

Abstract
A bioactivity-guided approach was taken to identify the acetylcholinesterase (AChE, EC 3.1.1.7) inhibitory agent in a Magnolia x soulangiana extract using a microplate enzyme assay with Ellman’s reagent. This permitted the isolation of the alkaloids taspine (1) and (−)-asimilobine (2), which were detected for the first time in this species. Compound 1 showed a significantly higher effect on AChE than the positive control galanthamine and selectively inhibited the enzyme in a long-lasting and concentration-dependent fashion with an IC50 value of 0.33 ± 0.07 μM. Extensive molecular docking studies were performed with human and Torpedo californica-AChE employing Gold software to rationalize the binding interaction. The results suggested ligand 1 to bind in an alternative binding orientation when compared to galanthamine. While this is located in close vicinity to the catalytic amino acid triad, the 1–AChE complex was found to be stabilized by (i) sandwich-like π-stacking interactions between the planar aromatic ligand (1) and the Trp84 and Phe330 of the enzyme, (ii) an esteratic site anchoring with the amino side chain, and (iii) a hydrogen-bonding network.
The enzyme acetylcholinesterase (AChE; EC 3.1.1.7) plays a central role in cholinergic brain synapses and neuromuscular junctions. Its major role is the termination of impulse transmission by rapid hydrolysis of the neurotransmitter acetylcholine (ACh). According to the cholinergic hypothesis,1 the selective and irreversible deficiency of cholinergic functions leads to memory impairment. The direct correlation of cholinergic deficit and the severity of dementia, e.g., in patients suffering from Alzheimer’s disease (AD) and other related diseases, is why these forms of memory impairment are treated symptomatically with AChE inhibitors (AChEIs). As knowledge of the mechanisms responsible for neurodegeneration in AD increases, it is anticipated that neuroprotective drugs will be developed to complement current symptomatic treatments. Research has shown a direct link between cholinergic impairment and altered amyloid precursor protein (APP) processing as a major pathogenetic event in AD.2 Beyond their well-known symptomatic effect in supplementing inadequate levels of ACh, there is evidence that AChEIs cause a selective muscarinic activation of α-secretase, induce the translation of APP mRNA, and may also restrict amyloid fiber assembly.3
Four AChEIs have been approved by the European and U.S. regulatory authorities: tacrine (Cognex), donepezil (Arizept), galanthamine (Reminyl), and rivastigmine (Exelon). Whereas tacrine and donepezil are synthetic in nature, galanthamine is a naturally occurring alkaloid from the genus Galanthus (Amaryllidaceae). Rivastigmine is a compound closely related to physostigmine representing the prototype AChEI from the seeds of Physostigma venenosum (Papilionaceae), although endowed with unfavorable pharmacokinetic properties. Huperzine A, a further alkaloid from the club moss Huperzia serrata (Lycopodiaceae), has been approved as drug for the treatment of AD in the People’s Republic of China.4 Several natural products has been found to act as potent anticholinesterase agents, among which there are a large number of alkaloids.5
The finding of novel lead structures with anticholinesterase potency is one of the most pressing pharmacological tasks in the search for improved drug candidates in view of the continuously growing number of patients suffering from dementia.6
In recent years, various computational data-mining approaches have been performed to bridge the gap between known structural features and pharmacological properties to offer a more focused procedure for ligand identification. AChE from Torpedo californica (Tc) has evolved as an important and essential tool for studying the structure and function of this enzyme. It consists of 537 amino acids and exhibits high homology with the human enzyme.7,8 Human AChE (hAChE) consists of 543 amino acid residues that form 12 β-sheets and 14 α-helices. A characteristic of AChE is a narrow gorge penetrating approximately 20 Å deep into the enzyme and there leading to its catalytic site. Altogether, 14 aromatic amino acid residues are involved in forming this gorge, which plays a key role in leading the substrate/product into/away from the active site by cation-stacking interactions. At the bottom of the gorge, an anionic binding site fixes the substrate to form ideal contacts with the catalytic triad, which consists of Ser203, His447, and Glu334.7,8
Numerous X-ray crystal structures of TcAChE complexed with various inhibitors have been added to the protein data bank (PDB)9 and have contributed in an essential way to the understanding of ligand–target binding interactions, the spatial arrangement of the binding sites, and the requirements for putative ligands. Previously, a structure-based pharmacophore model based on the structural knowledge of the TcAChE-galanthamine interactions (PDB entry 1QTI10) has been established in our group.11 The pharmacophore-based virtual screening of in-house-generated multiconformational 3D databases has aided in the identification of hitherto unknown nonalkaloid natural AChE inhibitors.11,12 However, for untapped natural sources or species, where sufficient information about the contained secondary metabolites is not available, the bioactivity-guided approach is still an indispensable method to access the biologically active principles of organisms.
