

Abstract
Background
Genetic background has a profound effect on inflammatory bowel disease. The Gpx1 and Gpx2 double knockout (GPX1/2-DKO) mice on a mixed C57BL/6 (B6) and 129S1/SvimJ (129) background had spontaneous ileocolitis. The DKO mice on a B6 background had mild ileocolitis. We characterized the 129 DKO mice to identify a genetic locus affecting disease severity.
Methods
We backcrossed B6;129 DKO mice to 129 and analyzed for ileocolitis penetrance and severity at N5, N7 and N10. By correlating disease severity with single-nucleotide polymorphism (SNP) markers, we identified a colitis locus.
Results
As early as 9 days of age, 129 DKO N5 and N10 mice showed disease signs and morbidity. The N10 DKO mice had the severest colitis with nearly complete penetrance and high morbidity compared with other generations or backgrounds. 129 DKO mice had elevated colonic KC and SAA3 expression, shorter colon length, and cecal E. coli overgrowth compared to B6 DKO mice. Analysis of the B6 loci in 129 N5, N7 and N10 cohorts pointed to a region of chromosome 2: 119 Mbp contributing to mild symptoms.
Conclusions
GPX1/2-DKO mice on 129 genetic background have the most aggressive colitis compared to B6;129 and B6 colonies. A B6 locus significantly contributing the resistance resides on chromosome 2: 119 Mbp. This region coincides with cytokine-deficiency-induced-colitis-susceptibility, Cdcs3, identified in the resistant B6 and sensitive C3H/HeJBir (C3Bir) with IL-10 deficiency. A three-way SNP analysis between 129, B6 and C3Bir locus points the major candidate genes to B2m, Dnajc17, Duox2, Pla2g4b, Pla2g4e, Pla2g4f and Slc30a4.
Keywords: mouse model, inflammatory bowel disease, glutathione peroxidase, innate immune system in IBD, genetic locus
INTRODUCTION
The mucosal epithelium of the gastrointestinal tract provides an essential barrier from the luminal microflora. Breaches of the barrier or disproportionate responses of the host to microflora are the basis of inflammation (1-3). We have previously reported that conventionally reared GPX1/2-DKO mice on a mixed B6 and 129 genetic background exhibit a microflora-dependent intestinal pathology and inflammation with mixed features of Crohn’s disease and ulcerative colitis (4). Disease is confined to the mucosa (UC-like), but can occur in both the ileum and the colon (CD-like) of DKO mice. The disease characteristics may stem from the consensus localization of the selenium-dependent glutathione peroxidases, GPX1 and GPX2, in the mucosal epithelium of both the ileum and colon (5).
The selenium-dependent Gpx gene family is a major component of the antioxidant arsenal. The closely related GPX1 and GPX2 isozymes are responsible for moderating intracellular hydroperoxide levels, principally alkyl hydroperoxides (6, 7) (8) (9, 10). They differ in that Gpx1 is expressed in virtually all cell types while Gpx2 is expressed predominantly in epithelial cells, including Paneth cells (5) (11). The cell types most visibly depleted in GPX1/2-DKO mice are Paneth cells of the ileum and the goblet cells of the ileum and colon. This depletion may be part of a pattern of elevated crypt apoptosis (12, 13). Both Gpx1 and Gpx2 may play a role to control apoptosis, since they are inducible by stress-response transcription factors; Gpx1 is regulated by p53 and Gpx2 is regulated by Nrf2 and p63 (14-16).
Similar to the germ-free mice of many mouse models for inflammatory bowel disease (IBD), germ-free GPX1/2-DKO did not have disease. The composition of the microflora profoundly affect the severity and duration of ileitis in GPX1/2-DKO mice, a feature that may be shared with human IBD (13, 17). In addition to environment, genetics plays a major role in IBD (18-20). We found that B6 GPX1/2-DKO mice had milder disease and little morbidity compared to the B6;129 mixed-strain GPX1/2-DKO colony. This morbidity was generally associated with the intensity of ileitis (21). Colitis was an intermittent feature, and was observed only in about 7% of B6 GPX1/2-DKO mice. Colitis was more frequent (20% of mice through 50 days of age) and occasionally associated with morbidity in the mixed-strain mice. This suggests that 129 mice harbor GPX1/2-DKO-induced colitis susceptibility alleles while B6 mice are a relatively resistant strain. A similar circumstance was observed in IL10-KO-induced colitis comparing these two strains (22). We set out to test our hypothesis that the 129 strain GPX1/2-DKO mice are more susceptible to ileocolitis than B6.
Identification of susceptibility genes has become a major goal for IBD research. One of the advantages of using mice is that reverse (transgenic manipulation) and classical (forward) genetic studies can be combined to identify potential IBD loci (23). This allows a means to reveal candidate IBD loci against a spectrum of known underlying genetic defects covering areas from defective barrier to immune dysfunction (24, 25). In humans, many IBD loci have been identified by genome-wide association studies (GWAS)(18). One of the IBD loci, IBD9, on chromosome 3p21, was identified with a marker rs9858542 (Thr3912Thr) located within an exon of the BSN. The IBD9 locus is one of the most convincing replicated regions containing GPX1, GHOA, DAG1, BSN, APEH, TRAIP, MSTR1, and MST1, which are in high linkage disequilibrium (26). Although the role of GPX1 in human IBD has not been established, it is clear that with combined deficiency of GPx1 and GPx2, mice develop spontaneous ileocolitis.
Considering the usefulness of rodent models to study human IBD, there are surprisingly few forward genetic studies to identify mouse IBD loci. Leiter, Sundberg and their associates have been the only group to study IBD genes in IL-10 null mice (25). They identified ten susceptibility loci, named cytokine-deficiency-induced colitis susceptibility (Cdcs) loci by comparing the colitis between the sensitive C3H/HeJBir (C3Bir) and resistant B6 mice (25, 27). Here, we have used forward genetics to identify a GPx-deficiency-associated colitis locus (Gdac), which differentiates disease severity between B6 and 129 strains of DKO mice.
MATERIAL AND METHODS
Mice
GPX1/2-DKO mice were generated by mating Gpx1tm1Ysh/tm1Ysh (Gpx1-KO) and Gpx2 tm2Coh/tm2Coh (Gpx2-KO) mice (13). The GPX1/2-DKO colony on a mixed B6;129 strain background was backcrossed to C57BL/6J to generate B6 (N8) and to 129S1/SvimJ to generate 129 N5, N7 and N10 colonies. The 129 N10 line was derived from the N5 line without selection until the incross phase when residual B6 contamination on chromosome 12 was bred out. The 129B6F1 was bred from the B6 N8 and 129N10 lines. The N7 line was separately derived from the N5 line by a marker-assisted approach to select for B6 alleles on chromosome 2: 60-160 Mbp. The N7 incrosses were set up to generate subcongenic lines, homozygous for regions of chromosome 2 around 60 mbp, 80 mbp, 99 mbp, 119 mbp and 142 mbp while eliminating B6 alleles from residual adjacent location and on other chromosomes. Incross-generation 1 and 2 had few mice homozygous in the subcongenic regions. By incross-generation 3-5, the homozygous state for the subcongenic regions was generally established and the surrounding heterozygosity was eliminated by SNP marker-assisted selection (Table 1).
Table 1.
SNP markers and their association with mouse health in N5 129 DKO
| Location (Mbp) |
SNP Refseq number |
B6/129 vs. 129/129 OR2 (95% C.I.2) |
P-value | B6/B6 vs. 129/129 OR (95% C.I.) |
P-value | Independence of health and genotype1 P-value |
|---|---|---|---|---|---|---|
| Chr2:60 | rs28024473 | Inf2 (0.16, Inf) | 0.46 | 0.00 (0.00, Inf) | 1 | 0.05 |
| Chr2:99 | rs273767973 | 0.32 (0.04, 2.06) | 0.23 | 0.00 (0.00, 0.85) | 0.03 | 0.04 |
| Chr2:119 | rs274243303 | 32.04 (3.23, 1679.98) | 0.0003 | Inf (7.26, Inf) | 5.10E-05 | 1.09E-06 |
| Chr2:138-143 | rs134662823 | 0.00 (0.00, 4.92) | 0.4 | 0.00 (0.00, 1.54) | 0.1 | 0.14 |
| Chr2:160 | rs28024473 | 1.48 (0.26, 8.42) | 0.71 | 1.00 (0.02, 16.30) | 1 | 0.87 |
| Chr5:127 | rs29544470 | 2.15 (0.14, 33.64) | 0.59 | 0.00 (0.00, Inf) | 1 | 0.59 |
| Chr5:150 | rs33464535 | 0.80 (0.14, 4.25) | 1 | 0.00 (0.00, 9.25) | 0.51 | 0.86 |
| Chr8:27 | rs32957570 | 0.00 (0.00, Inf) | 1 | 0.00 (0.00, 2.57) | 0.27 | 0.27 |
| Chr8:89 | rs32986909 | 0.69 (0.12, 3.78) | 0.72 | 0.29 (0.01, 5.43) | 0.61 | 0.8 |
| Chr11:69 | rs29440455 | 0.00 (0.00, Inf) | 1 | 0.00 (0.00, Inf) | 1 | 1 |
| Chr12:3.7 | rs29152550 | 0.88 (0.15, 5.08) | 1 | 0.34 (0.01, 4.57) | 0.6 | 0.8 |
| Chr12:29 | rs29223336 | Inf (0.05, Inf) | 0.34 | 0.00 (0.00, Inf) | 1 | 0.34 |
| Chr12:48 | rs29180099 | 0.43 (0.03, 4.58) | 0.62 | 0.00 (0.00, 19.70) | 1 | 0.75 |
| Chr13:44 | rs29738332 | 1.40 (0.22, 8.09) | 0.71 | 0.00 (0.00, Inf) | 1 | 0.71 |
| Chr17:24 | rs33583903 | 1.32 (0.16, 9.89) | 1 | 0.00 (0.00, 10.82) | 0.54 | 0.71 |
| Chr17:46 | rs33294015 | 1.29 (0.26, 6.58) | 0.74 | 0.00 (0.00, Inf) | 1 | 0.74 |
| Chr17:67 | rs33468298 | 0.55 (0.01, 7.95) | 1 | 0.00 (0.00, 67.29) | 1 | 1 |
| Chr19:39 | rs31111922 | 3.60 (0.17, 235.29) | 0.54 | 0.00 (0.00, 77.91) | 1 | 0.71 |
Fisher’s exact test. Because there are no B6 alleles present at several markers, 129/129 is the reference, except Chr2:119. This increases the significance in the genotype comparisons (from P=0.25 and P=0.001), but does not alter the overall test of independence and health.
