

Abstract
4-Formylbenzene diazonium hexafluorophosphate (FBDP) is a novel bench-stable crystalline diazonium salt that reacts selectively with tyrosine to install a bioorthogonal aldehyde functionality. Model studies with N-acyl-tyrosine methylamide allowed us to identify conditions optimal for tyrosine ligation reactions with small peptides and proteins. FBDP-based conjugation was used for the facile introduction of small molecule tags, poly(ethylene) glycol chains (PEGylation), and functional small molecules onto model proteins and to label the surface of living cells.
INTRODUCTION
Diazonium reagents have been used for selective modification of tyrosine(1–3) but are often not extensively applied to the modification of proteins and/or antibodies due to several limitations. For a precise site-specific modification of a protein, complete control over reagent stability, reactivity, and stoichiometry is required. Instability of the currently reported diazonium reagents makes it difficult to control the stoichiometry of the reaction and a high excess of the reagent is often required. Diazonium reagents are generally prepared from anilines in situ under strongly acidic conditions,(4) whereas the diazo coupling reaction with protein optimally proceeds under mildly basic conditions. The significant pH adjustment required is not necessarily possible with pH sensitive proteins. Perhaps the greatest barrier to the wide-spread use of diazonium labeling reagents is the prerequisite of in situ preparation just prior to use.(1, 2, 5, 6) This requirement limits their utility and access to a select group of practitioners. We herein present a practical protein modification method for the introduction of a bioorthogonal aldehyde group(7–11) based on the development of a novel bench-stable diazonium reagent.
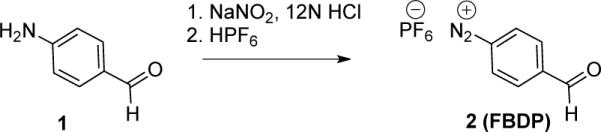
A bifunctional diazonium reagent was designed to satisfy two key requirements: 1) improved reagent stability while maintaining high reactivity in aqueous buffer and 2) ability to introduce a bioorthogonal functionality into the protein. The 4-formyl benzenediazonium with hexafluorophosphate (PF6−) or tetrafluoroborate (BF4−) counterions were chosen as suitable candidates (Scheme 1). Hexafluorophosphate(12) and tetrafluoroborate(13–16) were chosen as stabilizing counterions, and the aldehyde provides an excellent functional handle for the formation of oxime or hydrazone bonds.(7, 17, 18) The formyl group is essential for the stability of diazonium ion as it prevents cation formation at C4 and release of nitrogen gas; furthermore, it activates the N-N triple bond as an electrophile. The importance of the electron withdrawing substituents on the phenyl ring of diazonium reagent has been emphasized in several previous studies. Unlike many diazonium reagents, these reagents avoid the use of the potentially immunogenic NO2-substituted benzene.(5, 19–21)
Scheme 1.
Metal free and mild tyrosine modification reactions(1, 3, 22–26) are an attractive alternative to the commonly used lysine and cysteine modification protocols. The high abundance of lysine in a typical protein or antibody significantly complicates control of the stoichiometry and specificity of bioconjugation reactions. Cysteines, although less abundant, require reductive pre-treatment before bioconjugation. In addition, modification of cysteine may alter the stability and function of an antibody or other protein wherein disulfide linkages are key to stability.(27–29) Tyrosines are less common than lysine and do not form stabilizing linkages like cysteine. Introduction of the aldehyde functionality through tyrosine conjugation has an additional advantage: targeting modules containing hydrazine form stable but exchangeable hydrazone linkages with aldehyde that can later undergo site-specific exchange into the oxime linkage with a different targeting molecule, drug, or dye.(30, 31)
MATERIALS AND METHODS
General
1H NMR and 13C NMR spectra were recorded on Bruker DRX-600 (600 MHz), Varian Inova-400 (400 MHz), or Varian MER-300 (300 MHz) spectrometers in the stated solvents using tetramethylsilane as an internal standard. Chemical shifts were reported in parts per million (ppm) on the δ scale from an internal standard (NMR descriptions: s, singlet; d, doublet; t, triplet; q, quartet; h, heptet; m, multiplet; br, broad). Coupling constants, J, are reported in Hertz. Mass spectroscopy was performed by the Scripps Research Institute Mass Spectrometer Center. Analytical thin-layer chromatography and flash column chromatography were performed on Merck Kieselgel 60 F254 silica gel plates and Silica Gel ZEOprep 60 ECO 40–63 Micron, respectively. Visualization was accomplished with anisaldehyde or KMnO4. Unless otherwise noted, all the materials were obtained from commercial suppliers, and were used without further purification. All solvents were commercially available grade.
4-Formyl benzene diazonium hexafluorophosphate (2)
To a suspension of 4-aminobenzaldehyde polymer (5.00 g, 41.3 mmol) in 12N HCl (85 mL) was added the solution of NaNO2 (3.42 g, 49.5 mmol) in water (67 mL) at −10 °C. The resulting solution was stirred at −10 °C. After 1.5 h, 60% HPF6 in water (10.3 mL, 70.2 mmol) was added at −10 °C and stirred for 30 min. Then the reaction mixture was stirred at room temperature for 30 min. The resulting solids were collected by filtration, and washed with water and AcOEt to give 2 (FBDP) (4.77 g, 42%) as an off-white solid. 1H NMR (300 MHz, DMSO-d6): δ 10.2 (s, 1H), 8.85 (d, J = 5.2 Hz, 2H), 8.39 (d, J = 5.0 Hz, 2H). 13C NMR (75 MHz, acetone-d6): δ 190.32, 143.69, 133.39, 131.02, 119.41. 31P NMR (162 MHz, acetone-d6): δ −143.89 (h, JP-F = 707 MHz), 19F NMR (376 MHz, acetone-d6): δ −72.57 (d, JP-F = 707 MHz).HRMS: calcd for C7H5N2O+ (M+) 133.0402, found 133.0401.