In ongoing efforts to discover AChE inhibitors from nature, an in vitro assay screening and subsequent bioactivity-guided fractionation of active extracts helped in selecting plant material worth in-depth study. The alkaloid fraction of Magnolia x soulangiana was identified as having appreciable inhibitory potential against AChE. The aim of the current study was to (i) identify its bioactive constituent(s), (ii) determine the in vitro inhibitory effect against AChE and butyrylcholinesterase (BChE), and (iii) predict the ligand–target interactions in the enzyme’s binding site by means of docking studies.
Results and Discussion
Bioavtivity-Guided Approach
Some 130 CH2Cl2 and MeOH extracts of various angiosperms native to and cultivated in Tyrol, Austria, were screened for their AChE-inhibiting activity at a concentration of 1 mg/mL using a spectrophotometric enzyme assay based on Ellman’s method.13 Extracts of the leaves and bark of some Magnoliaceae showed significant inhibiting activity (Table S1, Supporting Information). Among the extracts investigated the methanol crude extract of the leaves of M. x soulangiana emerged as the most potent one. In a preliminary investigation an alkaloid fraction from the methanol extract of the leaves was prepared to determine a possibly AChE-inhibiting effect of these alkaloids in comparison to the nonalkaloid fraction. Whereas the latter showed no effect at a concentration of 100 μg/mL, the alkaloid fraction strongly inhibited AChE (Table S1, Supporting Information).
For the bioactivity-guided fractionation, an alkaloid-enriched extract (organic phase II) was prepared from the foliage of M. x soulangiana (see Experimental Section). This extract showed a high and stable AChE-inhibitory effect in the enzyme assay with an IC50 of 7.81 ± 1.22 μg/mL. The extract was fractionated using Sephadex gel chromatography with CH2Cl2 as eluant. The percentage of enzyme activity for each fraction (A1–A13) was calculated with a sample concentration of 10 μg/mL. Fraction A4 (190 mg), showing the highest inhibitory effect (96.45 ± 1.32%), was subjected to silica gel flash column chromatography to obtain 20.7 mg of a Dragendorff’s reagent-positive compound (subfraction B3). This was further purified by precipitation from MeOH to obtain 12.4 mg of an optically inactive isolate. The structure was identified as the tertiary base taspine 1 {1-(2-dimethylaminoethyl)-3,8-dimethoxychromeno[5,4,3-cde]chromene-5,10-dione}, deduced from mass fragmentation, 1D and 2D NMR experiments, and comparison with data in the literature.14 The molecular structure of 1 is characterized by high symmetry. The planar tetracyclic compound consists of two linked lactone rings; each is condensed again with an aromatic ring that is substituted with a methoxy group. One of the aromatic rings is bearing the dimethylaminoethyl moiety and thus represents a protoalkaloid.
A further alkaloid was obtained as a minor constituent from the alkaloid fraction (from subfraction B4; 3.4 mg). By comparison of its spectroscopic characteristics, optical rotation, and mass spectrometric data with previous reports,15 it was identified as the known aporphine alkaloid (−)-asimilobine (2) [1-methoxy-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinolin-2-ol (6aR)].
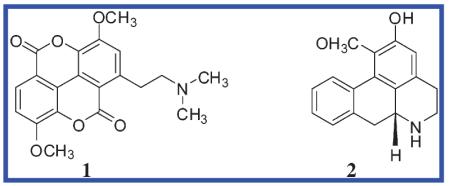
Previous researchers have investigated phytochemically M. x soulangiana (a hybrid of M. denudata and M. liliiflora).16 Lipophilic polyphenols, chromanols,17 melampolides,18 several lignans from the flower buds,19 and the neolignan denudatin A20 have been described as well as the benzylisoquinoline alkaloids anonaine, remerine, liriodenine,21 oxolaureline,22 and magnoflorine.23 Compounds 1 and 2 have not been described in the title species, but do occur in other representatives of the genus Magnolia.14,24 Whereas 2 is a well-known metabolite within the class of Magnoliopsida (e.g., in Nelumbonaceae, Menispermaceae, Magnoliaceae, and Annonaceae), compound 1 has been reported mainly from species in the Berberidaceae (e.g., in the genera Caulophyllum and Leontice) and the Euphorbiaceae (e.g., in the genus Croton). The biosynthesis of the unique structure 1 was discussed by Milanowski and coauthors,25 who suggested that the generation of compound 1 via magnoflorine is the biosynthetic endpoint of a variety of benzylisoquinoline-derived alkaloids from Croton lechleri. Although the family Euphorbiaceae is phylogenetically unrelated to the Magnoliaceae and Berberidaceae, the presence of similar benzylisoquinoline-derived alkaloids and the co-occurrence of 1 support this biosynthetic pathway.