OR means odds ratio; C.I. means confidence interval; Inf means infinite.
Two closely spaced SNPs (3K-64K bp) were used at these locations to avoid marker dropout. No discrepancies were found when both markers at the same location worked. The results are pooled for these marker sets, and the most reliable marker Refseq numbers are listed here.
Because homozygous GPX1/2-DKO dams cannot adequately foster their litters, we routinely bred homozygous DKO males with females of Gpx1+/−Gpx2−/− and Gpx1−/−Gpx2+/− genotypes. Homozygous male GPX1/2-DKO mice were weighed and checked for disease signs daily from 8 to 50 days of age and heterozygous male littermates were followed through weaning. Female DKO mice were followed to weaning, and then culled while female heterozygotes were kept for breeding. Mice analyzed in this study were maintained on irradiated LabDiet (LD) 5058 for breeders and LD 5043 for weaned pups (Purina, General Mills). To ensure the survival of the 129 N10 line and male Gpx1/2-DKO mice from N7 incross-generation 3-5, these mice were maintained on casein-, sucrose-, and corn oil-based defined diets containing AIN-76A micronutrients, formulated to mimic LD 5001 and 5020 (Harland-Teklad, Madison, WI; TD 06306 and TD 06307). The colonies are maintained at a standard research level. Sentinel mice are shipped quarterly to the University of Missouri Research Animal Diagnostic and Investigative Laboratory (Columbia, Missouri, USA) where they undergo serology tests for MHV, Sendai, PVM, REO 3, TMEV GDVII, Ectromelia, M. pulmonis, MNV, MPV, MMV, EDIM, and LCM, as well as microbiological, parasitological, and histopathological examination (Basic Necropsy Profile). Additionally, on an annual basis sentinel mice undergo serology tests for MAD 1, MAD 2, Polyoma, CARB, MCMV and Tyzzer’s (Comprehensive Plus Profile). Sentinel mice are tested once a year for Helicobacter spp. via PCR assay. Over the course of these studies, the sentinels tested negative for all except norovirus. The City of Hope IACUC approved animal usage for these studies.
Histology
A section of the distal ileum and the entire colon except the distal 1 cm were processed for histopathology analysis. Each tissue was scored for inflammation and pathology in a blinded fashion using a 14-point system as we described previously (13). These include lymphocytes and neutrophil infiltration (0-3 points), Paneth cell and goblet cell degranulation (0-2 points), epithelium reactivity such as crypt distortion (0-3 points), inflammatory foci (0-3 points), and apoptotic figures (0-3 points). The threshold for detection of acute inflammation corresponds to scores of 6-7 with these mice. Scores lower than 6 reflect findings of degranulation of Paneth and goblet cells, crypt distortion, and apoptosis either without acute inflammatory infiltration or limited to light monocyte and eosinophil infiltration [(12) and unpublished observations].
Quantitative PCR on rectum samples for KC and SAA3
Total RNA was isolated from the distal 1 cm of colon/rectum using the RNeasy kit (Qiagen, Valencia, CA). Quantitative real-time PCR (RT qPCR) was performed with the Eva qPCR SuperMix kit and detected with SYBR Green (BioChain Institute, Inc. Hayward, CA) using IQ5 (BioRad, Hercules, CA) instrument. Serum amyloid A (SAA) proteins are markers for acute phase responses and inflammation. SAA3 is expressed in mouse colon epithelial cells (28). The primer set for SAA3 are 5′-TGCCATCATTCTTTGCATCTTGA-3′ and 5′-CCGTGAACTTCTGAACAGCCT-3′ (28). KC/CXCL is a local tissue neutrophil-chemotactic factor (29, 30). The KC primers are 5′-GCTGGGATTCACCTCAAGAA-3′ and 5′-TGGGGACACCTTTTAGCATC-3′ (31). The mRNA levels were normalized against β-actin, which was amplified with forward 5′-GCTCCTCCTGAGCGCAAGT-3′ and reverse 5′-TCATCGTACTCCTGCTTGCTGAT-3′ primers. Each assay was done in triplicate.
Assay of Plasma IL6, TNF-αand lipopolysaccharide (LPS) levels
After asphyxiation, heparinized blood was collected from mice, by cardiac puncture, and centrifuged (3,000 × g for 5 min) to recover the plasma fraction. Plasma was aliquoted and frozen at −80°C until assay. LPS was assayed with a ToxinSensor Chromogenic LAL Endotoxin Assay kit (GenScript, Piscataway, NJ). IL-6 and TNF-α were assayed with AlphaLISA mouse kits (PerkinElmer Health Sciences, Waltham, MA). Each assay was done in either duplicate or triplicate.
Myeloperoxidase (MPO) assay
Sample processing for myeloperoxidase activity assay was done according to the method of Grisham et al. (32). The assay was performed with 3,3′,5,5′Tetramethylbenzidine (TMB) as the substrate following the described procedure (33). MPO activity, which was not inhibited by 0.1 mM Dapsone (lactoperoxidase inhibitor, Sigma), was converted to ng MPO equivalents/mg protein using purified bovine milk lactoperoxidase (Sigma) as a standard. The results were then multiplied by 100 and plotted.
Determination of E. coli colony forming unit in mouse cecum
Cecal microflora were characterized in 22-day-old pups or younger (16- to 21-day-old) sick mice when morbidity criteria dictated as we have done before (34). Briefly, the cecal content was cultured under aerobic conditions. Each sample was diluted 10,000 times the original sample volume with PBS, and then plated with the original volume on a Luria-Bertani (LB) plate at 37°C for 24 h and counted.
Identification of GPx-deficiency-associated-colitis (Gdac) loci
Ten 129 N5 mice were analyzed for single nucleotide polymorphisms (SNP) to identify B6 loci on the 129 background by the Jackson Laboratory Genome Scanning Service (Jackson Laboratory, Bar Harbor, ME). The marker panel of 141 markers covered the 19 autosomes at an average 20 Mbp interval. A smaller panel of 21 SNP markers (Table 1), focusing on the Chr2: 119 Mbp interval and other interesting regions, was constructed using the physical locations of the SNPs in the original panel. Surrogate SNPs were identified in the MGI SNP database (Jackson Laboratory, Bar Harbor, ME), and marker reagents were prepared by the Genetic Marker Core Facility of the City of Hope. SNPs were analyzed with a MassARRAY iPLEX Gold system (Sequenom Inc., San Diego, CA). This smaller panel was used to track the disposition of B6 alleles in backcrosses through N10 and in the incrosses at the N7 and N10 generations.
Statistics
For comparing time-to-event endpoints, such as survival time, the results were plotted with Kaplan-Meier curves and analyzed with the log-rank test. Analysis of variance (ANOVA) was used to compare the means ± standard deviations (SD). Mann-Whitney U test was used to evaluate the significance in differences among colon pathology scores. Levene’s test was used to test for homogeneity of variances, and Welch’s test was used to test the means when there are significant differences in variances. To analyze disease signs vs. alleles, mouse health and weights were evaluated from 8-22 days of age. Mice were first classified as “sick” or “well” based on presentation of persistent wet tail, diarrhea, perianal irritation and lethargy versus general absence of these disease signs. After stratification by health and sex, weight distribution of mice was analyzed and found to be highly significant based on prior division into well and sick categories (P=1×10−7; T-test, sick vs. well). Weight was used to classify rare cryptic mice that presented little outward disease signs but appeared abnormally small.
For QTL analysis of genotypes versus colon scores and other parameters the program, R/QTL, was used with the J/QTL interface (35). The physical positions of markers were translated into cM using nearby mapped genes or microsatellites. LOD thresholds were calculated from 2000 permutations.