(S,E)-2-acetamido-3-(3-((4-formylphenyl)diazenyl)-4-hydroxyphenyl)-N-methylpropanamide (5)
To a solution of N-acyl tyrosine methylamide 4 (20 mg, 0.0846 mmol) in 100 mM pH 7.0 NaH2PO4/Na2HPO4 buffer (2.83 mL) - DMSO (1.41 mL) was added the diazonium reagent 2 (25.9 mg, 0.0931 mmol) at room temperature. The resulting solution was stirred at room temperature for 45 min. After the reaction, water (2.82 mL) was added. The generated solid was filtered then washed with water and AcOEt to give 5 (31.0 mg, 99%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 10.9 (s, 1H), 10.1 (s, 1H), 8.16 (d, J = 8.6 Hz, 2H), 8.11 (d, J = 8.7 Hz, 2H), 7.93 (q, J = 4.7 Hz, 1H), 7.63 (d, J = 2.1 Hz, 1H), 7.32 (dd, J = 8.5, 2.2 Hz, 1H), 7.00 (d, J = 8.5 Hz, 1H), 4.45–4.37 (m, 1H), 2.94 (dd, J = 13.3, 5.0 Hz, 1H), 2.70 (dd, J = 13.5, 9.5 Hz, 1H), 2.56 (d, J = 4.6, 3H), 1.77 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ 192.59, 171.36, 168.93, 154.82, 154.14, 138.40, 137.05, 135.61, 130.65, 129.53, 123.08, 121.33, 117.94, 54.03, 36.82, 25.40, 22.40. HRMS: calcd for C19H21N4O4 (MH+) 369.1557, found 369.1556.
(S)-2-acetamido-3-(3-((E)-(4-((E)-(ethoxyimino)methyl)phenyl)diazenyl)-4-hydroxyphenyl)-N-methylpropanamide methylpropanamide (6)
To a suspension of compound 3 (15 mg, 0.041 mmol) in 100 mM pH 7.0 NaH2PO4/Na2HPO4 buffer (0.531 mL) - DMSO (1.54 mL) was added ethoxyamine hydrochloride 4 (4.37 mg, 0.045 mmol) at room temperature. The resulting suspension was stirred at room temperature for 2 h. After the reaction, water (4.00 mL) was added. The generated solid was filtered then washed with water and AcOEt to give 5 (17.2 mg, quant.) as a yellow solid.1H NMR (300MHz, DMSO-d6) (E:Z isomer ratio = 9 : 1): δ 10.9 (s, 1H), 8.33 (s, H), 8.13 (d, J = 8.5 Hz, 1H), 8.01 (d, J = 8.6 Hz, 2H), 7.93 (q, J = 4.5 Hz, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 2.0 Hz, 1H), 7.28 (dd, J = 8.4, 2.2 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 4.44–4.37 (m, 1H), 4.21 (q, J = 7.0 Hz, 2H), 2.94 (dd, J = 13.7, 5.1 Hz, 1H), 2.69 (dd, J = 13.6, 9.6 Hz, 1H), 2.56 (d, J = 4.6, 3H), 1.77 (s, 3H), 1.28 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, DMSO-d6): δ 171.39, 168.93, 153.30, 151.92, 147.74, 138.05, 134.38, 129.48, 127.71, 122.96, 122.57, 117.72, 69.30, 54.07, 36.84, 25.40, 22.40, 14.41. HRMS: calcd for C21H26N5O24 (MH+) 412.1979, found 412.1980.
General procedure of amino acid selectivity study
N-acyl methyl amides of amino acid were obtained as previously described.3 Equimolar solution of six amino acids N-acyl methyl amides (Tyr, His, Cys, Ser, Lys, Trp) was prepared in 0.1M Na2HPO4 buffer (pH 7, 7.4, 8). Final concentration of each amino acid was 100 μM (1 eq). 1 mL of the amino acid mixture solution was transferred into LCMS sample vial and 5 mL of FBDP solution in acetonitrile (20 mM solution, 1 eq) was added. Reaction mixture was vortexed gently and allowed to react at room temperature. Each reaction was performed independently at least 3 times. Aliquot of reaction mixture (5 μL) was analyzed by LCMS after 30 min of reaction time using Agilent LC/MSD SL mass spectrometer with Zorbax 300SP-C8, 5 μm, 4.6×50 mm column (Agilent). HPLC parameters were as follows: flow rate 0.5 ml/min, 5% mobile phase B over 2 min followed by a gradient to 5 to 95% B from 2 to 8 min, post time 4 min. Mobile phase A was 0.1% formic acid (v/v) in HPLC-grade H2O, and mobile phase B was 0.1% formic acid (v/v) in acetonitrile (v/v). API-ESMS data was collected for 115–1000 m/z positive and negative polarity. Data was analyzed using Agilent analysis software. MS TIC positive polarity trace was used to evaluate reaction selectivity. For clarity and quantification purposes traces corresponding to expected product ions were extracted from the MS TIC spectra (Refer to Supporting Information Figures S1–S6). Area under the peak was used to calculate content of each product (Refer to Supporting Information Tables S3, S4, S5).
Tocinoic acid modification procedure
To the 1 ml of 100 μM solution of tocinoic acid (Sigma-Aldrich) in 0.1 M Na2HPO4 buffer pH 7 or pH 8 was added 10 μL of 20 mM solution of FBDP in acetonitrile. Reaction was vortexed gently and allowed to react at room temperature. Aliquot of reaction mixture (5 μL) was analyzed by LCMS after 30 min of reaction time using Agilent LC/MSD SL mass spectrometer with Zorbax 300SP-C8, 5 μm, 4.6×50 mm column (Agilent). HPLC parameters were as follows: flow rate 0.5 ml/min, 5% mobile phase B over 2 min followed by a gradient to 5 to 95% B from 2 to 8 min, post time 4 min. Mobile phase A was 0.1% formic acid (v/v) in HPLC-grade H2O, and mobile phase B was 0.1% formic acid (v/v) in acetonitrile (v/v). API-ESMS data was collected for 115–1000 m/z positive and negative polarity. Data was analyzed using Agilent analysis software. MS TIC positive polarity trace was used to evaluate reaction selectivity. For clarity and quantification purposes traces corresponding to starting material and expected product ions were extracted from the MS TIC spectra (Refer to Supporting Information Figure S7). Area under the peak was used to calculate content of remaining starting material and product (Refer to Supporting Information Table S6). No N-terminal modification product has been found in the reaction mixture. LCMS: calcd for C37H49N10O11S2 (MH+) 873.2, found 873.2.