In the enzyme assay performed in this study, 1 showed a dose-dependent and long-lasting inhibitory effect on AChE, with an IC50 of 0.12 ± 0.03 μg/mL, or 0.33 ± 0.07 μM (Figure 1), thus being 10 times more potent than galanthamine, which served as the positive control in our assay (IC50 of 3.2 ± 1.0 μM). When hAChE was used for the in vitro test, similar inhibitory effects were determined, revealing 1 to be about twice as active as galanthamine (Table 1). Compound 1 showed no effect on BChE, thus acting as a selective inhibitor of AChE. In contrast to 1, compound 2 showed effects on neither AChE nor BChE (Table 1).
Figure 1.
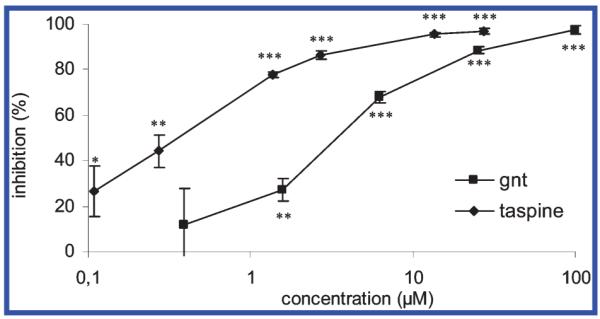
Microplate assay: inhibitory effects of taspine (1) and galanthamine on AChE. Statistical analysis: ***p < 0.001, **p < 0.01, *p < 0.05. Student’s t-test of absorption data after 30 min in comparison with medium control, n = 4.
Table 1.
In Vitro IC50 Values (μM) of Taspine (1) and (−)-Asimilobine (2) on AChE, hAChE, and BChE in Comparison with the Positive Controls Galanthamine (for AChE) and Tacrine (for BChE)
| AChEa | hAChEa | BChEa | |
|---|---|---|---|
| taspine (1) | 0.33 ± 0.07 | 0.54 ± 0.10 | >100 |
| (−)-asimilobine (2) | >100 | >100 | >100 |
| galanthamine | 3.20 ± 1.02 | 1.18 ± 0.31 | n.d. |
| tacrine | 0.22 ± 0.09 | n.d. | 0.04 ± 0.02 |
CI95
n.d. not determined
In Silico Approach
A further objective of this study was to predict how compound 1 interacted with the protein-binding site. Thus, 3D conformers of ligand 1 were calculated with Catalyst (Version 4.10, Accelrys, San Diego, CA), but none of them matched all the features of the pharmacophore model we elaborated previously, which is based on the cocrystal structure of TcAChE and galanthamine.11 To explore the underlying binding modes of the 1–AChE complex, extensive molecular docking studies were conducted.
We first assessed the ability of the docking program Gold26 to find accurate docking solutions for ligands that had previously cocrystallized with AChE. To this end, (−)-galanthamine was docked into the PDB entry 1W6R,27 and tacrine was docked into the binding site of the parent enzyme of PDB entry 1ACJ.28 Both ligands were found to be positioned identically to their experimentally derived binding positions into the enzyme by the docking algorithm. Accordingly, the Gold docking algorithm was well-suited for the experiments conducted.
