

Abstract
Sirtuins are NAD-dependent deacetylases that counter aging and diseases of aging. Sirtuin research has focused on SirT1, which deacetylates transcription factors and cofactors in the nucleus. More recent findings highlight SirT3 as a mitochondrial sirtuin that regulates metabolism and oxidative stress. This review focuses on new data linking SirT3 to management of reactive oxygen species from mitochondria, which may have profound implications for aging and late-onset diseases
Introduction
The molecular era of aging research can be said to have begun with the oxidative damage theory of aging in the 1950s (Harman, 1956). According to this theory, reactive oxygen species (ROS) generated by leakage of electrons during electron transport damage macromolecules within and outside of mitochondria. This effect would be cumulative with aging and lead to functional decline. This theory has followed a rocky path, and recent genetic studies provide evidence both in favor of it (Schriner et al., 2005) and against it (Pérez et al., 2009; Van Remmen et al., 2003). Clearly, other kinds of damage occur during aging, such as telomere shortening, protein aggregation, etc.(Garcia et al., 2007; Morimoto and Cuervo, 2009), but it remains possible that these are consequences of the primary trigger of cumulative oxidative stress.
A burgeoning area of aging research has involved genes that control the pace of aging, initially identified in lower organisms. Data indicates that these gene products, including FOXO and sirtuin proteins, can also slow aging phenotypes in mammals. It was suggested that a full understanding of the function of these genes in mammals might be a divining rod pointing to the most important causes of aging (Guarente, 2003).
Sirtuins are NAD+-dependent protein deacetylases that slow aging in lower organisms (Imai et al., 2000; Kaeberlein et al., 1999; Rogina and Helfand, 2004; Tissenbaum and Guarente, 2001; Viswanathan et al., 2005). Much data now indicate that mammalian sirtuins adapt animal physiology to dietary extremes, such as fasting and calorie restriction (Finkel et al., 2009). The mammalian Sir2 ortholog SirT1 has been shown to deacetylate scores of critical transcription factors in the nucleus to mediate a broad range of physiological effects (Haigis and Sinclair, 2010; Imai and Guarente, 2010). In general, pathways controlled by SirT1 govern two domains, metabolic adaptations and stress response. Thus, increases in SirT1 activity via transgenic or pharmacological activation may slow aging by a variety of criteria and combat diseases, such as diabetes (Banks et al., 2008), Alzheimer’s (Donmez et al., 2010) and cancer (Herranz et al., 2010). However, whole body SirT1 gain of function mice have not been reported to live longer on a normal chow diet, although they do show protection against aging-induced pathologies (Herranz et al., 2010).
Of the seven mammalian sirtuins, three are targeted to the mitochondrial matrix (Verdin et al., 2010) where they deacetylate and/or ADP-ribosylate enzymes to trigger metabolic adaptation to fuel sources such as amino acids and fatty acids during energy limitation (Table 1 and below). It has been argued that these metabolic adaptations, such as the oxidation of fatty acids to produce energy, may per se have salutary effects on cell maintenance (discussed in metabolism section, below).
Table 1.
Mitochondrial Sirtuin Targets
| Enzyme | Sirtuin | Pathway | Reference |
|---|---|---|---|
| Glutamate Dehydrogenase (GDH) | SirT3, SirT4 | Amino acid catabolism NADPH production |
(Haigis et al., 2006; Lombard et al., 2007; Schlicker et al., 2008) |
| Carbamoyl phosphate synthase 1 (CPS1) | SirT5 | Amino acid catabolism Urea Cycle |
(Nakagawa et al., 2009; Ogura et al., 2010) |
| Acetyl CoA synthase 2 (ACS2) | SirT3 | Acetate metabolism | (Hallows et al., 2006; Schwer et al., 2006) |
| Ornithine transcarbamoylase (OTC) | SirT3 | Amino acid catabolism Urea Cycle |
(Hallows et al., 2011) |
| 3-hydroxy-3-methylglutaryl CoA synthase 2 (Hmgcs2) | SirT3 | Ketogenesis | (Shimazu et al., 2010) |
| Long chain acyl-CoA dehydrogenase (LCAD) | SirT3 | β-oxidation | (Hirschey et al., 2010) |
| NADH quinone oxidoreductase (Complex I) | SirT3 | Oxidative Phosphoyrlation | (Ahn et al., 2008) |
| Succinate Dehydrogenase (Complex II) | SirT3 | Oxidative Phosphoyrlation | (Cimen et al., 2009; Lombard et al., 2007) |
| Isocitrate Dehydrogenase 2 (IDH2) | SirT3 | TCA cycle NADPH production |
(Schlicker et al., 2008; Someya et al., 2010) |
| Manganese Superoxide Disumutase (MnSOD or SOD2) | SirT3 | Antioxidant | (Qiu et al., 2010; Tao et al., 2010) |
| Cytochrome C | SIrT5 | Oxidative Phosphorylation Apoptosis |
(Schlicker et al., 2008) |
| Mitochondrial ribosomal protein L19 | SirT3 | Mitochondrial protein translation | (Yang et al., 2010) |
| Cyclophillin D (CypD) | SirT3 | Apoptosis Glycolysis |
(Hafner et al., 2010; Shulga et al., 2010) |
Beyond these metabolic functions, SirT3 has been recently shown to control the levels of ROS themselves by multiple mechanisms. The implications of these findings, if confirmed in a broad range of cell types, may redirect the focus of attention back to mitochondrial oxidative damage as a primary cause of aging, unite some of the central ideas on aging, and suggest novel ways to intervene pharmacologically.