RESULTS
129 genetic background increases the morbidity of GPX1/2-DKO mice
Genetic background has a strong influence on morbidity extent and timing in homozygous GPX1/2-DKO mice (Fig.1; Log-rank P<0.0001 for all comparisons except B6 vs. 129B6F1). Emergence of early morbidity (wasting or lethargy accompanied by wet tail and frank diarrhea) was one of the distinctive features of the 129 backcrosses (N5-10) in comparison with B6 and two mixed-strain colonies, 129B6F1 and B6;129 (Fig.1). 129 GPX1/2-DKO mice (N5 and N10) have disease onset as early as 9 days of age based on signs of wet tail (up to 15% of mice at 9 days) and runting (up to15%; wet tail and running-25% combined). This contrasts with the post-weaning morbidity observed in B6, 129B6F1 and the B6;129 colonies, illustrated best by the B6;129 DKO mice (Fig.1). Post-weaning morbidity involved wasting and lethargy was only intermittently associated with diarrhea in the mixed-strain colonies and never in B6. While the 129 GPX1/2-DKO mice tended to exhibit early morbidity, the extent was very different for the N5 and N10 backcross generations. Characterization of the 129 N5 GPX1/2-DKO cohort on LabDiet showed 34% morbidity through 50 days of age compared to 95% morbidity for the N10 cohort (Fig.1).
Figure 1.

Kaplan-Meier plot of GPX1/2-DKO mouse morbidity for different genetic backgrounds from 8 days of age. All mice were derived from breeders on LabDiet (9-10% fat). Numbers of mice in cohort are in parenthesis after each strain. Mouse colonies tested include B6, B6;129 mixed, 129B6F1, 129 N5 and 129 N10 (at backcross-generation 5 and 10, respectively). All comparisons between colonies are significantly different (log-rank P<0.0001, shown by symbols: *, **, ***) except B6 vs. 129B6F1.
Morbidity timing is associated with the severity of colitis
The extent and timing of morbidity seems to follow from different disease patterns exhibited among the colonies. Histology shows severe and prevalent crypt abscesses and ulceration (medium inflammation/pathology score of 9; Fig. 2 & 3) in the colon of nearly all 129 N10 mice by 22 days of age. B6 DKO mice have low inflammation/pathology scores (<6) reflecting largely gland apoptosis and hyperproliferation. The pathology rarely involves the deeper layers and the colon surface appears to be affected secondarily to the gland aberrations, differentiating this early onset disease from neonatal necrotizing enterocolitis (36). A comparison of colitis inflammation/pathology scores among the colonies at 22 days of age shows that only the 129 backcross cohorts (N5 and N10) had median scores of 6 or above, indicative of a high incidence of acute inflammation (Fig. 3). Due to the huge variance in the N5 set, there is no statistically significant difference between the 129 GPX1/2-DKO N5 and N10 cohorts. The test for homogeneity of variances was not significant. The high morbidity in the 129 N5 and N10 cohort occurs between 16-24 days of age, when they have severe colitis.
Figure 2.


Histology of the cecum, proximal and distal colon of B6 and 129 N10 GPX1/2-DKO mice. 2A. B6 DKO mice have very mild pathology in the cecum, proximal and distal colon as shown in Panels A, B and C, respectively. B6 DKO colon has sub-inflammatory disease with mucin depletion and high levels of apoptosis and mitosis in the glands. A representative 129 N10 DKO mouse with medium and severe inflammation is shown in Panels D-F and G-I, respectively, at 22 days of age (peak of colitis). Moderate pathology is found in some proximal colons of 129 DKO mice (Panel E). The 129 DKO colon often has neutrophil infiltration (black arrow), extensive exfoliation of crypt epithelial cells with epithelial restitution (white arrow), and a combination of crypt destruction and distortion.
2B. Detailed pathology of 129 DKO distal colon. Panel J shows sub-inflammatory pathology in a 129 DKO mouse (adjacent to the area in panel F). Distorted, mucin-depleted glands are shown with a juxtaposition of epithelial apoptosis (thin arrows) and mitosis (block arrows). Panel K shows an active abscess (wide white arrow), and a post-inflamed gland with remnants of exfoliated epithelial cells (wide black arrow) and compensating restitution (thin arrow) in a region of intense pathology. Typical of such regions, apoptosis and mitosis are no longer prominent in the adjacent less damaged glands.
Figure 3.
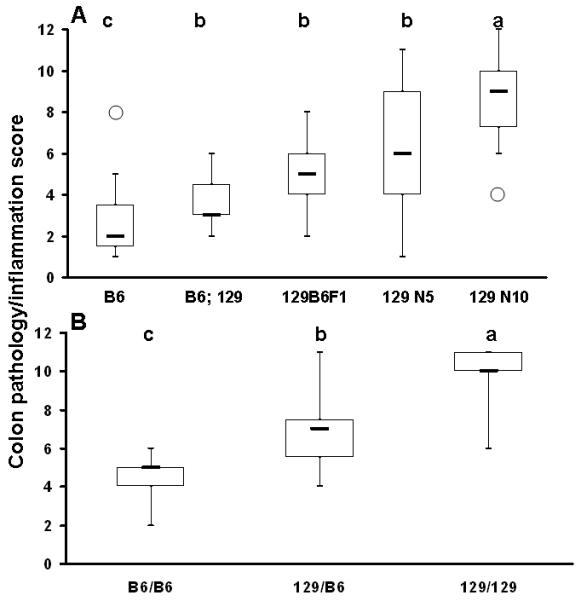
Box and whisker plot of colon inflammation/pathology scores of 22-day-old GPX1/2-DKO mice of different genetic backgrounds. Panel A. Colon scores analyzed in different DKO colonies. The open circles represent single outliers. The thick-horizontal bar is the median score. The number of mice studied in each colony is 15 for B6, 6 for B6;129, 18 for 129B6F1, 60 for 129 N5, and 18 for 129 N10 DKO. The letter shows difference in the median colitis scores, in the order of a>b>c., when α= 0.05.
Panel B. Colon inflammation/pathology score analyzed in 129 N5-N7 DKO mice stratified for B6 vs. 129 alleles on Chromosome 2: 118-119 Mbp (B6/B6, B6/129 and 129/129). The number of mice studied in each group is 13 for 129 N5 B6/B6, 14 for 129 N5 B6/129 and 6 for 129/129. The median colitis scores that differ are shown as a>b>c. The score of 129 N5 B6/B6 is higher than B6 colony, and the score of 129 N5 129/129 is significantly greater than 129 N10.
Colon pathology tends to precede ileitis in the DKO mice. In the B6;129 mixed-strain colony, ileitis generally commenced at 23-24 days of age and peaked at 40 days of age; while in B6, ileitis commenced and peaked even later, coincident with the late morbidity [Fig. 1 and (21)]. The ileitis in the 129 cohorts started earlier (19 days) than in the B6;129 cohort, and lagged behind colitis by several days with medium inflammation/pathology score of 6. However, moribund 129 GPX1/2-DKO mice through 22 days of age had minimal ileitis and there was little correlation for disease severity between the ileum and colon at this age (Pearson r=0.0003). In light of the generally late onset of ileitis and variability relative to disease signs and wasting in the 129 lines, we decided to focus the study on the colitis, which exhibits a penetrance of roughly 95% through 22 days of age in 129 N10 GPX1/2-DKO mice (17 of 18 animals; Fig 3A). We have noted that inflammation tends to be less severe or skips the proximal colon adjacent to the cecum, which is frequently inflamed (34). For 129 N10 mice on AIN diet, the median scores were 6.5, 4 and 9 in the cecum, proximal colon and distal colon, respectively; and a similar trend was noted for mice on LabDiet (Fig 2). The composite score emphasizes the worst pathology whether found in the proximal or distal colon and accounts for the range of the affected area (distal only; proximal only; both) in the foci category. This composite score was used to generate Fig. 3. Cecum was not routinely collected from the N5 cohort and the contemporary N10 cohort and was not considered in this analysis.
DKO mice have high incidence of apoptosis without ER stress in both 129 and B6 genetic backgrounds
We have previously reported a high incidence of crypt epithelial apoptosis in B6;129 and B6 DKO mice (13). Since 129 N5 and N10 DKO have more severe colitis, with massive crypt epithelial cell exfoliation and neutrophils transmigrating into crypt abscesses, we examined whether they had more cell death than B6 DKO mice. In B6, 35% of the colon glands display apoptosis, 12% with 2 or 3 bodies (44 mice; 40-50 crypts scored for each mouse). In the 129 strain, when crypts are ordered enough to score apoptosis, the results are not significantly different (40% glands with apoptosis, 12% with 2 or 3 bodies; 10 mice; minimum of 15 crypts per mouse).
Oxidative stress can increase ER stress and lead to susceptibility to colitis (37-39). We analyzed GPX1/2-DKO mice for signs of ER stress. However a hallmark of ER stress, the ratio of spliced Xbp1 to unspliced Xbp1 mRNA, was not elevated in the colons of 129 GPX1/2-DKO mice compared with that in 129 non-DKO (mean ± stdev of Xbp1spliced/Xbp1unspliced: 1.1±0.27 for DKO (5 mice); 1.63±0.6 for non-DKO (4 mice); P= 0.13; t-test) or B6 GPX1/2-DKO mice and B6 WT mice (2.5±0.5 for DKO; 1.8±0.5 for WT)(39).