In order to address the question as to whether the results obtained with TcAChE could be compared with hAChE, the ligand-binding domains of the PDB entries 1B41 (human enzyme), 1ACJ, and 1W6R (Tc variants) were overlaid using LigandScout software.29 The alignment of crucial amino acid residues of the active site (h/TcAChE: Trp86/84, Glu202/199, Ser203/200, and His447/440) resulted in an excellent superimposition with an rms value of only 0.271 (Figure 2), indicating that both binding pockets offered nearly identical conditions for ligand binding. As already known from crystallization experiments with TcAChE, the Phe330/Tyr337 residue has some conformational flexibility that is also recognized in our overlay. This conformational flexibility has a fundamental impact on the size and shape of the ligand-binding pocket. While in 1ACJ (originally containing tacrine as ligand), Phe330 and Trp86 are oriented nearly in a parallel conformation (flanking tacrine in a sandwich-like manner and forming ideal π–π stacking interactions), the galanthamine-containing entry 1W6R is slightly rotated. In the human enzyme, Tyr337 has a rotational freedom comparable to Phe330 in the Tc enzyme; however, as there is no structure with a cocrystallized ligand available, the definite orientation of Tyr337 remains open (Figure 2). As this flexibility is present in both hAChE and TcAChE, the Tc enzyme crystal structures may be regarded as useful tools for docking studies examining binding modes of hAChEI.
Figure 2.

Overlay of ligand-binding domains of hAChE (PDB: 1B41, stick style, highlighted) and TcAChE (PDBs: 1W6R, line style; 1ACJ, stick style) visualized with LigandScout.29 Galanthamine (from 1W6R) is shown in ball-and-stick style forming hydrogen-bonding interactions with two of the catalytic amino acids.
The impact of the conversion of Phe330 into Tyr337 on ligand binding is regarded to be negligible as docking studies suggest.30 In order to determine whether this finding is also applicable in the case of taspine (1), galanthamine, and tacrine, these ligands were docked in both their protonated and nonprotonated form into the active sites of the PDB complexes 1B41 (hAChE enzyme without cocrystallized ligand), 1W6R (TcAChE with a galanthamine-like ligand), and 1ACJ (TcAChE with tacrine).
Regarding the suitability of GoldScore to estimate the activity of the AChEIs investigated, it was observed that all of these compounds were highly ranked by the score, correctly indicating their in vitro activity. However, the algorithm is not suitable for quantitative activity prediction, as the order of ranking did not correspond to the measured activity (Table 2).
Table 2.
GoldScores for the Docking of Taspine (1), Galanthamine, and Tacrine in Their Protonated and Neutral Forms into Different AChE Crystal Structures
| ligand | 1B41 | 1W6R | 1ACJ |
|---|---|---|---|
| taspine | 43.78 | 61.12 | 62.23 |
| taspine-protonated | 49.70 | 66.10 | 58.59 |
| galanthamine | 48.34 | 76.00 | 65.63 |
| galanthamine-protonated | 49.76 | 75.78 | 67.58 |
| tacrine | 57.24 | 60.37 | 58.04 |
| tacrine-protonated | 54.11 | 50.31 | 48.97 |
In the case of taspine (1), the exchange of Phe330 for Tyr337 is not important for its binding orientation; in all experiments there was no interaction observed between the hydroxyl group of Tyr337 and the ligand. A detailed analysis of the docking results is provided in the Supporting Information.
Docking of Taspine (1) in TcAChE
The ligand-binding site of the PDB entry 1ACJ was selected for docking studies, as both tacrine (the original ligand to this complex) and compound 1 comprise a spacious, planar aromatic structure suggesting similar binding modes (Figure 3). As observed in the crystal structure, tacrine remains solely stacked between Trp84 and Phe330 (Figure 3A). No anchoring to the enzyme’s esteratic site could be observed. However, a water-mediated hydrogen-bonding network with several surrounding amino acids, e.g., Ser122, Asp72, accounts for the tight binding of tacrine to AChE (Figure 3B). For the docked 1–TcAChE complex three major interaction patterns were observed: (i) Compound 1 π-stacked on one side with Trp84 and on the other side with Phe330. Thus, the ideally placed and stabilized ligand 1 may be best seen in Figure 3C (violet π-π interactions). (ii) The cationic side chain of 1 occupied by a positively ionized feature (Figure 3D; blue sphere) reaches into the esteratic site (i.e., catalytically active site) anchoring at His440 (Figure 3D; green hydrogen bond donor), Ser200, and Glu199 (which is not part of the catalytic triad). (iii) A small hydrogen-bonding network (Figure 3D; red hydrogen bond acceptors) with active site water molecules and Ser122 further stabilized ligand binding. The multitude of observed interactions and the high GoldScore computed for the reported docking pose (56.35 vs 58.09 for tacrine) are good indicators for the accuracy of the determined binding orientation.
Figure 3.