Unity at last -- SirT3, calorie restriction, ROS and hearing loss
There has been some debate over the role of sirtuins in calorie restriction (CR), mainly centered on systems contrived in yeast, C. elegans and Drosophila to simulate rodent CR. In the case of mammalian sirtuins, considerable evidence has accumulated that by deacetylating many nuclear factors, SirT1 mediates many outputs of CR, including increased physical activity (Chen et al., 2005), increased protection against disease (Herranz and Serrano, 2010; Kume et al., 2010), regulation of central neuroendocrine control (Cohen et al., 2009), and longevity (Boily et al., 2008; Li et al., 2008). As mentioned above, mitochondrial sirtuins deacetylate (or ADP-ribosylate) mitochondrial metabolic enzymes that may be important for adaptation to CR.
But the clearest example relating CR, ROS, sirtuins and aging has come from a recent study on hearing loss in mice (FIGURE 1) (Someya et al., 2010). C57Bl/6 mice sustain cumulative oxidative damage to hair cells and spiral ganglia neurons of the inner ear cochlea, resulting in hearing loss in 12-month old animals. CR completely protected against both oxidative damage to these cells and hearing loss in wild type mice. However, SirT3−/− mice were completely refractory to protection by CR; i.e. CR mice sustained the same level of oxidative damage and hearing loss as ad libitum fed animals. Note that young SirT3−/− mice are generally indistinguishable from wild type mice by many physiological criteria (Lombard et al., 2007). Since CR is known to induce SirT3 protein levels in wild type mice (Hallows et al., 2011; Pérez et al., 2009; Shi et al., 2005), it is likely that this sirtuin can mediate ROS management, mitochondrial integrity, and sensory function, at least in those neurons that govern hearing.
Figure 1.
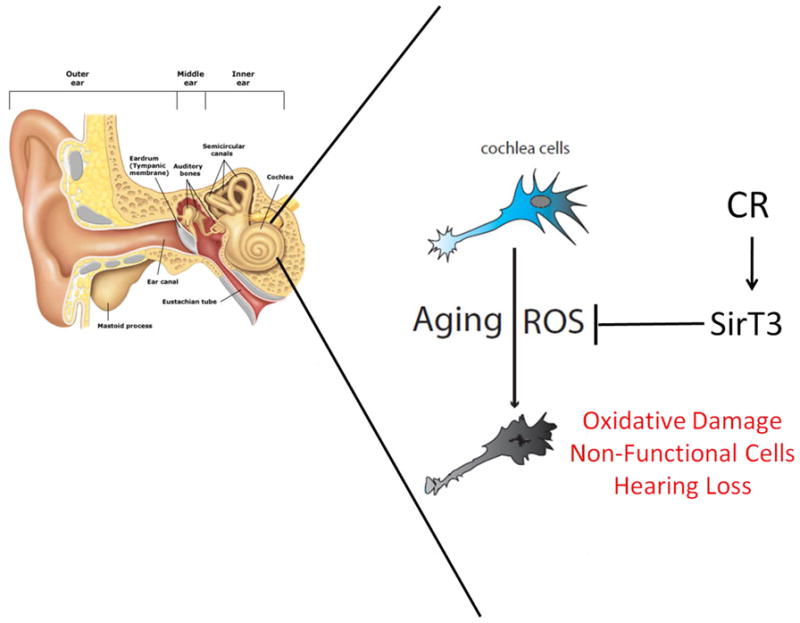
SirT3 is necessary for protection from hearing loss by calorie restriction. Aging induces ROS damage to inner ear cochlea neurons. In conditions of calorie restriction, SirT3 inhibits ROS mediated damage of cochlea neurons to delay hearling loss.