BSA modification
Method A (Stepwise)
To the 0.5 ml Eppendorf tube containing 99 μL of BSA solution (30 μM solution in 0.1M phosphate buffer pH 2–10) was added 1 μL or reagent solution (10 mM solution in CH3CN) and reaction mixture was vortexed briefly. Reaction was kept at room temperature for 30 minutes and the conversion was followed by visual observation (Figure S9) and UV-vis measurement at 340 nm that corresponds to the absorbance of diazene functionality (data not shown). The reaction products were purified using Zeba Spin desalting column (7,000Da MWC, Pierce) and the buffer was exchanged to PBS pH 5. In the 1.5 mL Eppendorf tube 99 mL of 30 μM solution of BSA-aldehyde in 0.1 M PBS pH 5 was treated with biotin hydrazide (1 μL of 100 mM solution in DMSO), overnight 4 °C, followed by removal of the excess of small molecule by Zeba Spin Desaltin column. The reaction products were tested in streptavidin ELISA (Figure S11) and characterized by MALDI-TOF (Figure S13).
Method B (One step diazo-coupling)
In the 0.5 ml Eppendorf tube were combined biotin hydrazide (10 μL of 100 mM solution in CH3CN; 1 eq), trifluoroacetic acid (10 μL of 200 mM solution in CH3CN; 2 eq) and FBDP (10 μL of 100 mM solution in CH3CN; 1 eq). The reaction mixture was mixed and allowed to react for 1 hr. Aliquot of the reagent was diluted and analyzed by LCMS (Figure S10) confirming formation of the hydrazide. To the 0.5 ml Eppendorf tube containing 99 μL of BSA solution (30 μM solution in 0.1M phosphate buffer pH 2–10) was added 1 μL or reagent solution (10 mM solution in CH3CN) and reaction mixture was vortexed briefly. Reaction was kept at room temperature for 30 minutes and reaction products were purified using Zeba Spin desalting column (7,000Da MWC, Pierce). The reaction products were tested in streptavidin ELISA (Figure S12) and characterized by MALDI-TOF and ESI-MS.
Streptavidin ELISA
Costar 96-well ELISA plate (Corning, Acton, MA) was coated with 100 ng of protein in 100 μL of PBS pH 7.4 and incubated overnight at 4 °C. The plate was washed with PBS containing 0.01% tween-20 (100 μL × 3). After blocking with 100 μL of 3% BSA/PBS, 0.01% tween-20 for 1 h at 37 °C and washing (100 μL of PBS containing 0.01% tween-20 × 5), 100 μL of streptavidin-HRP (Zymed, 1:3000 dilution in PBS) was added to each well and the plate was incubated for 1 hr at 37 °C. The plate was thoroughly washed with PBS/0.01% tween (100 μL × 5). Upon addition of the ABTS developing solution and incubation at room temperature for 10 min the absorbance was read at 405 nm. All measurements were done in triplicate and an average normalized result was graphed.
Chymotrypsinogen A modification and activity assay
Chymotrypsinogen was modified with 3.3 equivalents of FBDP at pH 8.0 following BSA modification procedure described above. Activity assay was performed essentially as described by Francis et al.1 In brief, an 114 μL aliquot of a reaction mixture for the modification of chymotrypsinogen (1.0 mg/mL, 38 μM total protein content) was treated with 2.6 μL of a solution of sequencing grade modified trypsin (Promega, 20 μg reconstituted with 200 μL of 50 mM acid). The activation of the zymogen was allowed to proceed for 10 minutes at room temperature before being purified and performing buffer exchange into 0.1 M Tris buffer (pH 7.6) using Zeba spin desalting column (Pierce). 40 μL aliquots of the activated protease were then added to 160 μL of 0.5 mM chymotrypsin substrate I, colorimetric (Suc- GGF-pNA, Calbiochem 230912) in 50 mM CaCl2, 20 mM Tris buffer, pH 7.6 (light green triangles). An analogous procedure was followed for reagent modified chymotrypsinogen A samples (light purple squares). In a negative control experiment, the enzyme was not activated with trypsin before addition of the tripeptide substrate (red squares), or the substrate was monitored with addition of only Tris buffer (blue diamonds). The progress of the reaction was monitored by UV-Vis spectrophotometry at 379 nm every 30 sec for 10 min (Figure S14). Each measurement was done in duplicate and the average value was plotted.
FBDP modification of trastuzumab antibody
To the 200 μL of trastuzumab solution in 0.1M phosphate buffer pH 8 (1.5 mg/ml) was added FBDP solution in CH3CN (2 μL of 20 μM solution). Reaction mixture was vortexed gently and allowed to react for 30 min. Reaction mixture turned yellow. The reaction was run through the Zeba spin desalting column (7K MWCO, Pierce) to remove excess FBDP and exchange into phosphate buffer pH 6.0. Then, 4 μL of 20 μM solution of biotin-oxyamine (Cayman Chemical, Cat.# 10009350) was added, reaction was vortexed and allowed to react at 4 °C for 18h. Excess of unconjugated biotin was removed using Zeba desalting column. Obtained conjugate was characterized by MALDI-TOF (Figure S23) and ELISA assays (Figure S22).