Two perspectives of tacrine bound to TcAChE (A and B) and two perspectives of taspine (1) docked into the bottom cavity of TcAChE revealing a binding mode comparable to tacrine (C and D) visualized with LigandScout.29 Color code: violet, π–π interactions; blue, positively ionized feature; green, hydrogen bond donor; red, hydrogen bond acceptor.
Docking of Taspine (1) and Tacrine into hBChE
In order to explain the potent binding of tacrine to BChE and determine why taspine (1) has no effect on the enzyme, both ligands were docked in their protonated form into the active site of hBChE (PDB entry 1P0M).9 The nonprotonated state of tacrine revealed a binding mode similar to that observed in the TcAChE active site (PDB entry 1ACJ). The ligand π–π stacks with Trp82 and forms hydrogen bonds with two active site water molecules (Figure 1A, Supporting Information). In the protonated form, it still aligns itself parallel to Trp82; however, it does so in a 180° flipped orientation. The protonated nitrogen atom forms ionic interactions with Glu197 (Figure 1B, Supporting Information). Generally, ionic interactions lead to tight binding of two interaction partners. This would explain the potent inhibition of BChE by tacrine.
While all tacrine solutions are nearly identical and reveal equal interactions with the protein, taspine (1) has obviously more options to spin around in the ligand-binding domain and does not orient itself parallel to Trp82. Only in two out of five docking solutions does the positive nitrogen atom interact with Glu197. When analyzing the top five ranked docking poses, no preferred orientation of taspine (1) in the BChE-binding pocket can be determined. Two of the different docking solutions are visualized in the Supporting Information (Figure S2).
Although the GoldScore for taspine (1) is similarly high to the one for tacrine, there seems to be no “ideal” binding geometry for taspine (1) to anchor tightly in the BChE-binding site. This observation could explain why taspine (1) is not an inhibitor for this enzyme.
Taken together, the molecular insight provided by the docking protocols offered an explanation for the strong binding affinity of compound 1 in the AChE-binding pocket. In addition to the hydrogen-bonding interaction of its cationic side chain with the esteratic site of the enzyme, it can be deduced that the sandwich-shape stabilization of the ligand’s planar tetracycle at the bottom of the aromatic gorge mainly contributes to the high inhibitory effect.
As a result of its known beneficial properties in the field of inflammation and for insect bites, viral infections, diarrhea, and wound-healing, taspine (1) has a long tradition in ethnomedical use as a well-tolerated main constituent of sangre de drago, i.e., Croton lechleri latex (Euphorbiaceae).31 This remedy has been used by indigenous tribes of South America for millennia. The major constituent 1 has been found to be responsible for wound-healing, cicatrizant, and anti-inflammatory effects32,33 without having a carcinogenic or tumor-promoting activity.33 Viewed in this perspective, the well-tried and safe profile of the pharmacological properties of 1 in combination with the findings of this study could make compound 1 an interesting AChE-inhibiting lead structure worthy of being studied more extensively as a potentially useful multipotent therapeutic agent for neurodegenerative diseases. Compound 1 also provides a novel anticholinesteratic scaffold from nature with a proposed binding interaction different from known AChEIs.
Experimental Section
General Experimental Procedures
Melting points were determined on a Kofler hot-stage microscope and are uncorrected. Optical rotations were measured on a Perkin-Elmer 341 polarimeter at 25 °C. The NMR spectra were recorded on a Bruker DRX300 at 300 K in CDCl3 and calibrated to the residual nondeuterated solvent signals. Mass spectra were acquired using an Esquire 3000 plus ion-trap mass spectrometer (Bruker Daltonics) equipped with electrospray ionization (ESI) in positive mode: spray voltage, 4.5 kV; sheath gas, N2, 30 psi; dry gas, N2, 6 L min−1, 350 °C; scanning range, m/z 50–1000. Column chromatography was performed under TLC monitoring using silica gel flash column chromatography (silica gel 60, 40–63 μm, Merck) and Sephadex LH-20 (20–100 μm, Pharmacia). TLC was performed on silica gel 60 F254 plates (0.25 mm, Merck). HPLC data were obtained on a Hewlett-Packard HP-1100 system, equipped with a photodiode array detector and auto sampler. The LC was fitted with a Zorbax SB C18 column (150 × 4.6 mm i.d., 3.5 μm particle size, Agilent) at a column temperature of 25 °C, a flow rate of 1.0 mL/min, and an injection volume of 10 μL. UV detection wavelength was set to 254 nm. The mobile phases consisted of (A) ammonium acetate buffer, 10 mM, pH 4.3 and (B) acetonitrile; linear gradient of 0 min 20% B; 13 min 27% B; 30 min 98% B. All chemicals and solvents were analytical grade.