Elevation of KC/CXCL and serum amyloid A (SAA)-3 gene expression in the colon of 129 GPX1/2-DKO mice but not non-DKO controls, B6 GPX1/2-DKO and B6 WT mice
Since 129 GPX1/2-DKO mouse colon usually has neutrophil infiltration, we analyzed the mRNA levels of the mouse neutrophil chemoattractant, KC/CXCL. The KC mRNA level is elevated 14X in the colon of 129 N7 (which has similar disease index as N5) DKO mice relative to non-DKO controls (Fig. 4A). B6 GPX1/2-DKO and WT colon had even lower levels of KC mRNA than the 129 non-DKO controls. A test for variance homogeneity was significant among the groups considered (P=0.047). Using the Welch test, the levels of KC/CXCL mRNA in the 129 GPX1/2-DKO were significantly different from other groups (P=0.008). There was no overlap in KC mRNA levels between the 129 GPX1/2-DKO cohort and the other sets; the lowest levels in 129 GPX1/2-DKO were at least 2-fold higher than the highest values in the non-DKO sibs and 10-fold higher than that in the B6 mice.
Figure 4.
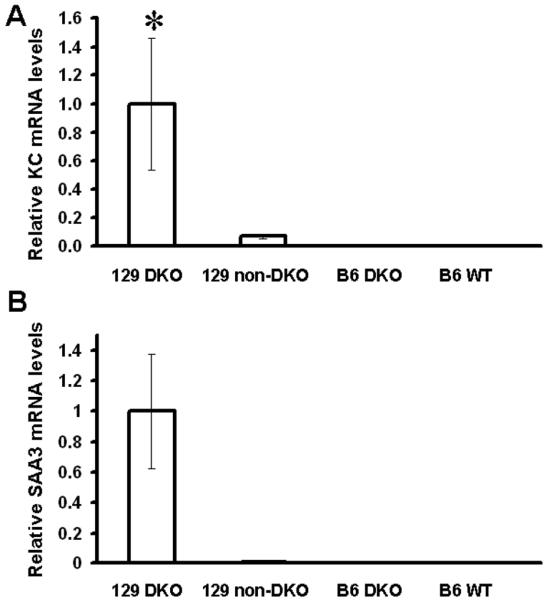
Relative mRNA levels of KC and SAA3 in mouse colon normalized against β-actin mRNA. Panel A shows mRNA levels of KC and Panel B shows mRNA levels of SAA3. The number of mice analyzed are five 129 GPX1/2-DKO, four 129 heterozygotes, four B6 GPX1/2-DKO, and two B6 WT. Error bars are standard errors of the means. A * symbol indicates significant difference from other groups.
Since neutrophil infiltration is a part of innate immunity responding to bacteria, and SAA3 is highly inducible by lipopolysaccharides, we also examined SAA3 expression in the colon (28). SAA3 mRNA is elevated ~100X in 129 GPX1/2-DKO mice compared to their non-DKO littermates as well as B6 DKO and WT mice (Fig. 4B). The test for variance homogeneity was non-significant. The high variance in the 129 GPx1/2-DKO mice precludes statistical significance even though there is no overlap in the SAA3 levels with the other groups (P=0.056).
Link of morbidity in GPX1/2-DKO mice to hypoglycemia and elevated plasma IL-6
In 70% of 129 N5-N10 DKO mice the transition from stable disease signs to a wasting and moribund condition was accompanied by apparent loss of appetite (empty stomach at necropsy) and borderline to frank hypoglycemia (fed glucose level of 20-86 mg/dL; N=16) compared to non-DKO controls (86-243 mg/dL, N=37), with a lone outlier at 50 mg/dL).
We reported hypothermia in B6;129 GPX1/2-DKO mice before (4). Here, we analyzed plasma IL-6, which has hypothermia-inducing and appetite-suppressing properties in mice (40, 41). Plasma IL-6 levels were notably elevated in 129 strain GPX1/2-DKO mice (Fig. 5A). N10 129 DKO mice had the highest median level compared to other groups; and unlike other groups, none of the N10 DKO mice had levels of ~0 pg/ml. However the variance homogeneity test in plasma IL-6 levels was significant and the high variation in IL-6 levels of 129 DKO mice causes difficulties in comparisons (P=0.03). No significant differences were found in any comparisons. However, the comparison of N7 DKO versus N7 non-DKO and B6 DKO was at the borderline of significance (P=0.059; 0.053, Welch test, N=18, 14 and 9, respectively). Age- and diet-matched B6 GPX1/2-DKO and heterozygous sibs had no detectable plasma IL-6 analyzed at the peak of inflammation in B6 DKO (N=9 and 3 respectively). When we correlated plasma IL-6 with colon histopathology, we found that 129 GPX1/2-DKO mice with scores of 7 and above tended to have elevated IL-6 (>3.5pg/ml). However, the overall correlation between colon scores and IL-6 levels in GPX1/2-DKO mice was only moderate (Pearson r =0.24). IL-6, KC and SAA3 levels correlate well among each other in the DKO mice (IL-6 vs. KC, r = 0.98; IL-6 vs. SAA3, r = 0.93; KC vs. SAA3, r = 0.85). None of these mice had elevated TNF-alpha in the plasma (data not shown).
Figure 5.
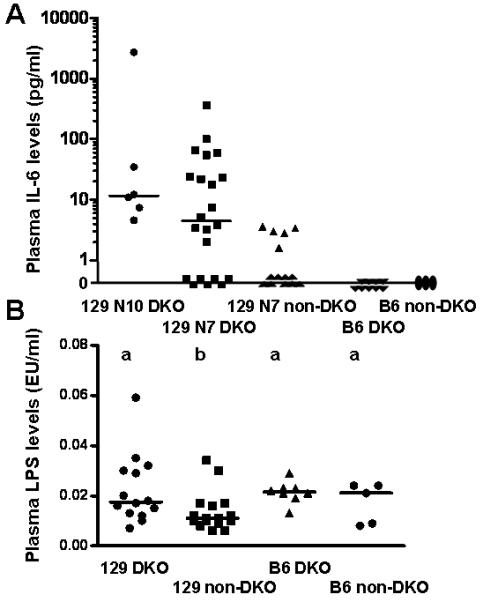
Scatter plots of plasma IL6 and LPS levels. Panel A. Plasma IL-6 levels from GPX1/2-DKO mice at peak of inflammation, i.e. 22-day-old 129 N7 and N10 mice and 50-day-old B6, as well as age-matched non-DKO controls. The number of mice in each group is: 6 for 129 N10 DKO, 22 for 129 N7 DKO, 14 for N7 non-DKO, 9 for B6 DKO and 3 for B6 non-DKO. No set is significantly different from the others. Panel B. Plasma LPS levels detected for the same groups. The levels in the 129 GPX1/2-DKO group are significantly greater than the heterozygous 129 mice but not the B6 groups (P=0.033, t-test). The means that differ are shown as a>b. The Y-axis is Endotoxin Unit/ml, which is generally converted to LPS concentration as 1 EU=100 pg LPS/ml. In both panels, the bars indicate the medians, and each symbol represents a mouse.
Leakiness, but no sepsis, in 129 GPX1/2-DKO mice
Since SAA3 is inducible by LPS and 129 GPX1/2-DKO mice have a very high level of SAA3 gene expression, we measured plasma LPS levels at the peak of pathology for each group. The plasma LPS level in 22-day-old 129 DKO mice is significantly higher than in non-DKO controls (P=0.033, t-test), but not different from 50-day-old B6 WT and B6 DKO mice (P=0.42 and 0.8, respectively; Fig. 5B). The variance homogeneity test was not significant. Wasting and elevated plasma IL-6 and SAA3 are not indicative of sepsis. The peak level detected in 129 GPX1/2-DKO mice (6 pg/ml) is well below the human plasma LPS levels with sepsis (50 ± 16 pg/ml) (42).
Identification of a GPx-deficiency-associated-colitis (Gdac) locus
We have documented through analysis of morbidity and colitis pathology the differences between 129 GPX1/2-DKO mice in the N5, incross-generation 2-4, and N10 cohorts (Fig. 1 and 3A). This difference appears to be part of a continuum of disease severity from B6 to 129 N10. The colitis in 129 GPX1/2-DKO N5 mice straddled the boundary between sub-inflammatory pathology and acute inflammation and showed a high variance. These observations are consistent with segregation of disease-modifying B6 alleles (Fig. 3A). While colitis in 129 and B6 Gpx1/2-DKO mice is very likely a complex trait, at N5 at least 90% of the genetic background would be of 129 strain. Using the Cdcs loci as a guide, the 129 N5 cohort could have 1 or 2 residual colitogenic loci with B6 alleles. Our premise was that individually or together, the 1 or 2 loci would have a B6 allele frequency high enough to suppress colitis in 35-50% of the 129 N5 cohort. The frequency estimate derives from 2 observations. One-half of the colon scores in the 125 N5 cohort are less than the minimum score of the N10 cohort, falling in the range of B6 scores. A survey of incross-generation 2 and 3 of 129 N5 shows that 39 of 113 animals are completely absent of disease signs.