It is striking that this example brings together major threads running through the history of modern research on aging. It would appear to resuscitate the importance of the oxidative damage theory and mitochondria in aging. Further, it suggests that CR may function, at least in part, by controlling oxidative damage, thereby preventing aging-induced decline. Finally, it shows how genes identified because they slow aging in lower organisms, sirtuins, can fit centrally into the control of oxidative damage by CR. The SirT3 divining rod has thus brought us back to familiar themes as we contemplate future interventions. Mechanisms by which SirT3 may govern production or levels of ROS are discussed in the next section.
Will these findings on hearing loss extend to other cell types, and, if so, what are the implications for the science of aging? The roles of ROS and oxidative damage in aging are likely important in the aging of post-mitotic cells, eg. neurons, skeletal muscle, and cardiac cells. Further, the up-regulation of SirT3 by CR has been observed in a variety of tissues (Hallows et al., 2011; Pérez et al., 2009; Shi et al., 2005), suggesting that this sirtuin may mediate a broad spectrum of tissue protection against ROS-induced aging. It will be important to test whether SirT3 gain of function, eg. transgenic mice that express CR levels of the protein, are protected against oxidative damage as they age. If so, it is possible that SirT3 activation will slow aging itself and many of its effects on health decline and disease. An important issue in considering SirT3 gain of function is whether ROS can be reduced or eliminated without compromising normal physiological functions of free radicals. For example, the induction of hypoxia inducible factor HIF-1α during embryogenesis during hypoxia is essential for development (Maltepe et al., 1997; Ryan et al., 1998) and numerous reports indicate that ROS plays a major role in hypoxic induction of HIF-1α (Bell et al., 2007; Brunelle et al., 2005; Guzy et al., 2005; Mansfield et al., 2005). ROS can also play a mitogenic role, are necessary for proper immune response, and can guide differentiation of tissue specific stem cells (Esposito et al., 2004; Pervaiz et al., 2009).
Mechanisms of protection against oxidative damage by SirT3
Recent studies have revealed a surprisingly large number of anti-oxidant functions for SirT3 in mitochondria. Accordingly, a large number of mitochondrial proteins implicated in the aging process are acetylated (Kim et al., 2006), and SirT3, but not the other mitochondrial sirtuins SirT4 and SirT5, deacetylate a substantial subset of mitochondrial proteins (Lombard et al., 2007). The study of Someya et al. proposed a specific mechanism by which SirT3 protected cochlea cells against oxidative damage (FIGURE 2). SirT3 deacetylated and activated the TCA cycle enzyme isocitrate dehydrogenase 2 (IDH2), which produces NADPH in the mitochondria. The elevated NADPH, in turn, is necessary for glutathione reductase, which converts reduced GSSG into glutathione, the cofactor used by mitochondrial glutathione peroxidase (GPX) to detoxify ROS. Interestingly, another target of SirT3, glutamate dehydrogenase (GDH) (Lombard et al., 2007; Schickler et al., 2008), also produces NADPH and may contribute to the increased pool of glutathione that is available for GPX to detoxify ROS within mitochondria.
Figure 2.

SirT3 protects against damage from mitochondrial derived ROS. ROS are primarily generated by complex I (matrix) and complex III (matrix and intermembrane space/cytosol). Superoxide (O2·) localized to the matrix are detoxified by manganese superoxide dismutase (SOD2) into hydrogen peroxide (H2O2), which is subsequently converted into water by glutathione peroxidase (GPX). GPX requires reduced glutathione (GSH) for enzymatic activity. Oxidized glutathione (GSSH) is reduced by glutathione reductase (GSR), which requires NADPH. NADPH is generated from NADP+ by isocitrate dehydrogenase 2 (IDH2). By activating SOD2 and IDH2 and inhibiting ROS generation by complex III and potentially complex I, SirT3 may be a major determinant of oxidative damage in cells.