Plant Material
Authenticated leaves and bark of Magnolia x soulangiana Soul. Bod. were collected in the Botanical Garden of Innsbruck. A voucher specimen (JR-20040518-A1) was deposited in the Herbarium of the Institute of Pharmacy/Pharmacognosy, Leopold-Franzens-University of Innsbruck, Austria.
Extraction and Isolation
For the in vitro screening of plant extracts, air-dried plant material of different organs (each 1.0 g) was powdered and extracted with 10 mL of CH2Cl2 in an ultrasonic bath for 20 min at room temperature; the solvent was evaporated in vacuo and the remaining plant material extracted with MeOH using the same procedure in order to obtain small amounts of a CH2Cl2 and MeOH extract. For the bioactivity-guided fractionation, 880 g of the air-dried and milled leaves of M. x soulangiana were premacerated with petroleum ether (3 L, 2 days, room temperature). The remaining plant material was moistened with 10% aqueous ammonium (500 mL) and extracted twice with CH2Cl2 (2 × 3.5 L, 2 × 2 days, room temperature). The plant material was filtered off, and the solvent was evaporated under reduced pressure. Water (500 mL) was added and acidified with 10% HCl to pH 2.5. The aqueous solution was extracted three times with 200 mL of CH2Cl2, respectively, to afford 12 g of an organic phase I. The remaining aqueous solution was basified with 10% aqueous ammonium (pH 9.0) and again extracted three times with 200 mL of CH2Cl2, respectively, to afford 510 mg of an organic phase II. The latter was fractionated using Sephadex column chromatography, 3.5 × 90 cm, and eluted with CH2Cl2(elution rate: 2 mL/min, yielding 13 fractions (A1–A13) by means of TLC monitoring (toluene–acetone–96% ethanol–25% aqueous NH3; 52:40:6:2). Fraction A4 (190 mg, 352–387 mL elution volume) was subjected to silica gel flash column chromatography (35 g, 1.2 × 40 cm) using a step gradient of CH2Cl2–MeOH–10% aqueous NH3 (98:2:0.2, 400 mL; 96:4:0.5, 250 mL; 90:10:0.5, 200 mL) to give four subfractions (B1–B4). Subfraction B3 (20.7 mg, 552–597 mL elution volume) was precipitated from MeOH and afforded a white, optically inactive powder (1; 12.4 mg) showing a positive Dragendorff’s reaction. By precipitation of sub-fraction B4 (4.2 mg; 597–622 mL elution volume) from acetone, a whitish, Dragendorff-positive substance (2; 3.4 mg) was obtained. Physical and spectroscopic data of isolates 1 and 2 agreed with those reported for taspine14 and (−)-asimilobine,15 respectively. Tabulated NMR data of 1 and 2 are included in the Supporting Information (Tables S2 and S3).
Microplate Assay
The AChE-inhibitory activity was determined using a modified Ellman’s method in a 96-well microplate assay as previously described.11,13 Electric eel AChE was used for the in vitro screening and the bioactivity-guided procedure due to the lack of commercially available electric ray enzyme (=TcAChE); however, all the residues exposed in the ligand-binding cavity are conserved in the two protein sequences, except for the exchange of Phe330 in TcAChE to Tyr in electric eel AChE.30 Since this is also the case in the human enzyme, AChE derived from electric eel is well-established for the assay screening. Electric eel AChE, hAChE (test concentration 0.022 U/mL each), horse serum butyrylcholinesterase (BChE; test concentration 0.011 U/mL), acetylthiocholiniodide, and 5,5′-dithiobis(2-nitrobenzoic acid) were purchased from Sigma-Aldrich Chemie Gmbh; positive controls galanthamine·HBr and tacrine·HCl.H2O were both from Tocris Cookson Ltd.
Statistical Analysis
The percentage of the enzyme inhibition was calculated by comparing the rates for the sample to the blank (containing 1% DMSO; n = 4) and analyzed with the Student’s t-test. The IC50 values were determined with probit analysis. For statistical processing, the SPSS 11.5 program package was used.