ErbB2 ELISA
ErbB2 binding ELISA was performed as described.2 In brief; Costar 96-well ELISA plates (Corning, Acton, MA) were coated with 25 ng of antigen (human ErbB2 or BSA) in 25 μL of PBS pH 7.4 and incubated overnight at 4 °C. The plate was washed with PBS containing 0.01% tween-20 (100 μL × 3). After blocking with 100 μL of 3% BSA/PBS, 0.01% tween-20 for 1 h at 37 °C and washing (100 μL of PBS containing 0.01% tween-20 × 5), 100ng/50 μL /well of trastuzumab, trastuzumab-CHO (trastuzumab modified only with FBDP, but not ligated with biotin-oxyamine) or trastuzumab-biotin(trastuzumab modified with FBDP followed by oxime ligation with biotin-oxyamine) construct or rituximab (negative control) solution was added and the plates were incubated for 1 hr at 37 °C. The plate was thoroughly washed with PBS/0.01% tween (100 μL × 5) followed by addition of 100 μL /well of secondary antibody solution (donkey anti-Human HRP, diluted 1:1000). The plate was incubated for 1h at 37 °C and washed with PBS/0.01% tween (100 μL × 5). Upon addition of the ABTS developing solution and incubation at room temperature for 20 min the absorbance was read at 405 nm. All measurements were done in triplicate and an average normalized result was graphed.
Streptavidin ELISA
In brief, Costar 96-well ELISA plates (Corning, Acton, MA) were coated with 200 ng of antigen (streptavidin or BSA) in 100 μL of PBS pH 7.4 and incubated overnight at 4 °C. The plate was washed with PBS containing 0.01% tween-20 (100 μL × 3). After blocking with 100 μL of 3% BSA/PBS, 0.01% tween-20 for 1 h at 37 °C and washing (100 μL of PBS containing 0.01% tween-20 × 5), 100ng/100 μL /well of trastuzumab, trastuzumab-CHO (trastuzumab modified only with FBDP, but not ligated with biotin-oxyamine) or trastuzumab-biotin (trastuzumab modified with FBDP followed by oxime ligation with biotin-oxyamine) construct, rituximab (negative control), biotinylated goat anti-rabbit antibody (positive control; Vector, Cat. # F1207) solution was added and the plates were incubated for 1 hr at 37 °C. The plate was thoroughly washed with PBS/0.01% tween (100 μL × 5) followed by addition of 100 μL /well of appropriate secondary antibody solution (AP goat anti-Human IgG F(ab)2 from Pierce, diluted 1:3000; or AP rabbit anti-goat IgG from Pierce, diluted 1:3000). The plate was incubated for 1h at 37 °C and washed with PBS/0.01% tween (100 μL × 5). Upon addition of the AP developing solution and incubation at room temperature for 20 min the absorbance was read at 405 nm. All measurements were done in triplicate and an average normalized result was graphed.
Cell Surface Labeling with FBDP
HeLa cells were harvested and washed twice with PBS pH 8.0. Washed cells were resuspended in PBS pH 8.0 to a final concentration of 5×106 cells/ml and 50 μL aliquots were transferred to the V-bottom non-TC treated plate followed by addition of FBDP (50 μL aliquots in PBS pH 8.0, 0.01% acetonitrile; final concentrations of FBDP were 100 μM, 10 μM and 1 μM). Cells were incubated with the reagent for 15 min at 37 °C. Cells were washed with PBS pH 6.0 (3 × 150 μL/well) followed by addition of 100 μL/well of 1 mM biotin-hydrazide (Sigma-Aldrich) in PBS pH 6.0 and incubation at room temperature for 30 min. Cells were washed with FACS buffer (3 × 150 μL/well; PBS pH 7.4, 1% BSA, 0.01% NaN3). Streptavidin-FITC (Jackson Laboratories) solution in FACS buffer was added to the cells (100 μL/well; 1:100 dilution) followed by incubation for 30 min at room temperature. The cells were washed with FACS buffer (5 × 150 μL/well) followed by Propidium Iodide staining for 15 min at room temperature (1 μg/ml; 100 μL/well). Cells were washed and transferred to the filter-top FACS tubes. Cells were analyzed on LSRII flow cytometer, 30000 events were collected. Data was analyzed usingFlowJo 8.7.1 and Microsoft Excel. All samples were tested in triplicate. Representative graphs from two independent experiments are shown (Figure S24).
RESULTS AND DISCUSSION
4-Formyl benzene diazonium hexafluorophosphate (FBDP, 2) and 4-formyl benzene diazonium tetrafluoroborate (FBDB) were prepared and reagent stability and reactivity were investigated. FBDP was synthesized on a gram scale from commercially available 4-formyl aniline polymer (Scheme 1). The aniline was treated with NaNO2 in concentrated HCl, followed by Cl− exchange to PF6− by the treatment with 60% HPF6 aqueous solution. The product precipitated from aqueous solution upon anion exchange making isolation straightforward. The FBDB reagent was prepared according to a previously reported procedure.(32, 33)
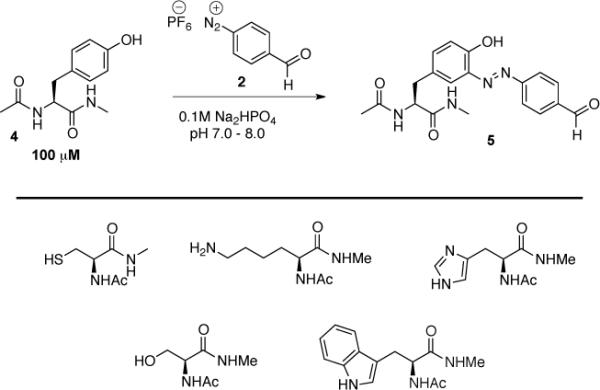
The reactivity profile of 4-formyl diazonium salts has not been extensively studied; only two reports were found in the literature and these concerned reactivity with phenol.(34, 35) Therefore a model reaction of the diazonium reagents with N-acyl tyrosine methylamide 4 was studied first. The reaction was performed in 67% sodium phosphate buffer/DMSO at room temperature. Buffer pH was varied from pH 5.0 to 8.0 (Scheme 2). The substrate concentration was 20 mM and only a small excess of the FBDP reagent was used (1.1 eq). FBDP reacted slowly with the model substrate at pH 5.0 and 6.0 providing the desired product 5 in 9% and 33% yields, respectively, after 12 h. In contrast, the reaction of FBDP at neutral pH and at mildly basic pH (pH 8.0), gave coupling product in excellent isolated yields of 96% and 99%, respectively. FBDP was stable and maintained excellent reactivity after more than 3 months of storage at 4 °C under air and/or after 1 week at room temperature under air. The isolated yields of the reaction at pH 8 performed with reagent stored at 4 °C and room temperature were 91% and 93%, respectively. In contrast, FBDB had good reactivity but much lower stability compared to FBDP. The isolated yield of the model reaction with freshly prepared FBDB was 84% under optimal pH conditions but only 21% after storing the reagent at room temperature for 1 week under air. Therefore FBDP was chosen for further studies.