Two other studies demonstrate that SirT3 deacetylates the critical anti-oxidant enzyme manganese superoxide dismutase (SOD2) in the mitochondrial matrix (FIGURE 2) (Qiu et al., 2010; Tao et al., 2010). Deacetylation of this enzyme resulted in increased specific activity and enhanced scavenging of ROS. Strikingly, the two studies revealed different lysines in SOD2 that were deacetylated by SirT3 (K53 and K68 in the Qiu et al. study, and K122 in the Tao et al., study). In both studies, mutating the lysine(s) to arginine gave rise to a hyperactive SOD2 that was not further enhanced by SirT3, suggesting that all of these lysines may be important. The Qiu et al. study also showed that CR itself resulted in the deacetylation and activation of SOD2, presumably by up-regulating SirT3. This finding may help explain why CR has consistently been associated with a reduction in ROS levels in mitochondria (Weindruch, 1988). It is important to note that the role of SirT3 on ROS detoxification through the generation of NADPH and activation of SOD2 is confined to the mitochondrial matrix and does not represent a boost in antioxidant capacity in the nucleus or cytosol. However, SirT3 does suppress ROS responsive pathways that are induced by cardiac hypertrophy in the cytosol such as RAS, AKT, and MAPK signaling (Sundaresan et al., 2009). Another example of SirT3 suppressing cytosolic ROS involves HIF-1α, as discussed below.
How might SirT3 influence ROS in the cytoplasm? It is intriguing that SirT3 has been shown to deacetylate numerous components of the electron transport chain, suggesting it may directly affect the production of ROS (FIGURE 2). For example, SirT3 was shown to deacetylate subunits of complex I (NADH dehydrogenase) (Ahn et al., 2008) and complex II (succinate dehydrogenase) (Cimen et al., 2009) while mitochondria from SirT3−/− MEFs had reduced complex I activity, complex II activity, and complex III activity (Ahn et al., 2008; Cimen et al., 2009; Kim et al., 2010). Crucially, complex III directs ROS to both the matrix and the cytoplasm (Muller et al., 2004). Thus, by regulating complex III SirT3 could affect cytoplasmic levels of ROS.
SirT3−/− cells are known to have reduced steady state ATP levels, which may result from a defect in deacetylating electron transport chain components (Ahn et al., 2008). While SirT3 has not been demonstrated to deacetylate subunits of the ATP generating complex V, it does physically interact with complex V subunits (Law et al., 2009). The net effect of SirT3 deficiency on electron transport may result in reduced efficiency of electron transfer within the chain thereby increasing the probability of electrons being transferred to molecular oxygen to generate ROS at the expense of ATP production. The fact that SirT3 has apparently evolved multiple mechanisms to reduce ROS hints of a close relationship between this sirtuin, CR and aging.
Finally, SirT3 and Sirt4 are necessary for nutrient sensitive protection from genotoxic stress facilitated by increased mitochondrial NAD+ levels (Yang et al., 2007). In addition, SirT3 deacetylates cyclophilin D to inhibit apoptosis induced by opening of the mitochondrial permeability transition pore (Hafner et al., 2010). Reduction of the ROS burden coupled with an increase in stress resistance may maintain cells during CR, such as the cochlea cells of the inner ear.
SirT3, HIF-1α and cancer
The first indication that SirT3 may function as a tumor suppressor was the observation that SirT3−/− mice develop mammary tumors at 24-months of age (Kim et al., 2010). Consistent with the idea that SirT3 is a tumor suppressor, SirT3−/− mouse embryo fibroblasts (MEFs) were easier to transform (requiring a single oncogene) compared with wild type MEFs (which also require inactivation of a tumor suppressor gene). The SirT3−/− cells also showed increased superoxide levels and greater chromosome instability when stressed. All these findings suggested that SirT3 suppresses tumor initiation in vivo by preventing chromosomal instability (FIGURE 3A).
Figure 3.

SirT3 acts as a tumor suppressor through the regulation of ROS. (A) SirT3 suppresses mitochondrial-generated ROS to reduce DNA damage and genomic instability. This may be important in the initiation of tumorigenesis. (B) By suppressing mitochondrial derived ROS, SirT3 also inhibits a signaling cascade resulting in the stabilization/activation of HIF-1α protein and activation of transcriptional targets that promote aerobic glycolysis, angiogenesis, and the progression of tumorigenesis.