Computational Study
The molecular structure of 1 was minimized using the default parameters of the CHARMM force field in order to reduce internal strain energy. Multiconformational models were generated by randomized conformational analysis using the Poling algorithm within Catalyst (FAST algorithm for conformer generation; maximum number of conformers: 100). The elaboration of the pharmacophore model is described in Rollinger et al.11
Overlay of hAChE/TcAChE Active Sites
To directly compare the 3D active site conformations of hAChE and TcAChE, three PDB files (1B41, 1ACJ, and 1W6R) were downloaded from the RCSB homepage (www.rcsb.org)9 and loaded into the LigandScout program.29 Generally, LigandScout is employed for automated structure-based pharmacophore model generation. Additionally, the program offers automated alignment of molecules, of molecules and pharmacophore models, and of pharmacophore models only. These functions offer the possibility to define several amino acid residues of the binding pocket as one ligand molecule and aligning it as a “binding pocket molecule”. When performing the alignment, LigandScout also computes a quantitative value for the match of the two binding pockets: the root-mean-square (rms) value. Thus, LigandScout offers a fast and convenient way to directly compare the electronic and spatial conditions present in two distinct ligand-binding domains. For each of the two AChE complexes, only essential amino acid residues forming the ligand-binding domain are selected as “ligand” (for 1B41: Trp86, Gly202, Ser203, Tyr337, and His447; for 1ACJ and 1W6R: Trp84, Gly199, Ser200, Phe330, and His440, respectively) and exported to the alignment panel.
Docking Experiments
For docking experiments, Gold 3.1 software was employed.26 Docking methods intend to predict the 3D structure of small molecules interacting with a protein-binding site. Generally, multiple binding orientations are regarded by the program and ranked according to a chosen scoring function. Gold uses a genetic algorithm employing energy functions partly based on conformational and nonbonded contact information from the Cambridge Structural Database for computing docking solutions. Additionally, full ligand flexibility and partial protein flexibility during docking as well as the customizable consideration of water molecules present in the binding pocket enable fine-tuned docking experiments according to the user’s requirements.
Overall, the default parameters of the program were employed. The protein atom types were determined by Gold, and the ligand atom types were assessed using the program Sybyl (Version 7.1, 2005, Tripos Inc.), ensuring a protonated state for the nitrogen atom. Furthermore, cocrystallized water molecules were included in the docking process by setting all water molecules in the binding pocket to “toggle” and “spin” employing our previously described perl script.34
Supplementary Material
Acknowledgment
This work was supported by the Austrian Science Fund (FWF No P18379) and by the “Nachwuchsförderung 05” of the Leopold Franzens-University of Innsbruck for J.M.R. The authors thank Inte:Ligand for providing the LigandScout software free of charge, and S. Sturm for LC-MS measurements.
Footnotes
Supporting Information Available: Table summarizing the AChE-inhibitory activity of plant extracts in the family Magnoliaceae (Table S1), evaluation of docking results (Figures S1 and S2), and tables with NMR data of taspine (1) and (−)-asimilobine (2) (Tables S2 and S3). This information is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).Bartus RT, Dean RL, Beer B, Lippa AS. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- (2).Zimmermann M, Gardoni F, Marcello E, Colciaghi F, Borroni B, Padovani A, Cattabeni F, Di Luca M. J. Neurochem. 2004;90:1489–1499. doi: 10.1111/j.1471-4159.2004.02680.x. [DOI] [PubMed] [Google Scholar]
- (3).Zimmermann M, Gardoni F, Di Luca M. Drugs Aging. 2005;22:27–37. doi: 10.2165/00002512-200522001-00003. [DOI] [PubMed] [Google Scholar]