Scheme 2.

We then studied formation of the oxime bond as a bioorthogonal linkage based on the introduction of the aldehyde handle. The reaction between the aldehyde and hydroxylamine hydrochloride (1.1 eq.) was performed in 25% 0.1 M Na2PO4 buffer/DMSO at room temperature (Scheme 2). As expected, the reaction proceeded smoothly and gave quantitative yield of 6 at pHs ranging from 5.0 to 8.0. Considering that a one-pot, three-component reaction would be more convenient for protein modification we evaluated the reaction of 4 with FBDP (1.1 eq.) for 1 h in 67% 0.1 M Na2PO4 buffer/DMSO at room temperature. Hydroxylamine hydrochloride (1.1 eq.) was added into the reaction mixture, and the reaction was stirred for 2 h. The one-pot reaction gave 6 in 93% isolated yield. Hydrazines could be used rather than hydroxylamine but the reaction time had to be extended. Reactions with methylhydrazine, acylhydrazine and benzohydrazide gave 92%, 85%, and 87% isolated yields, respectively. The alternative protocol involving premixing of the aldehyde and hydrazine or of the hydroxylamine components followed by diazonium coupling was also studied (see supporting information). After reaction between FBDP and hydroxylamine hydrochloride in the presence of AcOH in DMSO for 2 h, the resultant solution was added to 4 in 0.138 M Na2PO4 buffer (pH 8.0). This protocol provided 47% isolated yield of 6.
Our observations suggested that use of FBDP in the pH range of 7 to 8 would be optimal for protein bioconjugation. Therefore we conducted a selectivity study(25) with N-acyl methylamides of the amino acids potentially reactive with FBDP under the conditions closely resembling the intended protein modification reaction (Scheme 3). An equimolar mixture of N-acyl methylamides of tyrosine, histidine, tryptophan, lysine, serine, and cysteine (100 μM concentration of each amino acid) in 0.1 M Na2PO4 buffer at pHs of 7.0, 7.4, and 8.0 were treated with 1 equivalent of FBDP solution in acetonitrile (final content of acetonitrile in reaction mixture <1%), and the reaction progress was monitored by LCMS. Preferential diazo coupling to tyrosine was observed after 30 minutes at room temperature with average 95.3% conversion to diazene product 5 at pH 8.0, 89.1% conversion at pH 7.4, and 87.2% conversion at pH 7.0. The other amino acids were less reactive with histidine (1% at pH 8.0, 5.5% at pH 7.4, 7% at pH 7.0) more reactive than tryptophan (2% at pH 8.0, 3.2% at pH 7.4, 3.4% at pH 7.0) with unoxidized cysteine being the least reactive (1.7% at pH 8.0, 2.2% at pH 7.4, 2.2% at pH 7.0) (see supporting information). No reaction was observed with the lysine and serine amides. Thus, high tyrosine selective labeling is achieved at the modestly basic pH of 8. To further study selectivity, we examined the labeling efficiency and specificity using a more complex peptide. Reaction with a 100 μM solution of a the cyclic peptide tocinoic acid 7 proceeded cleanly with 2 equivalents of FBDP in 30 minutes to give only the tyrosine-modified adduct 8 in 88.5% and 91.4% conversions at pHs of 7.4 and 8.0, respectively (Scheme 4). No modification at the N-terminal amine was observed. Based on these results we expected that the FBDP reagent would preferentially modify surface-exposed tyrosine residues on proteins.
Scheme 3.
Scheme 4.

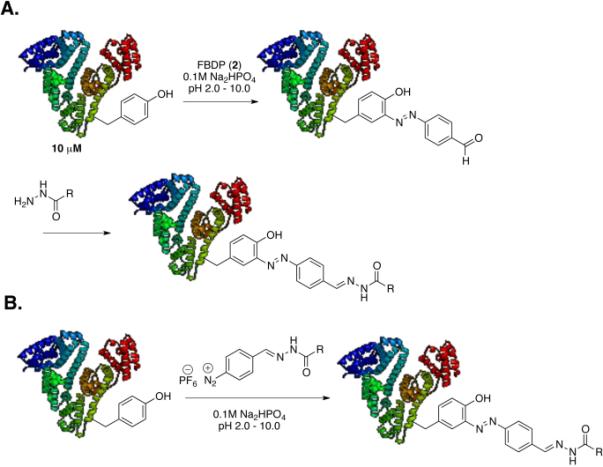
We then tested the FBDP reagent in reactions with model proteins including bovine serum albumin (BSA), human serum albumin (HSA), and chymotrypsinogen A. These proteins contain 21, 19, and 4 tyrosines, respectively. The one-pot vs. stepwise introduction of the biotin tag to BSA was studied with 30 μM BSA in 0.1 M Na2PO4 (pH 2.0–10) using 3.3 eq. of FBDP reagent (Figure 1). In the stepwise reaction BSA was treated with FBDP reagent for 30 minutes at room temperature, excess reagent was removed by gel filtration, and the protein exchanged into pH 6.0 buffer. Aldehyde containing protein was then treated with excess biotin-hydrazide for 18 h at 4 °C and excess biotin was removed by gel filtration. In the one-pot reaction, biotin-hydrazide was pre-mixed with FBDP solution in acetonitrile in the presence of 2 equivalents of TFA for 1 hour (hydrazide formation was confirmed by LCMS). Subsequently, 3.3 equivalents of the obtained reagent were added to the 30 μM BSA solution in 0.1 M Na2PO4 (pH 2.0–10). The reaction was allowed to proceed for 30 min at room temperature, and then excess of reagent was removed by gel filtration. Formation of diazene adduct was observed visually (upon reaction, the solution turns yellow) and by UV-vis spectroscopy (characteristic absorption at 340 nm). Biotin conjugation was confirmed by MALDI-TOF, ESI-MS and a biotin-streptavidin ELISA assay (Figure 2). The stepwise protocol resulted in better conjugation efficiency than did the one-pot reaction. Only mono-conjugated product was observed by ESI-MS (See Supporting Information). This labeling strategy is versatile since a stock of aldehyde-containing protein can be prepared and used for conjugation with a variety of oxyamine and/or hydrazine tags.