Additional insight into the role of SirT3 as a tumor suppressor came from two studies examining the relationship between SirT3 and the transcription factor hypoxia inducible factor, HIF-α. As mentioned above, ROS mediated stabilization of HIF-1α is one major adaptive response to hypoxia. In one study, the inactivation of SirT3 by shRNA in several tumor cell lines gave rise to higher ROS and HIF-1α activation in normoxia and hyper-activation of HIF-1α in hypoxic conditions. This is consistent with the idea that SirT3 suppresses ROS, which are necessary for the hypoxic induction of HIF-1α. Moreover, transgenic over-expression of SirT3 prevented the induction of HIF-1α by hypoxia (Bell et al., 2011). This finding is interesting because HIF-1α is known to enhance the growth of tumors by adapting them to hypoxic conditions which occur during tumor development (Majmundar et al., 2010). These adaptations include induction of aerobic glycolysis for biosynthetic intermediates (Warburg effect) (Vander Heiden et al., 2009), and induction of angiogenesis to bring additional glucose and oxygen to the tumor. In fact, xenografts of tumor cells with normal, reduced or elevated SirT3 levels were generated, and the tumors with reduced SirT3 were the largest, while the tumors with the over-expressed SirT3 were the smallest (Bell et al., 2011). Interestingly, antioxidant treatment was able to normalize the size of the tumors lacking SirT3. Moreover, inducing SirT3 expression after initiation of tumor formation sufficed to arrest tumor growth, suggesting that SirT3 is important not only in the initiation of tumors, as suggested above, but in the maintenance and progression of tumors (FIGURE 3B).
In the other study, SirT3−/− MEFs consumed more glucose, generated more lactate, had higher levels of glycolytic intermediates, and decreased levels of some TCA cycle intermediates (Finley et al., 2011). This data is consistent with altered cellular metabolism that is similar to the Warburg effect in cancer cells. Consistent with this metabolic reprogramming, transcriptional profiling of SirT3−/− MEFs demonstrated hyper activation of HIF-1α targets, which was reversed in the presence of shRNA’s targeting HIF-1α. In a complementary finding, overexpression of SirT3 reversed the Warburg effect in different breast cancer cell lines. The above data demonstrate that SirT3 suppresses the Warburg effect through the ROS-HIF-1α axis. In addition, the deacetylation of cyclophillin D by SirT3 may contribute to this suppression because it also promotes the dissociation of hexokinase II from mitochondria (Shulga et al., 2010), thereby decreasing the entry of glucose into glycolysis. It will be important to determine how generally SirT3-mediated control of ROS governs the Warburg effect in other kinds of tumors.
Finally, a broad survey indicated SirT3 gene deletion and reduction in protein levels in human breast and ovarian carcinomas (40%) and other tumors (20–30%) compared to non-cancer control (Finley et al., 2011; Kim et al., 2010). These data suggest that SirT3 is an important tumor suppressor in human cancers. While it has been suggested that aging may be the price for zealous tumor suppression early in life (eg. by p53) (Tyner et al., 2002), the above findings suggests that the tumor suppressor SirT3 involves no such trade off and actually links tumor suppression to a slowing of aging.
SirT3 and metabolism
As alluded to above, SirT3 promotes mitochondrial oxidative metabolism of amino acids and fatty acids during energy limitation. To wit, SirT3 mediated deacetylation and activation of Acetyl CoA synthase 2 (ACS2) allows incorporation of acetate into central metabolism, and deacetylation and activation of long chain acyl-CoA dehydrogenase (LCAD) promotes β-oxidation of fatty acids (Hirschey et al., 2010), which may be reinforced by SirT3 mediated regulation of the LKB1-AMPK axis (Pillai et al., 2010; Shi et al., 2010). Moreover, this could explain why SirT3 modulates susceptibility to lipotoxicity (Bao et al., 2010). Likewise, deacetylation of GDH allows amino acids to be converted into αKG for metabolism. A recent proteomics screen identified many potential SirT3 mitochondrial protein substrates, including ornithine transcarbamoylase (Hallows et al., 2011). Deacetylation of this urea cycle enzyme by SirT3 activates the urea cycle for disposal of ammonia when amino acids are catabolically stripped of their carbon. The fact that SirT5 was already known to deacetylate and activate another urea cycle enzyme, CPS1 (Nakagawa et al., 2009; Ogura et al., 2010), reinforces the importance of the urea cycle in sirtuin-mediated adaptations to energy limitation. Starvation also induces the liver to synthesize ketones to help bridge energy deficits for the brain, and SirT3 was also shown to deacetylate and activate 3-hydroxy-3-methylglutaryl CoA synthase 2 (Hmgcs2), the rate-limiting enzyme for the synthesis of the ketone, β-hydroxybuterate (Shimazu et al., 2010).