- (4).Jiang H, Luo X, Bai D. Curr. Med. Chem. 2003;10:2231–2252. doi: 10.2174/0929867033456747. [DOI] [PubMed] [Google Scholar]
- (5).Houghton PJ, Ren Y, Howes M-J. Nat. Prod. Rep. 2006;23:181–199. doi: 10.1039/b508966m. [DOI] [PubMed] [Google Scholar]
- (6).Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Steinhilber D, Schubert-Zsilavecz M, Roth HJ. Medizinische Chemie-Targets und Arzneistoffe. Deutscher Apotheker Verlag; Stuttgart: 2005. pp. 119–121. [Google Scholar]
- (8).Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- (9).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bartolucci C, Perola E, Pilger C, Fels G, Lamba D. Proteins. 2001;42:182–191. doi: 10.1002/1097-0134(20010201)42:2<182::aid-prot50>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- (11).Rollinger JM, Hornick A, Langer T, Stuppner H, Prast H. J. Med. Chem. 2004;47:6248–6254. doi: 10.1021/jm049655r. [DOI] [PubMed] [Google Scholar]
- (12).Rollinger JM, Mock P, Zidorn C, Ellmerer EP, Langer T, Stuppner H. Drug Discovery Technol. 2005;2:185–193. doi: 10.2174/1570163054866855. [DOI] [PubMed] [Google Scholar]
- (13).Ellman GL, Courtney D, Andres V, Featherstone RM. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- (14).Talapatra B, Chauduri PK, Talapatra SK. Phytochemistry. 1982;21:747–750. [Google Scholar]
- (15).Achenbach H, Renner C, Addae-Mensah I. Liebigs Ann. Chem. 1982:1623–1633. and references therein. [Google Scholar]
- (16).Meyer FH, Hecker U, Höster HR, Schroeder F-G. Fitschen Gehölzflora. 10th ed. Quelle und Meyer; Heidelberg: 1994. pp. 10–15. [Google Scholar]
- (17).Lichtenthaler HKZ. Pflanzenphysiol. 1965;53:388–403. [Google Scholar]
- (18).El-Feraly FS. Phytochemistry. 1983;22:2239–2241. [Google Scholar]
- (19).Abdallah OM. Phytochemistry. 1993;34:1185–1187. [Google Scholar]
- (20).Song Q, Fronczek FR, Fischer NH. Acta Crystallogr. 2001;E57:1105–1106. [Google Scholar]
- (21).Ziyaev R, Shtonda NI, Sturua MD, Abdusamatov A, Tsakadze DM. Chem. Nat. Comp. 1999;35:366–367. [Google Scholar]
- (22).Ziyaev R, Abdusamatov A, Yunusov S. Yu. Khim. Prir. Soedin. 1975;11:528–529. [Google Scholar]
- (23).Slavik J, Slavikova L. Collect Czech. Chem. Commun. 1995;60:1034–1041. [Google Scholar]
- (24).Furmanowa M, Jozefowicz J. Acta Soc. Bot. Pol. 1980;49:527–535. [Google Scholar]
- (25).Milanowski DJ, Winter REK, Elvin-Lewis MPF, Lewis WH. J. Nat. Prod. 2002;65:814–819. doi: 10.1021/np000270v. [DOI] [PubMed] [Google Scholar]
- (26).Gold 3.1; available from the Cambridge Crystallographic Data Center (CCDC) [Accessed April 2006]; www.ccdc.cam.ac.uk/products/life_sciences/gold/
- (27).Greenblatt HM, Guillou C, Guenard D, Badet B, Thal C, Silman I, Sussman JL. J. Am. Chem. Soc. 2004;126:15405–15411. doi: 10.1021/ja0466154. [DOI] [PubMed] [Google Scholar]
- (28).Harel M, Schalk I, Ehret-Sabatier L, Bouet F, Goeldner M, Hirth C, Axelsen PH, Silman I, Sussman JL. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9031–9035. doi: 10.1073/pnas.90.19.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wolber G, Langer T. J. Chem. Inf. Model. 2005;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]; [Accessed April 2006];LigandScout Vers. 1.02. 2006 available from Inte:Ligand GmbH, Austria ( www.inteligand.com)
- (30).Mizutani MY, Itai A. J. Med. Chem. 2004;47:4818–4828. doi: 10.1021/jm030605g. [DOI] [PubMed] [Google Scholar]
- (31).Jones KJ. Alternat. Compl. Med. 2003;9:877–896. doi: 10.1089/107555303771952235. [DOI] [PubMed] [Google Scholar]
- (32).Perdue GP, Blomster RN, Blake DA, Farnsworth NR. J. Pharm. Sci. 1979;68:124–126. doi: 10.1002/jps.2600680145. [DOI] [PubMed] [Google Scholar]
- (33).Vaisberg AJ, Milla M, Planas MC, Cordova JL, de Agusti ER, Ferreyra R, Mustiga MC, Carlin L, Hammond GB. Planta Med. 1989;55:140–143. doi: 10.1055/s-2006-961907. [DOI] [PubMed] [Google Scholar]
- (34).Schuster D, Maurer E, Laggner C, Nashev L, Wilckens T, Langer T, Odermatt A. J. Med. Chem. 2006;49:3454–3466. doi: 10.1021/jm0600794. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.