Using whole genome SNP analysis on ten 129 N5 pups selected from 3 of the 6 founder breeder cages for this genomic project (representing 3 sires and 5 dams from N5 incross generation 3), 13 loci were identified with B6 alleles. The loci are displayed along with colitogenic Cdcs and Dssc loci identified by studying genetic differences between B6 and C3Bir IL-10 KO mice (Cdcs) and standard B6 and C3Bir (Dssc) (Fig. 6 and Supplemental Table 1)(25, 43). B6 alleles on chromosomes 1, 3 and 15 were found in only one of ten mice and in a heterozygous state, thus were not consider as candidates for disease modifying loci or tracked again until N7, incross-generation 3 (Supplemental Table 2). B6 alleles on chromosomes 2, 4, 8, 12, 13, 17 and 19 occurred in nearly half of the mice (some homozygous) and were considered as good candidates for colitogenic loci. A high frequency of B6 alleles on chromosome 4 was expected due to our deliberate selection to retain the silent λLIZ transgene (B6/N Big Blue® mice) for later in vivo mutation assessment (21).
Figure 6.
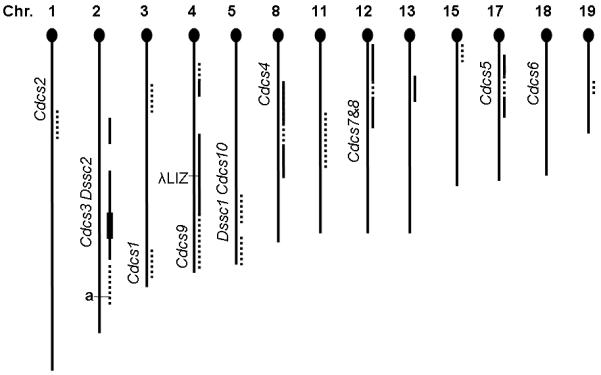
Distribution and incidence of B6 alleles in the 129 N5 cohort, represented by 10 offspring, sampling 5 breeder females and 3 breeder males in comparison with colitogenic loci detected in 2 other studies (complete data in Supplemental Table 1). Chromosomes are listed in order by number and oriented with the telomeric centromere (= 0 Mbp) at the top. The dashed or solid lines to the right of the chromosome stick map indicate loci where B6 alleles were detected in the 129 N5 cohort. Loci with low representation of B6 alleles (1-4 mice; almost all heterozygous) are shown by dashes, and those heavily represented sites (5 or more; some homozygous B6 alleles) shown by solid lines. The left side of the stick map is marked with the location of Cdcs (Cytokine-deficiency-induced colitis susceptibility) and Dssc (Dextran-sodium-sulfate colitis) loci along with the non-agouti gene and the λLIZ transgene. The chromosome 2 B6 loci (solid line) in 129 N5 were detected in 7-8 mice with 1-4 homozygous B6 (4 at 119 Mbp, thickened line). This analysis was based on the GWAS performed on the first set of 129 N5 mice (Supplemental Table 1), thus did not include the new loci on Chromosome 1 and 3 identified in 129 N7 mice (Supplemental Table 2).
The 129 N5 incross-generation 4 population was expanded to a cohort of 42 GPX1/2-DKO mice from the founders mentioned above. A smaller SNP panel was constructed to track B6 alleles on chromosomes 2, 4, 5, 8, 11, 12, 13, 17 and 19 (Table 1). Analysis across the 129 N5 colony showed that stratification by presence or absence of the λLIZ transgene on chromosome 4 did not affect the proportions of sick vs. well mice (79 mice with transgene and 34 without; P=0.83, Fisher’s exact test). Tests of independence of disease and genotype for markers flanking the transgene did not achieve significance (Chromosome 4: 3.65 Mbp, P= 0.63; 23.21 Mbp, P=0.42; 67.44 Mbp, P=0.03; 122.49 Mbp, P=1; 142.9 Mbp, P=0.27; Fisher’s exact test). This suggests that the high frequency of B6 alleles on chromosome 4 is related to selection of the λLIZ transgene and does not affect disease. Therefore, only the presence or absence of the transgene was tracked in mice during additional backcrosses. Association testing of the B6/129 alleles versus symptoms and body weight suggested that loci on chromosome 2 near 119 Mbp partially governed the severity of colitis between 129 and B6 (P=1.09E-6) (Fig 7 and Table 1). Proximal markers, at 60 Mbp and 99 Mbp, and distal markers, including SNPs at 138-143 Mbp and 160 Mbp, did not show association with symptoms (P≥ 0.003; Bonferroni adjusted value for multiple testing).
Figure 7.
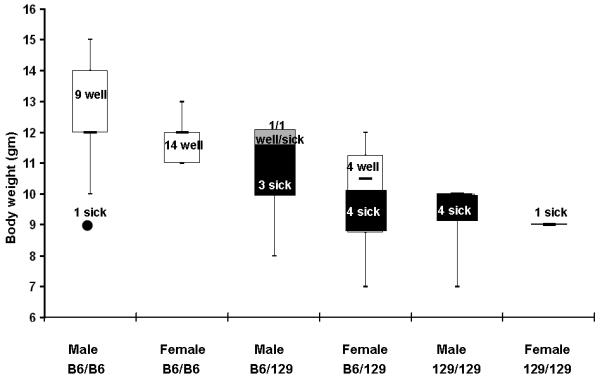
Stratification of the mouse health status, body weight and the genotype at Gdac locus. Forty-two 129 N5 DKO mice were checked for symptoms of wet tail/diarrhea, perianal alopecia, perianal ulceration and lethargy from 8-22 days of age and weighed near 22 days of age. Mice were grouped by the genotype at chromosome 2: 119 Mbp, Gdac locus. In each group, mice were characterized either sick (black portion of the box and whisker plot, N=12) or well (white portion of the plot, N=28). The gray portion of male B6/129 box indicates one well and one sick mouse at 12 gm. Sick mice had consecutive days of symptoms and a median body weight of 9.5 gm (range 7-12 gm, no outliers). Well mice had one or at most two days of intermittent symptoms and a median body weight of 12 gm (range 11-15 gm, one outlier at 9 gm: 1.5X>IQR). Due designation as an outlier by virtue of low body weight, one male B6/B6 mouse was re-classified as sick for association analysis (black circle). The designations by health status and weight corresponded to colon inflammation/pathology scores shown in Fig. 3B.
The backcross to N7 with selection for chromosome 2 loci eliminated some of the residual background. But relative to N5, incross-generation 1 and 2 of the N7 backcross represented a step backwards; the distribution of alleles at chromosome 2: 119 mbp from 19 DKO offspring was 2 B6/B6, 13 B6/129 and 4 129/129. Evaluating survival on LabDiet out to 22 days of age (15 of 19) did not produce a statistically significant result versus a cohort of 19 diet-matched N10 mice (9 of 19) (P=0.12; Log-Rank). All 4 of the morbid mice from the N7 cohort were 129/129 at chromosome 2: 119 mbp. When these are removed and the results re-analyzed, the difference in the survival outcome is significant (P=0.0016; Log-rank). The median composite colon histopathology scores were 5 for the B6/B6 mice, 7 for the B6/129 mice and 10.5 for the 129/129 mice. The genotypes from N5 and N7 complemented each other; N5 supplied more scores from B6/B6 mice and N7 supplied more scores from 129/129 mice (Fig. 3B). Two groups of mice could be roughly distinguished in the N7 cohort by health. The healthier N7 group, which included 2 B6/B6 mice and 8 B6/129 mice, had a median body weight of 9.5 gm and median colitis score of 6. The sicker group, which included 4 129/129 mice and 5 B6/129 mice, had a median body weight of 6.1 gm and a median colitis score of 10. The body weights and colitis scores are significantly different (for weight, P=0.0018, T-test; for colitis score, P=0.0004, Mann-Whitney). Pooling results from N5 and N7 produced nearly identical results in a test of independence of health and genotype (results not shown).
An analysis of colon histopathology of DKO 129 N10, 129 N5-N7 and B6 mice suggests that stratification of alleles at 119 Mbp (129/129 vs. B6/129 vs. B6/B6) accounts for a large portion of the variation in colon histopathology scores between the B6 and 129 strains with additive inheritance (Fig. 3B). 129 GPX1/2-DKO N5 mice B6/B6 at chromosome 2:119 Mbp had colon scores near that of the B6 strain but still significantly higher (P=0.0047; Mann-Whitney). GPX1/2-DKO mice heterozygous at chromosome 2: 119 Mbp had a mean colon histopathology score intermediate between B6 and 129 N10 (median 6.5, range 4-9; or 6.5∓1.8 for mean ± stdev) but significantly different from 129 N10 (P= 0.02) and B6 (P= 0.0035). N5-7 mice with 129/129 DNA across the 119 Mbp interval had a median score of 10 (or 10 ± 2 for mean ± stdev; significantly different from all other groups; P≤0.004) (Fig. 3B). Running the combined N5-N7 data set in R/QTL using colon scores (proximal, distal and composite scores vs. markers on chromosomes 2, 5, 8, 11,12,13,17 and 19) gave similar results to the analyses based on mouse health and body weight for N5 and N5-N7. Chromosome 2: 125-126 Mbp (119-130 Mbp; Bayesian credible interval, 0.95) was identified with peak LOD scores of 6.35-7.01 (P≤0.001, P=0.004, P=0.001; proximal, distal and composite scores, respectively; Supplemental Fig. 1-3). Thus, we give the chromosome 2: 119-130 Mbp region the provisional name, Gdac1. This interval accounted for 34%, 37.5% and 27% of the variation in proximal, distal and composite scores, respectively, when other loci accounted for a maximum of 6% each of the variation.