These metabolic adaptations all allow the organism to use fuels that might otherwise be stored (fat and amino acids) or ignored (acetate) and globally shift energy production away from carbohydrate catabolism. How might this be beneficial for health and longevity? It was suggested that garnering energy from fat might per se reduce ROS production (Guarente, 2008). This follows, because some of the electrons derived from oxidation of fatty acids feed into the electron transport chain via FADH2 (and not NADH) thus bypassing one of the sources of ROS production, complex I. Of course, this strategy does not bypass the other source of ROS production, complex III. However, the induction of SirT3 might do so, since this sirtuin appears to suppress ROS from complex III (Bell et al., 2011).
Do SirT1 and SirT3 cooperate in calorie restriction?
Given the strong data that both SirT1 and SirT3 are required for the responses to CR, is there any single, unifying pathway that links these two sirtuins? Interestingly, SirT3 expression has been linked to the activity of the coactivator PGC-1α via the estrogen related receptor alpha (ERRα), which binds to the SirT3 promoter (Giralt et al., 2011; Kong et al., 2010). SIRT1 has been convincingly shown to deacetylate PGC-1α to increase its potential to activate transcription (Nemoto et al., 2005; Rodgers et al., 2005). For example, in calorie restriction, SirT1 protein levels and NAD+ levels increase in muscle and white adipose tissue, and this results in an increase in mitochondrial biogenesis by the activated PGC-1α (Nisoli et al., 2005). It is thus tempting to draw a linear pathway in which CR triggers an increase in SirT1 activity, thereby activating PGC-1α and SirT3 expression (FIGURE 4). This strategy would coordinate the CR response between the nucleus and the mitochondria, and might be a core mechanism driving the anti-aging effects of this dietary regimen.
Figure 4.

Model by which SirT1 and SirT3 cooperate during calorie restriction. In conditions of decreased caloric intake, SirT1 protein levels and activity are increased, leading to the deacetylation and activation of PGC-1α. Transactivation of ERRα by PGC-1α increases transcription of SirT3, resulting in a decrease in ROS and the promotion of healthy aging.
However, it is also possible that the activity of SirT3 would be induced by an increase in the mitochondrial NAD+/NADH ratio during CR (Guarente, 2000; Nakagawa et al., 2009) even without an increase in the levels of this sirtuin. The observed increase in SirT3 protein levels may only magnify such an increase in activity. One test of the model in FIGURE 4 would be determining whether CR can induce expression of SirT3 in tissues knocked out for SirT1. If not, this would confirm the model linking SirT1 to SirT3 expression.
Conclusion
Sirtuins have been proposed to be regulators of aging and diseases of aging. For the nuclear SirT1, there is strong data supporting disease mitigation and slowing of aging by several criteria, although life span extension has not yet been observed. This latter finding may be because SirT1 has so many important functions in mammalian physiology that global up-regulation can exert opposing effects. Alternatively, global up-regulation may indeed slow aging globally, but not affect the proximal cause limiting life span in the mouse strains tested.
In contrast, SirT3 is a mitochondrial protein (Onyango et al., 2002; Schwer et al., 2002), and knockout mice present with no obvious phenotypes, at least as young animals. However, more subtle analyses have shown that CR induces SirT3 levels resulting in lower ROS and oxidative damage to mitochondria. So far, most of the evidence linking SirT3 to these processes involves loss of function, which triggers greater ROS, mitochondrial oxidative damage, and loss of protection by CR. Cell-based studies have recently provided the first evidence that gain of function has the potential to exert opposite effects (Bell et al., 2011). It will be important to bolster these studies with transgenic mice that exhibit increased SirT3 expression.
Interestingly, SirT3 is the only sirtuin for which a human polymorphism has been associated with extreme longevity. This allele is reported to create an enhancer in intron 5 of the SirT3 gene, and is highly enriched in a long-lived population in southern Italy (Bellizzi et al., 2009; Bellizzi et al., 2005; Rose et al., 2003). It will be important to carry out comprehensive analyses of SirT3 polymorphisms in other populations to secure the link between SirT3 and human longevity. Finally, a recent paper indicates that SirT3 is critical in protecting mouse preimplantation embryos against oxidative damage (Kawamura et al., 2010). Testing whether sirtuins play roles in early embryogenesis or even gametogenesis is fertile ground for study, on the hunch that anti-aging mechanisms protecting the soma of adults may also maintain youthfulness of the species in transitioning from one generation to the next.
