007;83A:954–964. doi: 10.1002/jbm.a.31282. [DOI] [PubMed] [Google Scholar]
- 15.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson DM, Li L, Fisher S, Pearce VP, Shay JW, Wright WE, Cavanagh HD, Jester JV. Invest Ophthalmol Visual Sci. 2005;46:470–478. doi: 10.1167/iovs.04-0528. [DOI] [PubMed] [Google Scholar]
- 17.Allen-Hoffmann BL, Rheinwald JG. Proc Natl Acad Sci U S A. 1984;81:7802–7806. doi: 10.1073/pnas.81.24.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabatini LM, Allen-Hoffmann BL, Warner TF, Azen EA. In Vitro Cell Dev Biol. 1991;27:939–948. doi: 10.1007/BF02631121. [DOI] [PubMed] [Google Scholar]
- 19.White PD, Chan WC. Basic principles. In: Chan WC, White PD, editors. Fmoc solid phase peptide synthesis: a practical approach. Oxford University Press Inc; New York: 2000. [Google Scholar]
- 20.Schmidt DR, Kao WJ. J Biomed Mater Res, Part A. 2007;83A:617–625. doi: 10.1002/jbm.a.31270. [DOI] [PubMed] [Google Scholar]
- 21.Hahn MS, Miller JS, West JL. Adv Mater. 2005;17:2939–2942. [Google Scholar]
- 22.Zhu JM, Tang C, Kottke-Marchant K, Marchant RE. Bioconjugate Chem. 2009;20:333–339. doi: 10.1021/bc800441v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beamish JA, Zhu JM, Kottke-Marchant K, Marchant RE. J Biomed Mater Res, Part A. 2010;92A:441–450. doi: 10.1002/jbm.a.32353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Decker C. Polym Int. 1998;45:133–141. [Google Scholar]
- 25.Lin S, Sangaj N, Razafiarison T, Zhang C, Varghese S. Pharm Res. 2011;28:1422–1430. doi: 10.1007/s11095-011-0378-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teixeira AI, Abrams GA, Bertics PJ, Murphy CJ, Nealey PF. J Cell Sci. 2003;116:1881–1892. doi: 10.1242/jcs.00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massia SP, Hubbell JA. J Cell Biol. 1991;114:1089–1100. doi: 10.1083/jcb.114.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts C, Chen CS, Mrksich M, Martichonok V, Ingber DE, Whitesides GM. J Am Chem Soc. 1998;120:6548–6555. [Google Scholar]
- 29.Maheshwari G, Brown G, Lauffenburger DA, Wells A, Griffith LG. J Cell Sci. 2000;113:1677–1686. doi: 10.1242/jcs.113.10.1677. [DOI] [PubMed] [Google Scholar]
- 30.Lee KY, Alsberg E, Hsiong S, Comisar W, Linderman J, Ziff R, Mooney D. Nano Lett. 2004;4:1501–1506. doi: 10.1021/nl0493592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burdick JA, Anseth KS. Biomaterials. 2002;23:4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 32.Ingber DE. FASEB J. 2006;20:811–827. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 33.Kumagai H, Tajima M, Ueno Y, Yuhko GH, Ohba M. Biochem Biophys Res Commun. 1991;177:74–82. doi: 10.1016/0006-291x(91)91950-h. [DOI] [PubMed] [Google Scholar]
- 34.Shin H, Jo S, Mikos AG. J Biomed Mater Res. 2002;61:169–179. doi: 10.1002/jbm.10193. [DOI] [PubMed] [Google Scholar]
- 35.Griffith LG, Lopina S. Biomaterials. 1998;19:979–986. doi: 10.1016/s0142-9612(97)00185-3. [DOI] [PubMed] [Google Scholar]
- 36.Heymann B, Grubmuller H. Chem Phys Lett. 1999;307:425–432. [Google Scholar]
- 37.Kong HJ, Polte TR, Alsberg E, Mooney DJ. Proc Natl Acad Sci U S A. 2005;102:4300–4305. doi: 10.1073/pnas.0405873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.