Gdac1 is in the vicinity of Cdcs3 and Dssc2, possibly replicating Cdcs3 (25, 43). We analyzed the genetic difference among the strains used to map Cdcs3 (B6 vs. C3Bir) and the strains/colonies used in this study. Among 36 genes exhibiting nonsynonymous coding SNPs in the region of chromosome 2:118-130 Mbp, B2m, Dnajc17, Duox2, Pla2g4b, Pla2g4f and Scl30a4 stand out as plausible candidates for colitogenic genes based on the assumption of replication (Supplemental Table 3).
Subcongenic analysis
In addition to mapping, we are establishing subcongenics dividing the initially identified chromosome interval (60-143 Mbp) that harbored many B6 alleles. In N7 incross-generation 3-5, subcongenics were largely homozygous for the selected markers and residual heterozygosity at other loci was eliminated. We also used the opportunity to test whether these mice have only 129 genes at all other loci by performing GWAS on a set of ten pups, sampling from all 6 sires and 9 of 12 dams used to generate the subcongenics (Supplemental Table 2). In addition to the loci tracked by our marker panel, we detected 3 concentrations of B6 alleles on chromosome 1 and 3. While these may have been missed in our original N5 screen, it is also possible that these were picked up in backcrosses to N10. To ensure survival of the male DKO subcongenic stock and to resolve morbidity issues in the N10 colony, all 129 mice were placed on AIN diet. Kaplan-Meier analysis of survival by 35 days of age showed distinct differences between the N7 subcongenics and N10 (18 of 20 survived in N7; 9 of 41 survived in N10; P≤ 0.0001; Log-rank test). Additionally, the cecum, proximal and distal colon scores for N7 mice were significantly lower than the N10 (scores of 4, 2 and 4 in N7 vs. 6, 4 and 9 in N10; P≤0.0023; Mann Whitney test). Assay of MPO in total mid-colon showed that N10 mice tended to have higher levels than N7 mice, although it is not statistically significant (Fig. 8A).
Figure 8.
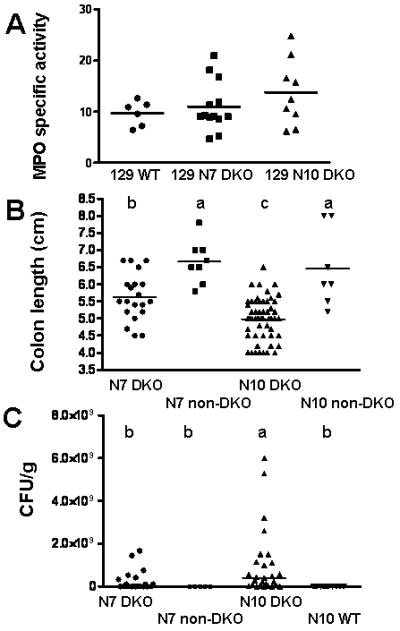
Scatter plots of subcongenic N7 with N10 129 GPX1/2-DKO mice. Panel A. Specific activity of myeloperoxidase (MPO) in the mid-colon sections of 129 mice. Each symbol represents a mouse for all panels. The number of mouse in each group is 6 for WT, 13 for N7 DKO and 9 for N10 DKO. The horizontal line is the mean. Although the mean for each group is in the order of N10 DKO > N7 DKO > WT, this difference is not statistically significant. Panel B. Total colon length measured in N7 and N10 129 mice. The bar represents the mean. The number of mice in each group is 20 for N7 DKO (incross-generation 3-5), 8 for N7 non-DKO which are sibs of DKO, 54 for N10 DKO and 7 for N10 non-DKO. All mice were on AIN diet and analyzed at 22 days of age or morbidity. The median colon length that differs is shown as a>b>c (P≤ 0.03). Panel C. Colony forming unit of E. coli cultured from the cecum of 129 mice. The bacterial counts were determined by the number of colony forming unit (CFU) per gm of cecal content in 129 strain mice fed AIN diet. The number of mice in each group is 20 for N7 DKO, 5 for N7 non-DKO, 27 for N10 DKO and 12 for N10 WT. The horizontal bar represents the median. The groups that differ are shown as a>b (P≤ 0.03, unpaired t test with Welch’s correction).
Several parameters studied in the N7 subcongenics and N10 strains were examined in R/QTL. Colon length was differentiated among N7 DKO, N10 DKO and heterozygous mice (Fig. 8B; P≤ 0.03 for comparisons except heterozygous N7 vs. heterozygous N10). Analysis of colon length vs. genotype in N7 suggested involvement of the same chromosome 2 region indicated earlier (peak LOD score-3.7; P=0.011; 23% variance accounted at chromosome 2 and peak 5% at other loci; Supplemental Fig. 4). Over growth of E. coli in the cecum of N7 and N10 DKO mice was found to be a marker of disease [Fig. 8C and (34)]. When colony forming units/gm (CFU/g) cecal content was used as the phenotype, the same region was identified with LOD score of 4.24 (P=0.38; 28% variance accounted on chromosome 2 and 12% each by chromosomes 11 and 13; Supplemental Fig. 5). An evaluation of health versus genotype also produced only suggestive evidence (P=0.048) and colon scores and MPO levels failed to identify any loci (Supplemental Fig. 6 and results not shown). This may be due to the low frequency of mice with severe disease in this group through 22 days of age (4 of 44 vs. 40 of 90 for N10).
DISCUSSION
The results emphasize that lack of GPX in the GI tract is only a moderate predisposing factor for spontaneous ileocolitis. The consensus pathology from GPX-null seems to be crypt epithelium apoptosis and compensating hyperproliferation (13). B6 and B6;129 mixed-strain GPX1/2-DKO mice did not differ in the extent of these conditions (21). However, the tendency for ileocolitis was much stronger in the mixed-strain and in 129 DKO mice. Assessment of the extent of apoptosis in 129 GPX1/2-DKO mice is complicated by disease severity, as in many glands there is depletion of the epithelium by exfoliation. In areas where scoring was possible, we did not find apoptosis to be more prominent. We note that severely inflamed regions rarely have apoptosis or mitosis even in the less damaged glands. This may be due to the limits on the extent of apoptosis before cell death causes barrier breakdown and triggers inflammation (44). In B6, inflammation is largely limited to recruitment of monocytes, while in B6;129 and 129 GPX1/2-DKO mice this progresses to neutrophil infiltration and extends to uniform colon involvement in 129 (21). The differences between B6 and 129 GPX1/2-DKO mice seem to be based on acute immune signaling rather than effects directly attributable to oxidative stress. This may explain why the genetic analysis appears to replicate the findings from an IL-10 KO IBD model, which is based on immune dysfunction.
Loss of appetite and increased metabolic burden due to disease may explain the tendency for the ileocolitis-associated moribund condition in mice particularly during the active growth phase. Detection of elevated plasma IL-6 levels in the young diseased 129 DKO mice would support the notion that colitis is linked to suppressed appetite and high morbidity (40, 41).
We anticipated that the 129 genetic background would increase ileocolitis incidence and severity in GPX1/2-DKO mice, and were surprised by the specific increase in colitis severity. The early disease signs of diarrhea, lethargy and wasting suggest that microflora may be inducing the pathology that is the root of disease at the earliest stages of colonization (45, 46). The timing of disease in the GPX1/2-DKO pups and its absence in non-DKO littermates suggests that the microflora in question would qualify as commensal. We recently found overgrowth of E. coli in the cecum of 45% of N7 DKO mice (incross-generation 4-5) and 77% of N10 DKO mice (34). Despite the carnage found in the colon of these mice, the low plasma LPS levels suggest only modest increase in gut leakiness without sepsis. While the response in the colon is inappropriate, presumably exaggerated by increased oxidative stress, it seems to involve a well-orchestrated innate immune response. Analysis of the Gdac1 locus may provide an explanation for the difference in disease response between B6 and 129 in the colon.
The tendency for early disease onset, which is associated with more aggressive colitis, was evident in the 129 N5 cohort. This suggests that some colitis-mitigating B6 alleles were lost by N5, which was the expected outcome. For example, the B6 alleles of the colitogenic Cdcs1 and Cdcs2 loci may have been represented in only a single heterozygous mouse in the 10 N5 GWAS-tested mice, while there was a relative concentration of B6 alleles on chromosome 2 from 60 Mbp to 143 Mbp.