Acknowledgments
Work from the authors lab is supported by grants from the NIH and the Glenn Foundation for Medical Research. We apologize to those whose work was not cited due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, Kon N, Knight C, Matsumoto M, Gutiérrez-Juárez R, Rossetti L, Gu W, Accili D. SirT1 Gain of Function Increases Energy Efficiency and Prevents Diabetes in Mice. Cell Metabolism. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free radical biology & medicine. 2010;49:1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Emerling BM, Ricoult SJH, Guarente L. SirT3 suppresses hypoxia inducible factor 1[alpha] and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011 doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GRS, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. The Journal of Cell Biology. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellizzi D, Covello G, Di Cianni F, Tong Q, De Benedictis G. Identification of GATA2 and AP-1 Activator elements within the enhancer VNTR occurring in intron 5 of the human SIRT3 gene. Molecules and cells. 2009;28:87–92. doi: 10.1007/s10059-009-0110-3. [DOI] [PubMed] [Google Scholar]
- Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, et al. A novel VNTR enhancer within the SIRT3 gene, a human homologue of SIR2, is associated with survival at oldest ages. Genomics. 2005;85:258–263. doi: 10.1016/j.ygeno.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C, Moffat C, Crawford S, Saliba S, Jardine K, et al. SirT1 Regulates Energy Metabolism and Response to Caloric Restriction in Mice. PloS one. 2008;3:e1759. doi: 10.1371/journal.pone.0001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell metabolism. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Chen D, Steele AD, Lindquist S, Guarente L. Increase in Activity During Calorie Restriction Requires Sirt1. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of Succinate Dehydrogenase Activity by SIRT3 in Mammalian Mitochondria. Biochemistry. 2009;49:304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen DE, Supinski AM, Bonkowski MS, Donmez G, Guarente LP. Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction. Genes & development. 2009;23:2812–2817. doi: 10.1101/gad.1839209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 Suppresses β-Amyloid Production by Activating the α-Secretase Gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Esposito F, Ammendola R, Faraonio R, Russo T, Cimino F. Redox Control of Signal Transduction, Gene Expression and Cellular Senescence. Neurochemical Research. 2004;29:617–628. doi: 10.1023/b:nere.0000014832.78725.1a. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley Lydia WS, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira Paula I, Cardoso Sandra M, Clish Clary B, et al. SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1[alpha] Destabilization. Cancer cell. 2011;19:416–428. doi: 10.1016/j.ccr.2011.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CK, Wright WE, Shay JW. Human diseases of telomerase dysfunction: insights into tissue aging. Nucleic Acids Research. 2007;35:7406–7416. doi: 10.1093/nar/gkm644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Hondares E, Villena JA, Ribas F, Díaz-Delfin J, Giralt M, Iglesias R, Villarroya F. PGC-1α controls the transcription of the SIRT3 gene, an essential component of the thermogenic brown adipocyte phenotype. Journal of Biological Chemistry. 2011 doi: 10.1074/jbc.M110.202390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes & development. 2000;14:1021–1026. [PubMed] [Google Scholar]
- Guarente L. Ageless Quest: One Scientist’s Search for the Genes That Prolong Youth. Cold Spring Harbor Laboratory Press; 2003. [Google Scholar]
- Guarente L. Mitochondria--A Nexus for Aging, Calorie Restriction, and Sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell metabolism. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging. 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy Andrew J, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic [beta] Cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annual Review of Pathology: Mechanisms of Disease. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallows WC, Yu W, Smith BC, Devires MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, et al. Sirt3 Promotes the Urea Cycle and Fatty Acid Oxidation during Dietary Restriction. Molecular Cell. 2011;41:139–149. doi: 10.1016/j.molcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: A Theory Based on Free Radical and Radiation Chemistry. 1956. pp. 298–300. [DOI] [PubMed] [Google Scholar]
- Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun. 2010;1:1–8. doi: 10.1038/ncomms1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Serrano M. SIRT1: recent lessons from mouse models. Nat Rev Cancer. 2010;10:819–823. doi: 10.1038/nrc2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S-i, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends in Pharmacological Sciences. 2010;31:212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes & development. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Uchijima Y, Horike N, Tonami K, Nishiyama K, Amano T, Asano T, Kurihara Y, Kurihara H. Sirt3 protects in vitro-fertilized mouse preimplantation embryos against oxidative stress-induced p53-mediated developmental arrest. J Clin Invest. 