Figure 1.
Diazonium-based biotinylation of proteins.
Figure 2.
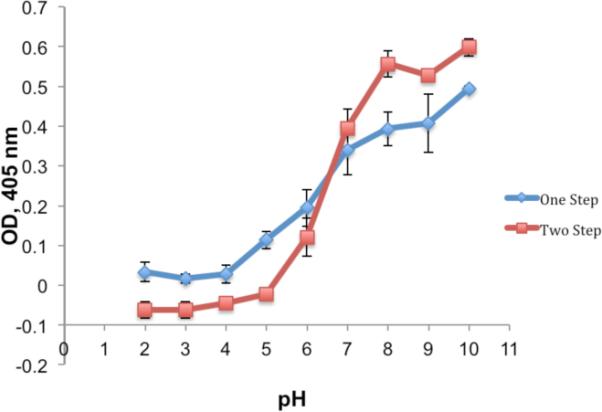
Diazonium-based biotinylation efficiency as a function of pH.
We next evaluated FBDP modification of chymotrypsinogen A using the stepwise protocol; modification proceeded smoothly at pH 8.0 with 3.3 equivalents of FBDP and did not affect activity of the enzyme (see supporting information). We then used the reaction with HSA to evaluate the influence of conditions including pH (7.0, 7.4, 7.6, 7.8, 8.0, 8.4), reaction time (30 min, 1h, 2h, 3h, 4h), temperature (4 °C, 25 °C, 37 °C), reagent excess (1–100 equiv.), and protein concentration (2.5 – 20 mg/ml). Factors that influenced the reaction the most were reaction temperature (reaction slowed at 4 °C and yielded only ~0.7 modifications/protein in 30 min) and amount of excess reagent used (over 20 equivalents of reagent provided >2 modifications/protein in 30 min at pH 8.0). A pH range of 7.4–8.0 was optimal and provided an average of ~1 modification/protein. At pH 8.0, an extended reaction time (>1h) resulted in incorporation of ~1.5 modifications/protein. Protein concentration had no effect on reaction outcome and provided ~1 modification/protein for all tested protein concentrations. Optimal reaction conditions are therefore 10 equivalents of FBDP at pH 8.0 for 30 min at room temperature (see supporting information for further details of these experiments). Since the introduction of poly(ethylene) glycol chains (PEGylation) is widely used to modify the pharmacokinetic properties of proteins, we studied the potential of this approach in protein PEGylation(36, 37). This was achieved by simply reacting FBDP-modified HSA with 5K linear poly(ethylene) glycol –hydrazide (PEG-hydrazide) at pH 5.0 for 18 h; exclusively mono-PEGylated product was formed, however in low conversion in this unoptimized study (see supporting information). Although the reaction was incomplete after 18 h, separation of homogeneous product from starting material was straightforward and starting material could be recycled. For comparison, reaction with 10 equivalents of 5k PEG-NHS for the same amount of time resulted in a mixture of mono-, bis-, tri-, and tetra-PEGylated products and starting material.
Our protein modification results are in accord with studies published by other research groups and provide further support to the observation that diazonium salts react preferentially with surface exposed tyrosine residues. Therefore some degree of control over the reaction selectivity can be expected with this approach.
Since antibodies constitute perhaps the most important class of proteins in bioconjugation chemistry, we studied the potential of FBDP labeling of the antibody trastuzumab.(38) Antibody was modified using the optimized two-step protocol with 10 equivalents of FBDP (pH 8.0, 30 min, rt) and 20 equivalents of biotin-oxyamine (pH 6.0, 4 °C, o/n). Chemically modified trastuzumab was found to incorporate an average of 1.8 biotin tags per antibody molecule according to MALDI-TOF characterization. ESI-MS characterization revealed adducts containing one, two and three modifications per antibody molecule. Chemical modification did not affect ErbB2 recognition of trastuzumab and the biotin tag was readily detected through binding to streptavidin (Figure 3).
Figure 3.

ELISA evaluation of the FBDP modified trastuzumab and binding to ErbB2 and streptavidin.
Considering that diazocoupling reaction with FBDP takes place rapidly and under relatively mild conditions, we have tested its potential for cell surface labeling of live cells. Freshly harvested HeLa cells were treated with FBDP for 15 min at 37 °C followed by conjugation of the aldehyde tag to the biotin hydrazide and detection of biotin with streptavidin-FITC. Toxicity of the FBDP modification procedure was monitored by staining cells with propidium iodide. This study demonstrated that human cells were efficiently labeled in a concentration dependent fashion using 1–100 μM FBDP (Figure 4) and that even at the highest reagent concentration used, we did not observe any significant toxicity(see supporting information for full details).
Figure 4.

HeLa cells labeling with FBDP. (a) Schematic representation of the labeling procedure; (b) FITC histogram overlay of unlabeled cells (red), cells treated with 100 uM FBDP (blue) or 10 uM FBDP (green) or 1 uM FBDP (orange); (c) Representative dot plots of cells labeled with 100 uM FBDP (I) and unlabeled cells (II).