Analysis of colitis scores from the 129 N5-N7 mice compared to B6 GPX1/2-DKO and 129 N10 GPX1/2-DKO mice supports the claim for the presence of B6 colitis-resistant alleles in 129 N5-N7. Since 129 N10 DKO mice presented more severe colitis and greater morbidity than N5 DKO, we took advantage of the relatively low number of B6 alleles present in the 129 N5 colony to identify the B6 colitis-resistance alleles. By correlating colitis pathology scores and SNP analysis, we found B6 alleles in the vicinity of 119 Mbp on chromosome 2 are most likely to protect mice from more severe colitis and morbidity observed in N10 DKO mice. As this is based on both association analysis of health status with P-values far exceeding significance (1.5E-6 < 2.5E-3; Bonferroni adjusted P value for α=0.05) and LOD scores of 6.35-7.01 for colon score analysis, the results stand up even with the specter of unaccounted loci in the backcross stock. Unaccounted loci do potentially diminish the value of the N7 stock as a reagent and add uncertainty as to whether every aspect of disease is affected by this locus.
We noted that the Gdac1 locus coincides with Cdcs3, a quantitative trait locus identified in crosses between C3Bir and B6, both incorporating IL-10 KO alleles (25, 47). Cdcs1, the major effect locus found in the B6;C3Bir IL10-KO mice, was replicated in studies on Gnai2 deficient mice (24). Farmer et al. characterized the effects of the B6 allele of the Cdcs3 locus as recessive and epistatic with Cdcs1. The absence of allelic Cdcs1 in the N5 generation and a different impact of GPX-deficiency relative to IL-10 may explain why we would characterize the effect as additive and a major effect locus. Also, the results from B6;C3Bir IL-10 KO, Gnai2 and dextran sodium sulfate-induced colitis studies suggest that colitis is governed by at least 13 genes in mice scattered throughout the genome present in complex loci (24, 27, 43, 48). We noticed that 6 of these loci might have been sites of residual B6 alleles in 129 N5 (Cdcs1, 2, 3, 4, 5, 9) although the frequency of B6 alleles corresponding to Cdcs1 (chromosome 3) and Cdcs2 (chromosome 1) were too low to account for the phenotype of the cohort. Introgression of the IL10-KO allele into B6 and 129 strains suggests that immunity gene alleles exist between the B6 and 129 strains affecting colitis severity similarly to B6 vs. C3Bir (22).
If Gdac1 is a replicate of Cdcs3, the three-way comparison of non-synonymous SNPs suggests 7 attractive candidate genes out of 37 genes within a chromosome 2:119-130 Mbp region (Supplemental Table 3). They are B2m, Dnajc17, Duox2, Pla2g4b, Pla2g4e, Pla2g4f, Slc30a4. Colon β2-microglobulin (B2m) expression increases as mice approach birth and the gene product is involved in MHC and NK function (49). Duox2 protein generates superoxide and is involved in innate immune responses (50). The phospholipase A2 type IV genes are involved in membrane remodeling and might play a role in repair of damage during inflammation, immune signaling and in macrophage phagocytosis (51). A nonsense codon in the zinc transporter, Slc30a4, causes a condition known as lethal milk (52). A milder phenotype might play some role in GPX1/2-DKO pathology. The gene product of Dnajc17 may be a heat shock protein. Heat-shock proteins have roles in immune function (53). In summary, we have found GPX1/2-DKO mice on 129 genetic background have severe spontaneous colitis achieving maximum penetrance around 16 days of age. These mice have elevated plasma IL-6 levels and increased colonic KC and SAA3 gene expression, consistent with the presence of acute inflammation. By comparing the phenotype and B6 alleles present in 129 N5 and 129 N10, we have identified a putative colitis-associated locus, Gdac1, present in chromosome 2. B6 alleles in this locus concur with milder colitis observed in 129 N5 DKO mice compared with 129 N10 DKO.
Supplementary Material
Supplemental Figure 1. LOD scores for the 129 N5 cohort, genotypes vs. proximal colon histopathology scores. The peak and the 95% confidence interval are shown as drop-down lines. The LOD plot is shown with distances in cM. These were converted back to Mbp using reference genetically mapped genes and microsatellite markers.
Supplemental Figure 2. LOD scores for the 129 N5 cohort, genotypes vs. distal colon histopathology scores.
Supplemental Figure 3. LOD scores for the 129 N5 cohort, genotypes vs. composite colon scores.
Supplemental Figure 4. LOD scores for the 129 N7 cohort, genotypes vs. colon length.
Supplemental Figure 5. LOD scores for the 129 N7 cohort, genotypes vs. E. coli colonies cultured from mouse cecal content.
Supplemental Figure 6. LOD scores for the 129 N7 cohort, genotypes vs. animal health based on disease activity index modified to account for growth arrest as well as weight loss.
Supplemental Table 1. Genome-wide scan for B6 and 129 loci performed on ten 129 N5 mice using 142 single nucleotide polymorphism (SNP) markers. The first column shows the chromosome number, SNP marker, and the source of the original sequence (M- MIT, N- Novartis, C- Celera). The 2nd column shows the marker position at Mbp on each chromosome. The navy blue cells are sites homozygous for 129 DNA, the yellow cells are heterozygous sites, and the light-blue cells are homozygous B6 sites. The grey cells are sites failed to discriminate the two alleles.
Supplemental Table 2. Genome-wide scan for 129 and B6 loci performed on ten 129 N7 mice (incross-generation 4 and 5) using 142 SNP markers. A similar panel of SNP markers was used as in Supplemental Table 1. The % at the 1st row indicates the % of 129 DNA in each mouse.
Supplemental Table 3. List of genes in the candidate interval on chromosome 2 (Gdac1) with non-synonymous single nucleotide polymorphisms, B6 vs. 129 and C3Bir. Only those genes have different SNP between B6/129 and B6/C3Bir are included.
ACKNOWLEDGEMENTS
The authors thank Sofia Loera and Tina Montgomery of the Anatomic Pathology Core for tissue processing and the High Throughput Screening Core for AlphaLISA reading (P30 CA33572). No competing financial interests exist for all authors. Sources of Support: NIH R01CA114569 (FFC).
REFERENCES
- 1.Neish AS, Gewirtz AT, Zeng H, et al. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science. 2000;289:1560–1563. doi: 10.1126/science.289.5484.1560. [DOI] [PubMed] [Google Scholar]
- 2.Vaishnava S, Behrendt CL, Ismail AS, et al. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slack E, Hapfelmeier S, Stecher B, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esworthy RS, Aranda R, Martin MG, et al. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G848–855. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 5.Esworthy RS, Swiderek KM, Ho YS, et al. Selenium-dependent glutathione peroxidase-GI is a major glutathione peroxidase activity in the mucosal epithelium of rodent intestine. Biochim Biophys Acta. 1998;1381:213–226. doi: 10.1016/s0304-4165(98)00032-4. [DOI] [PubMed] [Google Scholar]
- 6.Chu FF, Doroshow JH, Esworthy RS. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J Biol Chem. 1993;268:2571–2576. [PubMed] [Google Scholar]
- 7.de Haan JB, Bladier C, Griffiths P, et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem. 1998;273:22528–22536. doi: 10.1074/jbc.273.35.22528. [DOI] [PubMed] [Google Scholar]
- 8.Esposito LA, Kokoszka JE, Waymire KG, et al. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic Biol Med. 2000;28:754–766. doi: 10.1016/s0891-5849(00)00161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson RM, Ho YS, Yu DY, et al. The Effect of Disruption of Genes for Peroxiredoxin-2, Glutathione Peroxidase-1 and Catalase on Erythrocyte Oxidative Metabolism. Free Radic Biol Med. 2009 doi: 10.1016/j.freeradbiomed.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peskin AV, Low FM, Paton LN, et al. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 11.Brigelius-Flohe R, Muller C, Menard J, et al. Functions of GI-GPx: lessons from selenium-dependent expression and intracellular localization. Biofactors. 2001;14:101–106. doi: 10.1002/biof.5520140114. [DOI] [PubMed] [Google Scholar]
- 12.Esworthy RS, Yang L, Frankel PH, et al. Epithelium-specific glutathione peroxidase, Gpx2, is involved in the prevention of intestinal inflammation in selenium-deficient mice. J Nutr. 2005;135:740–745. doi: 10.1093/jn/135.4.740. [DOI] [PubMed] [Google Scholar]
- 13.Chu FF, Esworthy RS, Chu PG, et al. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 2004;64:962–968. doi: 10.1158/0008-5472.can-03-2272. [DOI] [PubMed] [Google Scholar]
- 14.Tan M, Li S, Swaroop M, et al. Transcriptional activation of the human glutathione peroxidase promoter by p53. J Biol Chem. 1999;274:12061–12066. doi: 10.1074/jbc.274.17.12061. [DOI] [PubMed] [Google Scholar]
- 15.Yan W, Chen X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J Biol Chem. 2006;281:7856–7862. doi: 10.1074/jbc.M512655200. [DOI] [PubMed] [Google Scholar]