2010;120:2817–2828. doi: 10.1172/JCI42020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Molecular cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F, Chang Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PloS one. 2010;5:e11707. doi: 10.1371/journal.pone.0011707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume S, Uzu T, Horiike K, Chin-Kanasaki M, Isshiki K, Araki S-i, Sugimoto T, Haneda M, Kashiwagi A, Koya D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. The Journal of Clinical Investigation. 2010;120:1043–1055. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law IKM, Liu L, Xu A, Lam KSL, Vanhoutte PM, Che CM, Leung PTY, Wang Y. Identification and characterization of proteins interacting with SIRT1 and SIRT3: implications in the anti-aging and metabolic effects of sirtuins. PROTEOMICS. 2009;9:2444–2456. doi: 10.1002/pmic.200800738. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu W, McBurney MW, Longo VD. SirT1 Inhibition Reduces IGF-I/IRS-2/Ras/ERK1/2 Signaling and Protects Neurons. Cell Metabolism. 2008;8:38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Molecular and cellular biology. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Molecular Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-[alpha] activation. Cell metabolism. 2005;1:393–399. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI, Cuervo AM. Protein Homeostasis and Aging: Taking Care of Proteins From the Cradle to the Grave. 2009. pp. 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Liu Y, Van Remmen H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates Carbamoyl Phosphate Synthetase 1 and Regulates the Urea Cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. SIRT1 Functionally Interacts with the Metabolic Regulator and Transcriptional Coactivator PGC-1α. Journal of Biological Chemistry. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, et al. Calorie Restriction Promotes Mitochondrial Biogenesis by Inducing the Expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- Ogura M, Nakamura Y, Tanaka D, Zhuang X, Fujita Y, Obara A, Hamasaki A, Hosokawa M, Inagaki N. Overexpression of SIRT5 confirms its involvement in deacetylation and activation of carbamoyl phosphate synthetase 1. Biochemical and biophysical research communications. 2010;393:73–78. doi: 10.1016/j.bbrc.2010.01.081. [DOI] [PubMed] [Google Scholar]
- Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 2009;8:73–75. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervaiz S, Taneja R, Ghaffari S. Oxidative stress regulation of stem and progenitor cells. Antioxidants & redox signaling. 2009;11:2777–2789. doi: 10.1089/ars.2009.2804. [DOI] [PubMed] [Google Scholar]
- Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD Blocks Cardiac Hypertrophic Response via Activation of the SIRT3-LKB1-AMP-activated Kinase Pathway. 2010. pp. 3133–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell metabolism. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1[alpha] and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose G, Dato S, Altomare K, Bellizzi D, Garasto S, Greco V, Passarino G, Feraco E, Mari V, Barbi C, et al. Variability of the SIRT3 gene, human silent information regulator Sir2 homologue, and survivorship in the elderly. Experimental gerontology. 2003;38:1065–1070. doi: 10.1016/s0531-5565(03)00209-2. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. Embo J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CFW, Steegborn C. Substrates and Regulation Mechanisms for the Human Mitochondrial Sirtuins Sirt3 and Sirt5. Journal of Molecular Biology. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, et al. Extension of Murine Life Span by Overexpression of Catalase Targeted to Mitochondria. 2005. pp. 1909–1911. [DOI] [PubMed] [Google Scholar]
- Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Fan GQ, Xiao SD. SIRT3 reduces lipid accumulation via AMPK activation in human hepatic cells. Journal of digestive diseases. 2010;11:55–62. doi: 10.1111/j.1751-2980.2009.00416.x. [DOI] [PubMed] [Google Scholar]
- Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. The Journal of biological chemistry. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, et al. SIRT3 Deacetylates Mitochondrial 3-Hydroxy-3-Methylglutaryl CoA Synthase 2 and Regulates Ketone Body Production. Cell Metabolism. 2010;12:654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. Journal of cell science. 2010;123:894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 Mediates Reduction of Oxidative Damage and Prevention of Age-Related Hearing Loss under Caloric Restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et al. Sirt3-Mediated Deacetylation of Evolutionarily Conserved Lysine 122 Regulates MnSOD Activity in Response to Stress. Molecular cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang T-T, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. 2003. pp. 29–37. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E, Hirschey MD, Finley LWS, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends in Biochemical Sciences. 2010;35:669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan M, Kim SK, Berdichevsky A, Guarente L. A Role for SIR-2.1 Regulation of ER Stress Response Genes in Determining C. elegans Life Span. Developmental Cell. 2005;9:605–615. doi: 10.1016/j.devcel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Weindruch RaW, Roy L. The Retardation of Aging and Disease by Dietary Restriction. Charles C Thomas; 1988. [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Cimen H, Han M-J, Shi T, Deng J-H, Koc H, Palacios OM, Montier L, Bai Y, Tong Q, Koc EC. NAD+-dependent Deacetylase SIRT3 Regulates Mitochondrial Protein Synthesis by Deacetylation of the Ribosomal Protein MRPL10. 2010. pp. 7417–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]