In summary, we have synthesized a novel and unusually stable 4-formyl benzenediazonium hexafluorophosphate salt that can be used for tyrosine-selective modification of peptides and proteins to introduce bioorthogonal aldehyde tags suitable for classical oxime and hydrazide ligations. The hexafluorophosphate counterion was essential for stability of the diazonium salt and allows preparation of the reagent as a crystalline solid on a multigram scale. This convenient new reagent should facilitate the preparation of a wide variety of peptide and protein conjugates and may be utilized for cell surface labeling. Through an agreement with Sigma-Aldrich, FBDP is now commercially available (product number: L-511382).
Supplementary Material
ACKNOWLEDGMENT
This study was supported by US National Institutes of Health grant AI 095038 and The Skaggs Institute for Chemical Biology.
ABBREVIATIONS
- FBDP
4-formyl benzene diazonium hexafluorophosphate
- FBDB
4-formyl benzene diazonium tetrafluoroborate
- BSA
bovine serum albumin
- HSA
human serum albumin
Footnotes
Supporting Information. Detailed syntheses and analytical data of molecules, coupling protocols, selectivity studies, protein and cell labeling studies, and assay details are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Hooker JM, Kovacs EW, Francis MB. Interior surface modification of bacteriophage MS2. Journal of the American Chemical Society. 2004;126:3718–9. doi: 10.1021/ja031790q. [DOI] [PubMed] [Google Scholar]
- (2).Schlick TL, Ding Z, Kovacs EW, Francis MB. Dual-surface modification of the tobacco mosaic virus. Journal of the American Chemical Society. 2005;127:3718–23. doi: 10.1021/ja046239n. [DOI] [PubMed] [Google Scholar]
- (3).Jones MW, Mantovani G, Blindauer CA, Ryan SM, Wang X, Brayden DJ, Haddleton DM. Direct peptide bioconjugation/PEGylation at tyrosine with linear and branched polymeric diazonium salts. Journal of the American Chemical Society. 2012;134:7406–13. doi: 10.1021/ja211855q. [DOI] [PubMed] [Google Scholar]
- (4).Roglans A, Pla-Quintana A, Moreno-Manas M. Diazonium salts as substrates in palladium-catalyzed cross-coupling reactions. Chemical reviews. 2006;106:4622–43. doi: 10.1021/cr0509861. [DOI] [PubMed] [Google Scholar]
- (5).Hooker JM, Datta A, Botta M, Raymond KN, Francis MB. Magnetic resonance contrast agents from viral capsid shells: a comparison of exterior and interior cargo strategies. Nano letters. 2007;7:2207–10. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]
- (6).Le Callonnec F, Fouquet E, Felpin FX. Unprecedented substoichiometric use of hazardous aryl diazonium salts in the Heck-Matsuda reaction via a double catalytic cycle. Organic letters. 2011;13:2646–9. doi: 10.1021/ol200752x. [DOI] [PubMed] [Google Scholar]
- (7).Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl. 2009;48:6974–98. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hudak JE, Yu HH, Bertozzi CR. Protein glycoengineering enabled by the versatile synthesis of aminooxy glycans and the genetically encoded aldehyde tag. Journal of the American Chemical Society. 2011;133:16127–35. doi: 10.1021/ja206023e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Iyer G, Pinaud F, Xu J, Ebenstein Y, Li J, Chang J, Dahan M, Weiss S. Aromatic aldehyde and hydrazine activated peptide coated quantum dots for easy bioconjugation and live cell imaging. Bioconjugate chemistry. 2011;22:1006–11. doi: 10.1021/bc100593m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hudak JE, Barfield RM, de Hart GW, Grob P, Nogales E, Bertozzi CR, Rabuka D. Synthesis of heterobifunctional protein fusions using copper-free click chemistry and the aldehyde tag. Angew Chem Int Ed Engl. 2012;51:4161–5. doi: 10.1002/anie.201108130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Key JA, Li C, Cairo CW. Detection of cellular sialic acid content using nitrobenzoxadiazole carbonyl-reactive chromophores. Bioconjugate chemistry. 2012;23:363–71. doi: 10.1021/bc200276k. [DOI] [PubMed] [Google Scholar]
- (12).Han S-Y, Kim Y-A. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron. 2004;60:2447–2467. [Google Scholar]
- (13).Cygler MP, M., Elofson RM. The crystal structure of benzenediazonium tetrafluoroborate, C6H5N2+•BF4−. Canadian Journal of Chemistry. 1982;60:2852–2855. [Google Scholar]
- (14).Ebrahimi N, Zarchi MAK. Iodination of Stable Aromatic Diazonium Salt Using Crosslinked Poly (4-vinylpyridine)-Supported Iodide. Journal of Applied Polymer Science. 2011;124:2807–2813. [Google Scholar]
- (15).Mahouche-Chergui S, Gam-Derouich S, Mangeney C, Chehimi MM. Aryl diazonium salts: a new class of coupling agents for bonding polymers, biomacromolecules and nanoparticles to surfaces. Chemical Society reviews. 2011;40:4143–66. doi: 10.1039/c0cs00179a. [DOI] [PubMed] [Google Scholar]
- (16).Hari DP, Schroll P, Konig B. Metal-free, visible-light-mediated direct C-H arylation of heteroarenes with aryl diazonium salts. Journal of the American Chemical Society. 2012;134:2958–61. doi: 10.1021/ja212099r. [DOI] [PubMed] [Google Scholar]
- (17).Dirksen A, Hackeng TM, Dawson PE. Nucleophilic catalysis of oxime ligation. Angew Chem Int Ed Engl. 2006;45:7581–4. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- (18).Dirksen A, Dawson PE. Rapid oxime and hydrazone ligations with aromatic aldehydes for biomolecular labeling. Bioconjugate chemistry. 2008;19:2543–8. doi: 10.1021/bc800310p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. Dual-surface-modified bacteriophage MS2 as an ideal scaffold for a viral capsid-based drug delivery system. Bioconjugate chemistry. 2007;18:1140–7. doi: 10.1021/bc070006e. [DOI] [PubMed] [Google Scholar]