- 16.Banning A, Deubel S, Kluth D, et al. The GI-GPx gene is a target for Nrf2. Mol Cell Biol. 2005;25:4914–4923. doi: 10.1128/MCB.25.12.4914-4923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez-Medina M, Aldeguer X, Lopez-Siles M, et al. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm Bowel Dis. 2009;15:872–882. doi: 10.1002/ibd.20860. [DOI] [PubMed] [Google Scholar]
- 18.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 19.Thompson NP, Driscoll R, Pounder RE, et al. Genetics versus environment in inflammatory bowel disease: results of a British twin study. BMJ. 1996;312:95–96. doi: 10.1136/bmj.312.7023.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed FE. Role of genes, the environment and their interactions in the etiology of inflammatory bowel diseases. Expert Rev Mol Diagn. 2006;6:345–363. doi: 10.1586/14737159.6.3.345. [DOI] [PubMed] [Google Scholar]
- 21.Lee DH, Esworthy RS, Chu C, et al. Mutation accumulation in the intestine and colon of mice deficient in two intracellular glutathione peroxidases. Cancer Res. 2006;66:9845–9851. doi: 10.1158/0008-5472.CAN-06-0732. [DOI] [PubMed] [Google Scholar]
- 22.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gondo Y, Fukumura R, Murata T, et al. Next-generation gene targeting in the mouse for functional genomics. BMB Rep. 2009;42:315–323. doi: 10.5483/bmbrep.2009.42.6.315. [DOI] [PubMed] [Google Scholar]
- 24.Borm ME, He J, Kelsall B, et al. A major quantitative trait locus on mouse chromosome 3 is involved in disease susceptibility in different colitis models. Gastroenterology. 2005;128:74–85. doi: 10.1053/j.gastro.2004.10.044. [DOI] [PubMed] [Google Scholar]
- 25.Bristol IJ, Farmer MA, Cong Y, et al. Heritable susceptibility for colitis in mice induced by IL-10 deficiency. Inflamm Bowel Dis. 2000;6:290–302. doi: 10.1002/ibd.3780060407. [DOI] [PubMed] [Google Scholar]
- 26.Latiano A, Palmieri O, Corritore G, et al. Variants at the 3p21 locus influence susceptibility and phenotype both in adults and early-onset patients with inflammatory bowel disease. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.21176. [DOI] [PubMed] [Google Scholar]
- 27.Bleich A, Buchler G, Beckwith J, et al. Cdcs1 a major colitis susceptibility locus in mice; Subcongenic analysis reveals genetic complexity. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reigstad CS, Lunden GO, Felin J, et al. Regulation of serum amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PLoS One. 2009;4:e5842. doi: 10.1371/journal.pone.0005842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armstrong DA, Major JA, Chudyk A, et al. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J Leukoc Biol. 2004;75:641–648. doi: 10.1189/jlb.0803370. [DOI] [PubMed] [Google Scholar]
- 30.Yan XT, Tumpey TM, Kunkel SL, et al. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Invest Ophthalmol Vis Sci. 1998;39:1854–1862. [PubMed] [Google Scholar]
- 31.von Kockritz-Blickwede M, Rohde M, Oehmcke S, et al. Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am J Pathol. 2008;173:1657–1668. doi: 10.2353/ajpath.2008.080337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grisham MB, Benoit JN, Granger DN. Assessment of leukocyte involvement during ischemia and reperfusion of intestine. Methods Enzymol. 1990;186:729–742. doi: 10.1016/0076-6879(90)86172-r. [DOI] [PubMed] [Google Scholar]
- 33.Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, et al. Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol. 2003;29:206–212. doi: 10.1165/rcmb.2002-0152OC. [DOI] [PubMed] [Google Scholar]
- 34.Esworthy RS, Smith DD, Chu F-F. A strong impact of genetic background on gut microflora in mice. International Journal of Inflammation. 2010;2010 doi: 10.4061/2010/986046. http://www.sage-hindawi.com/journals/iji/aip.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Broman KW, Wu H, Sen S, et al. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 36.Hsueh W, Caplan MS, Qu XW, et al. Neonatal necrotizing enterocolitis: clinical considerations and pathogenetic concepts. Pediatr Dev Pathol. 2003;6:6–23. doi: 10.1007/s10024-002-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holtz WA, Turetzky JM, Jong YJ, et al. Oxidative stress-triggered unfolded protein response is upstream of intrinsic cell death evoked by parkinsonian mimetics. J Neurochem. 2006;99:54–69. doi: 10.1111/j.1471-4159.2006.04025.x. [DOI] [PubMed] [Google Scholar]
- 38.Heazlewood CK, Cook MC, Eri R, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008;5:e54. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leon LR. Hypothermia in systemic inflammation: role of cytokines. Front Biosci. 2004;9:1877–1888. doi: 10.2741/1381. [DOI] [PubMed] [Google Scholar]
- 41.Leon LR, White AA, Kluger MJ. Role of IL-6 and TNF in thermoregulation and survival during sepsis in mice. Am J Physiol. 1998;275:R269–277. doi: 10.1152/ajpregu.1998.275.1.R269. [DOI] [PubMed] [Google Scholar]
- 42.Tani T, Hanasawa K, Kodama M, et al. Correlation between plasma endotoxin, plasma cytokines, and plasminogen activator inhibitor-1 activities in septic patients. World J Surg. 2001;25:660–668. doi: 10.1007/s002680020028. [DOI] [PubMed] [Google Scholar]
- 43.Mahler M, Bristol IJ, Sundberg JP, et al. Genetic analysis of susceptibility to dextran sulfate sodium-induced colitis in mice. Genomics. 1999;55:147–156. doi: 10.1006/geno.1998.5636. [DOI] [PubMed] [Google Scholar]
- 44.Schulzke JD, Ploeger S, Amasheh M, et al. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci. 2009;1165:294–300. doi: 10.1111/j.1749-6632.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- 45.Savage DC, Dubos R, Schaedler RW. The gastrointestinal epithelium and its autochthonous bacterial flora. J Exp Med. 1968;127:67–76. doi: 10.1084/jem.127.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis CP, McAllister JS, Savage DC. Microbial colonization of the intestinal epithelium in suckling mice. Infect Immun. 1973;7:666–672. doi: 10.1128/iai.7.4.666-672.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farmer MA, Sundberg JP, Bristol IJ, et al. A major quantitative trait locus on chromosome 3 controls colitis severity in IL-10-deficient mice. Proc Natl Acad Sci U S A. 2001;98:13820–13825. doi: 10.1073/pnas.241258698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Buhr MF, Mahler M, Geffers R, et al. Cd14, Gbp1, and Pla2g2a: three major candidate genes for experimental IBD identified by combining QTL and microarray analyses. Physiol Genomics. 2006;25:426–434. doi: 10.1152/physiolgenomics.00022.2005. [DOI] [PubMed] [Google Scholar]
- 49.Park YK, Franklin JL, Settle SH, et al. Gene expression profile analysis of mouse colon embryonic development. Genesis. 2005;41:1–12. doi: 10.1002/gene.20088. [DOI] [PubMed] [Google Scholar]
- 50.Geiszt M, Lekstrom K, Brenner S, et al. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J Immunol. 2003;171:299–306. doi: 10.4049/jimmunol.171.1.299. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh M, Tucker DE, Burchett SA, et al. Properties of the Group IV phospholipase A2 family. Prog Lipid Res. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Huang L, Gitschier J. A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nat Genet. 1997;17:292–297. doi: 10.1038/ng1197-292. [DOI] [PubMed] [Google Scholar]
- 53.Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. LOD scores for the 129 N5 cohort, genotypes vs. proximal colon histopathology scores. The peak and the 95% confidence interval are shown as drop-down lines. The LOD plot is shown with distances in cM. These were converted back to Mbp using reference genetically mapped genes and microsatellite markers.
Supplemental Figure 2. LOD scores for the 129 N5 cohort, genotypes vs. distal colon histopathology scores.
Supplemental Figure 3. LOD scores for the 129 N5 cohort, genotypes vs. composite colon scores.
Supplemental Figure 4. LOD scores for the 129 N7 cohort, genotypes vs. colon length.
Supplemental Figure 5. LOD scores for the 129 N7 cohort, genotypes vs. E. coli colonies cultured from mouse cecal content.
Supplemental Figure 6. LOD scores for the 129 N7 cohort, genotypes vs. animal health based on disease activity index modified to account for growth arrest as well as weight loss.
Supplemental Table 1. Genome-wide scan for B6 and 129 loci performed on ten 129 N5 mice using 142 single nucleotide polymorphism (SNP) markers. The first column shows the chromosome number, SNP marker, and the source of the original sequence (M- MIT, N- Novartis, C- Celera). The 2nd column shows the marker position at Mbp on each chromosome. The navy blue cells are sites homozygous for 129 DNA, the yellow cells are heterozygous sites, and the light-blue cells are homozygous B6 sites. The grey cells are sites failed to discriminate the two alleles.
Supplemental Table 2. Genome-wide scan for 129 and B6 loci performed on ten 129 N7 mice (incross-generation 4 and 5) using 142 SNP markers. A similar panel of SNP markers was used as in Supplemental Table 1. The % at the 1st row indicates the % of 129 DNA in each mouse.
Supplemental Table 3. List of genes in the candidate interval on chromosome 2 (Gdac1) with non-synonymous single nucleotide polymorphisms, B6 vs. 129 and C3Bir. Only those genes have different SNP between B6/129 and B6/C3Bir are included.