- (20).Datta A, Hooker JM, Botta M, Francis MB, Aime S, Raymond KN. High relaxivity gadolinium hydroxypyridonate-viral capsid conjugates: nanosized MRI contrast agents. Journal of the American Chemical Society. 2008;130:2546–52. doi: 10.1021/ja0765363. [DOI] [PubMed] [Google Scholar]
- (21).Hooker JM, O'Neil JP, Romanini DW, Taylor SE, Francis MB. Genome-free viral capsids as carriers for positron emission tomography radiolabels. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. 2008;10:182–91. doi: 10.1007/s11307-008-0136-5. [DOI] [PubMed] [Google Scholar]
- (22).Joshi NS, Whitaker LR, Francis MB. A three-component Mannich-type reaction for selective tyrosine bioconjugation. Journal of the American Chemical Society. 2004;126:15942–3. doi: 10.1021/ja0439017. [DOI] [PubMed] [Google Scholar]
- (23).McFarland JM, Joshi NS, Francis MB. Characterization of a three-component coupling reaction on proteins by isotopic labeling and nuclear magnetic resonance spectroscopy. Journal of the American Chemical Society. 2008;130:7639–44. doi: 10.1021/ja710927q. [DOI] [PubMed] [Google Scholar]
- (24).Guo HM, Minakawa M, Ueno L, Tanaka F. Synthesis and evaluation of a cyclic imine derivative conjugated to a fluorescent molecule for labeling of proteins. Bioorganic & medicinal chemistry letters. 2009;19:1210–3. doi: 10.1016/j.bmcl.2008.12.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ban H, Gavrilyuk J, Barbas CF., 3rd Tyrosine bioconjugation through aqueous ene-type reactions: a click-like reaction for tyrosine. Journal of the American Chemical Society. 2010;132:1523–5. doi: 10.1021/ja909062q. [DOI] [PubMed] [Google Scholar]
- (26).Lorenzi M, Puppo C, Lebrun R, Lignon S, Roubaud V, Martinho M, Mileo E, Tordo P, Marque SR, Gontero B, Guigliarelli B, Belle V. Tyrosine-targeted spin labeling and EPR spectroscopy: an alternative strategy for studying structural transitions in proteins. Angew Chem Int Ed Engl. 2011;50:9108–11. doi: 10.1002/anie.201102539. [DOI] [PubMed] [Google Scholar]
- (27).McAuley A, Jacob J, Kolvenbach CG, Westland K, Lee HJ, Brych SR, Rehder D, Kleemann GR, Brems DN, Matsumura M. Contributions of a disulfide bond to the structure, stability, and dimerization of human IgG1 antibody CH3 domain. Protein science : a publication of the Protein Society. 2008;17:95–106. doi: 10.1110/ps.073134408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Traxlmayr MW, Wozniak-Knopp G, Antes B, Stadlmayr G, Ruker F, Obinger C. Integrin binding human antibody constant domains--probing the C-terminal structural loops for grafting the RGD motif. Journal of biotechnology. 2011;155:193–202. doi: 10.1016/j.jbiotec.2011.06.042. [DOI] [PubMed] [Google Scholar]
- (29).Almagro JC, Raghunathan G, Beil E, Janecki DJ, Chen Q, Dinh T, LaCombe A, Connor J, Ware M, Kim PH, Swanson RV, Fransson J. Characterization of a high-affinity human antibody with a disulfide bridge in the third complementarity-determining region of the heavy chain. Journal of molecular recognition : JMR. 2012;25:125–35. doi: 10.1002/jmr.1168. [DOI] [PubMed] [Google Scholar]
- (30).Takaoka Y, Tsutsumi H, Kasagi N, Nakata E, Hamachi I. One-pot and sequential organic chemistry on an enzyme surface to tether a fluorescent probe at the proximity of the active site with restoring enzyme activity. Journal of the American Chemical Society. 2006;128:3273–80. doi: 10.1021/ja057926x. [DOI] [PubMed] [Google Scholar]
- (31).Wakabayashi H, Miyagawa M, Koshi Y, Takaoka Y, Tsukiji S, Hamachi I. Affinity-labeling-based introduction of a reactive handle for natural protein modification. Chemistry, an Asian journal. 2008;3:1134–9. doi: 10.1002/asia.200800057. [DOI] [PubMed] [Google Scholar]
- (32).Lai L-L, Ho C-H, Lin Y-J, Wang E, Liu Y-H, Wang Y, Lin Y-C, Cheng K-L. Synthesis and Study of an N,N-Disubstituted 4-[(4-Aminophenyl)diazenyl]benzaldehyde. Helvetica Chimica Acta. 2002;85:108–114. [Google Scholar]
- (33).Haque AM, Kim K. Aldehyde-functionalized benzenediazonium cation for multiprobe immobilization on microelectrode array surfaces. Langmuir : the ACS journal of surfaces and colloids. 2011;27:882–6. doi: 10.1021/la104270b. [DOI] [PubMed] [Google Scholar]
- (34).Lan Y-X, Qiu Y-G, Lin W-S, Yan S, Na G, Biao G. Synthesis of 2- (4-Formylphenylazo)-7- (4-Antipyrylazo)-1,8-Dihydroxynaphthalene-3,6-Disulphonic Acid. Journal of Shenyang Institute of Chemical Technology. 2004;3:165–168. [Google Scholar]
- (35).Kuvshinova S, Zavyalov A, Burmistrov V, Aleksandriiskii V, Koifman O. Mesogenic 4-alkoxy-2-hydroxy-4-formylazobenzenes. Russian Journal of Organic Chemistry. 2006;42:393–395. [Google Scholar]
- (36).Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2008;22:315–29. doi: 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- (37).Bailon P, Won CY. PEG-modified biopharmaceuticals. Expert opinion on drug delivery. 2009;6:1–16. doi: 10.1517/17425240802650568. [DOI] [PubMed] [Google Scholar]
- (38).Goldenberg MM. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clinical therapeutics. 1999;21:309–18. doi: 10.1016/S0149-2918(00)88288-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.